

Abstract
Thorough screening of cancer-specific biomarkers, such as DNA mutations, can require large amounts of genomic material; however, the amount of genomic material obtained from some specimens (such as biopsies, fine-needle aspirations, circulating-DNA or tumor cells, and histological slides) may limit the analyses that can be performed. Furthermore, mutant alleles may be at low-abundance relative to wild-type DNA, reducing detection ability. We present a multiplex-PCR approach tailored to amplify targets of interest from small amounts of precious specimens, for extensive downstream detection of low-abundance alleles. Using 3 ng of DNA (1000 genome-equivalents), we amplified the 1 coding exons (2-11) of TP53 via multiplex-PCR. Following multiplex-PCR, we performed COLD-PCR (co-amplification of major and minor alleles at lower denaturation temperature) to enrich low-abundance variants and high resolution melting (HRM) to screen for aberrant melting profiles. Mutation-positive samples were sequenced. Evaluation of mutation-containing dilutions revealed improved sensitivities after COLD-PCR over conventional-PCR. COLD-PCR improved HRM sensitivity by approximately threefold to sixfold. Similarly, COLD-PCR improved mutation identification in sequence-chromatograms over conventional PCR. In clinical specimens, eight mutations were detected via conventional-PCR-HRM, whereas 12 were detected by COLD-PCR-HRM, yielding a 33% improvement in mutation detection. In summary, we demonstrate an efficient approach to increase screening capabilities from limited DNA material via multiplex-PCR and improve mutation detection sensitivity via COLD-PCR amplification.
A vast collection of markers have been identified as important determinants of cancer type, treatment course, acquired resistance, and survival. However, the ability to screen for an extensive panel of markers can be difficult when the specimen sample is low in DNA concentration or tumor abundance. For example, biopsies, fine needle aspirations, circulating tumor cells, plasma-circulating DNA, buccal swab specimens, filter paper-collected blood, and histological slide sections may contain important genetic markers; however, the DNA that can be retrieved from such samples may be low in concentration. As a result, the number of biomarkers that can be analyzed is limited and the amount of information that can be obtained is restricted. Whole genome amplification (WGA) is often used to overcome sample limitations.1–6 Although it is broadly useful, WGA often requires a minimum of 10 ng of DNA per reaction if one is to capture intratumoral heterogeneity and low-abundance events.7 For target-specific applications, multiplex-PCR pre-amplification of the target of interest is an alternative that has certain advantages over WGA, depending on subsequent sample analysis (discussed herein).
For downstream mutation screening, high resolution melting (HRM) curve analysis is simple, rapid, and inexpensive to perform,8–22 and exhibits high sensitivity for scanning for unknown low-abundance mutations and variants. The reported sensitivity of HRM is largely determined by fragment length, sequence composition, mutation identity, PCR quality, and equipment.8–22 Although recent publications report the ability to detect <1% mutant in wild-type DNA,23,24 most applications of HRM-based assays exhibit a detection capability of approximately 5% to 10% mutant among wild-type alleles.14,17,18,24–28
Although HRM mutation scanning is highly sensitive and efficient, HRM lacks the ability to identify the specific nucleotide change; this is a particularly important issue when mutations or variants are not known a priori and are likely to occur at any position within the amplicon sequence. Furthermore, HRM is generally more sensitive than conventional Sanger sequencing analysis of PCR amplicons; as such, discrete identification of low-abundance mutations that are unknown a priori requires additional analyses. Pyrosequencing could potentially be used to screen for low-abundance mutations (lower limit of 2% to 5%); however, it is most efficient in sequencing short reads of predefined regions or mutational hotspots. Confirmation by single-strand conformation polymorphism, denaturing gradient gel electrophoresis, or high pressure liquid chromatography (HPLC) fractionation, followed by either band selection or targeted collection of a specific peak, and final analysis via Sanger sequencing, may be performed, although these methodologies are often time-intensive. Similarly, more complex approaches such as digital PCR, single molecule sequencing, or cloning followed by sequencing22 could also be applied.
We recently demonstrated that amplifying DNA with COLD-PCR (co-amplification of major and minor alleles at lower denaturation temperature) before screening with HRM allows for the detection of low-abundance mutations with increased sensitivity and the ability to specifically identify the mutation via Sanger Sequencing.29 COLD-PCR is a recently-developed PCR-based approach to enrich low-abundance mutations and minor allele variants.30 COLD-PCR often reduces the limitations in low-abundance mutation detection and identification by enriching unknown mutations at any position within the amplicon through the use of a critical denaturation temperature during PCR amplification. The critical denaturation temperature (Tc) is lower than standard denaturation temperatures and preferentially denatures heteroduplexed molecules (those formed by hybridization of mutant and wild type sequences) and amplicons possessing mutations that lower the amplicon melting temperature (Tm), such as G:C>A:T or G:C>T:A. Minor allele enrichment by COLD-PCR has been demonstrated in combination with several downstream approaches such as Sanger sequencing,29–31 denaturing high-performance liquid chromatography/Surveyor (Transgenomic Inc., Omaha, NE),30 MALDI-TOF,30 Pyrosequencing,30,32 real-time TaqMan analyses,33 single-strand conformation polymorphism,34 PCR-based mutation-specific restriction enzyme digestion,35 and HRM.29,36 When combined with HRM, COLD-PCR can improve mutation detection and identification by approximately sixfold to 20-fold.29
In this study, we present a multiplex-PCR approach that can be specifically tailored to targets of interest and low-abundance alleles when DNA obtained from clinical samples is limited. The multiplex-PCR amplification that we present here is selected to amplify all exons and splice regions of the tumor suppressor gene TP53 in a single step. Subsequent application of COLD-PCR allows for the enrichment of low-abundance mutations, increasing the sensitivity of the assay and the accuracy of mutation detection. We demonstrate that subsequent screening of COLD-PCR amplicons with HRM allows for the identification of aberrant melting profiles in a rapid and inexpensive manner29; downstream sequencing of these products allows for the discrete identification of individual mutations.
Materials and Methods
DNA and Tumor Samples
Genomic DNA from cell lines with defined TP53 mutations (T47D, SNU-182, HCC2157, MDA-MB-435, and HCC2218; Table 1) was purchased from American Type Culture Collection (ATCC); the cell line SW480 was purchased from ATCC and genomic DNA was extracted from cultured cells. Genomic male DNA (Promega Corporation, Madison, WI) served as the wild-type control.
Table 1.
Cell-Line DNA Used to Evaluate the Efficacy of COLD-PCR Enrichment
| TP53 | Amplicon | Cell-line | Mutation (nt) | Mutation (aa) |
|---|---|---|---|---|
| Exon 6 | 6A | T47D | c.580C>T | p.L194F |
| Exon 7 | 7 | HCC2157 | c.742C>T | p.R248W |
| Exon 8 | 8A | MDA-MB-435 | c.797G>A | p.G266E |
| Exon 8 | 8B | HCC2218 | c.847C>T | p.R283C |
| Exon 9 | 9 | SW480 | c.925C>T | p.P309S |
Clinical colorectal (N = 12) and glioma tumor tissue specimens (N = 12), snap-frozen in liquid nitrogen within 1 to 2 hours following surgery, were obtained from the Massachusetts General Hospital Tumor Bank and from the Division of Neurosurgery at the Beth Israel – Deaconess Medical Center, respectively. Paired formalin-fixed paraffin-embedded (FFPE) histology slides were also obtained for the glioblastoma specimens. Patient specimens were used according to Internal Review Board approval and standards.
Following manual macrodissection of the tissue, genomic DNA was isolated from the colorectal specimens using the DNeasy tissue kit (Qiagen Inc., Valencia, CA) according to manufacturer's instructions. Genomic DNA quality was assessed and quantified using the NanoDrop1000 spectrophotometer (Thermo Scientific, Inc., Wilmington, DE); purified DNA was stored at −80°C.
Glioblastoma tumors were independently analyzed from both frozen and FFPE forms. For each, histological analysis of slides was completed before tissue selection and genomic extraction. Frozen glioblastoma tumor specimens were embedded in Tissue-Tek Optimal Cutting Temperature (OCT) Compound (Sakura Finetek Inc., Torrance, CA). The OCT compound is composed of water soluble glycol and resins, and is an appropriate specimen matrix for cryostat sectioning at temperatures of −10°C and lower. At the Brigham and Women's Hospital, Specialized Histology Longwood Core facility, several 4 μmol/L and 10 μmol/L sections were cut from the OCT-embedded tissue for (1) staining with hematoxylin and eosin, and (2) genomic DNA extractions, respectively. Stained sections were examined and the percent tumor content was determined. Samples containing ≥70% to 80% tumor content were selected for analysis. OCT was removed via three washes with 5 ml of phosphate buffered saline (PBS) before isolating genomic DNA with the DNeasy tissue kit (Qiagen Inc.).
For the FFPE glioblastoma tumor specimens, approximately five 4 μmol/L sections were selected from unstained histological slides. Genomic DNA was isolated from the FFPE sections using a QIAamp DNA FFPE tissue kit (Qiagen Inc.) following manufacturer's protocols. For both sets of genomic DNA (from frozen and FFPE specimens), quality was assessed and quantified using the NanoDrop1000 spectrophotometer (ThermoFisher Scientific, Inc.). Purified DNA was stored at −80°C.
The protocol presented below was first validated for varying amounts of snap-frozen tumor DNA as starting material. To determine and assure method sensitivity, the assays were evaluated using three different starting amounts of DNA, 30 ng, 10 ng, and 3 ng. For each starting amount, serial dilutions were created; multiplex-PCR was performed, followed by conventional PCR and COLD-PCR in separate analyses, and finally screened via HRM. The amplification was robust in all cases (data not shown). As clinical patient samples are precious and often in limited amounts, we proceeded to develop the approach using as little DNA as necessary. For all remaining reactions, 3 ng of DNA (∼1000 genome equivalents) was used.
Multiplex-PCR for TP53
Multiplex-PCR for TP53 was designed using guidelines presented by Fredriksson et al.37 Seven primer sets were used to amplify all exons and splice regions of TP53 (Table 2). Amplifications (reaction volume 15 μL) were performed on a MasterCycler EP (Eppendorf Inc., Hauppauge, NY) with thermocycling conditions performed as described in Table 3. Multiplex-PCR was performed using a high-fidelity polymerase (Phusion DNA Polymerase; Thermo Scientific Inc., Lafayette, CO) to prevent PCR error as follows: 1X manufacturer-supplied high-fidelity buffer, 2.0 mmol/L MgCl2, 0.2 mmol/L dNTPs, 0.1 μmol/L each primer (14 in total), 5 U/μL Phusion high fidelity polymerase, and 3 ng of DNA. Exonuclease I (1 μL) was added to each multiplex-PCR product and incubated at 37°C for 30 minutes, followed by 95°C for 5 minutes, to degrade any remaining unincorporated primers.
Table 2.
Multiplex TP53 Primer Sequences from Fredriksson et al.37
| Exon | Sequence | Size (bp) | Tm°C | Tc°C | Ta°C |
|---|---|---|---|---|---|
| Multiplex primers | |||||
| 2-3 | Forward-5′-ATGCTGGATCCCCACTTTTC-3′⁎ | 350 | |||
| Reverse-5′-GACCAGGTCCTCAGCCC-3′⁎ | |||||
| 4 | Forward-5′-GACAAGGGTTGGGCTGG-3′⁎ | 486 | |||
| Reverse-5′-CCAAAGGGTGAAGAGGAATC-3′⁎ | |||||
| 5-6 | Forward-5′-TCTTTGCTGCCGTCTTCC-3′⁎ | 517 | |||
| Reverse-5′-AGGGCCACTGACAACCAC-3′⁎ | |||||
| 7 | Forward-5′-TGCTTGCCACAGGTCTCC-3′⁎ | 235 | |||
| Reverse-5′-GTCAGAGGCAAGCAGAGGC-3′⁎ | |||||
| 8-9 | Forward-5′-GGACAGGTAGGACCTGATTTCC-3′⁎ | 441 | |||
| Reverse-5′-AAACAGTCAAGAAGAAAACGGC-3′⁎ | |||||
| 10 | Forward-5′-AACTTGAACCATCTTTTAACTCAGG-3′⁎ | 243 | |||
| Reverse-5′-GGAATCCTATGGCTTTCCAAC-3′⁎ | |||||
| 11 | Forward-5′-AGGGGCACAGACCCTCTC-3′⁎ | 222 | |||
| Reverse-5′-AGACCCAAAACCCAAAATGG-3′⁎ | |||||
| Nested primers | |||||
| 2 | Forward-5′-GCAGCCAGACTGCCTTCCG-3′ | 134 | 90.6 | 89.6 | 57 |
| Reverse-5′-GTGGGCCTGCCCTTCCAAT-3′ | |||||
| 3 | Forward-5′-gtaaaacgacggccagtTGGGACTGACTTTCTGCT-3′† | 108 | 86.8 | 85.9 | 58 |
| Reverse-5′-tcccgcgaaattaatacgacGCCCAACCCTTGTCCTTA-3′† | |||||
| 4a | Forward-5′-GTCCTCTGACTGCTCTTTTCACCC-3′ | 181 | 90.8 | 89.8 | 60 |
| Reverse-5′-GGTGTAGGAGCTGCTGGTGC-3′ | |||||
| 4b | Forward-5′-CCCGTGGCCCCTGCACC-3′ | 186 | 88.8‡ | 87.8 | 70 |
| Reverse-5′-AGCCAGCCCCTCAGGGCAA-3′ | |||||
| 5a | Forward-5′-TGTGCCCTGACTTTCAACTCTGTCTC-3′ | 120 | 88.8 | 87.7 | 68 |
| Reverse-5′-GGGTGTGGAATCAACCCACAGC-3′ | |||||
| 5b | Forward-5′-TTCCACACCCCCGCCCG-3′ | 148 | 94.5 | 93.5 | 63 |
| Reverse-5′-GCCCTGTCGTCTCTCCAGCC-3′ | |||||
| 6a | Forward-5′-GCCTCTGATTCCTCACTGATTG-3′ | 129 | 86.9 | 86.0 | 57 |
| Reverse-5′-TAGGGCACCACCACACTATG-3′ | |||||
| 6b | Forward-5′-TGCGTGTGGAGTATTTGGATGAC-3′ | 105 | 88.5 | 87.5 | 57 |
| Reverse-5′-CCCTCCTCCCAGAGACCC-3′ | |||||
| 7 | Forward-5′-CCAAGGCGCACTGGCCTCA-3′ | 185 | 91.0 | 90.0 | 57 |
| Reverse-5′-GCCAGTGTGCAGGGTGGCAA-3′ | |||||
| 8a | Forward-5′-TGCCTCTTGCTTCTCTTTTC-3′† | 128 | 88.8 | 88.0 | 57 |
| Reverse-5′-CTTTCTTGCGGAGATTCTCTTC-3′† | |||||
| 8b | Forward-5′-GAACAGCTTTGAGGTGCGTGTTT-3′ | 155 | 91.4 | 90.5 | 57 |
| Reverse-5′-TGGTCTCCTCCACCGCTTC-3′ | |||||
| 9 | Forward-5′-GGTGCAGTTATGCCTCAGAT-3′ | 176 | 87.2 | 86.1 | 57 |
| Reverse-5′-GTTAGACTGGAAACTTTCCACTTGATA-3′ | |||||
| 10 | Forward-5′-ATATACTTACTTCTCCCCCTCCTCTGTTGC-3′ | 172 | 92.8 | 91.8 | 70 |
| Reverse-5′-TAGGGCCAGGAAGGGGCTGA-3′ | |||||
| 11 | Forward-5′-CTCACTCATGTGATGTCATCTCT-3′ | 165 | 88.1 | 87.2 | 57 |
| Reverse-5′-GGGAGGCTGTCAGTGGG-3′ |
Nested primers were used to amplify the length of each TP53 exon via both conventional and COLD-PCR. Nested Exon 3 primers were modified to include M13 and Tag1 linker sequences (lower case font) in order to increase the product length and allow sequence analysis. The annealing (Ta), amplicon melting temperature (Tm), and critical denaturation temperature (Tc) of COLD-PCR are presented for PCR reactions performed with the Phusion high-fidelity polymerase system and a 1X concentration of LCGreen+ dye (Idaho Technologies Inc.). When necessary (Exons 4, 5, 6, and 8), two amplicons were analyzed in order to screen the entire length of the exon; primer sets are differentiated by “a” and “b” labels.
Fredriksson et al.37
Bastien et al.23
DMSO was added at 5% to lower the amplicon melting temperature.
Table 3.
PCR Thermocycling Conditions
| PCR type | Step | Conditions |
|---|---|---|
| Multiplex-PCR | Initial denaturation | 98°C for 30 s |
| Thermocycling: 35 cycles | 98°C for 10 s | |
| 55°C for 20 s | ||
| 72°C for 20 s | ||
| Extension | 72°C for 15 s | |
| Nested conventional PCR | Initial denaturation | 98°C for 30 s |
| Thermocycling: 35 cycles | 98°C for 10 s | |
| Ta⁎ for 30 s | ||
| 72°C for 10 s | ||
| Nested fast-COLD-PCR | Initial denaturation | 98°C for 30 s |
| Stage 1 cycling: 5 cycles | 98°C for 10 s | |
| Ta⁎ for 20 s, fluorescent reading | ||
| 72°C for 10 s | ||
| Stage 2 cycling: 20 cycles | Tc⁎ for 10 s | |
| Ta⁎ for 20 s, fluorescent reading | ||
| 72°C for 10 s | ||
| Stage 3 cycling: 10 cycles | 98°C for 10 s | |
| Ta⁎ for 20 s, fluorescent reading | ||
| 72°C for 10 s | ||
| Melting curve | Ramping 0.2°C/s, 60°C to 90°C |
Annealing temperatures and critical denaturation temperatures are defined in Table 2.
To determine the sensitivity of our approach, 3 ng genomic DNA from cell lines (T47D, SNU-182, HCC2157, MD-MBA-435, HCC2218, and SW480) was serially diluted into wild-type DNA and amplified by the TP53 multiplex-PCR approach. These sets of serial dilutions served for estimating method sensitivity in exons 6, 7, 8, and 9. The specific mutant fractions examined were: 0.1%, 0.25%, 0.5%, 1.0%, 2.0%, 3.0%, 4.0%, 5.0%, 6.0%, 8.0%, and 10% mutant-to-wild-type ratios. In addition, several replicates of wild-type human genomic DNA (0% mutant; male G1471, Promega Corp) were amplified and evaluated in parallel. Serial dilutions of the mutant cell lines were used in downstream assays to validate method sensitivity and enrichment selectivity. Additionally, 3 ng of colon tumor (N = 12), as well as paired frozen and FFPE glioblastoma tumor (N = 12 for each respectively) genomic DNA were amplified by the TP53 multiplex-PCR method, along with additional genomic male reference DNA (n = 6). Subsequently, conventional PCR amplification was applied for each TP53 exon.
Conventional PCR-HRM and Sequence Analysis
The 10 coding exons (exons 2 through 11) of TP53 were amplified in 14 individual reactions from the multiplex-PCR products (Table 2). The primers selected amplified each exon of TP53 including the intron-exon splicing regions. Two amplicons were required to span the entire length of the exon and splice region of four exons (exons 4, 5, 6, and 8). Primers were designed or selected23 such that the PCR amplicons would possess a single melting domain, amplify efficiently, and generate amplicons of less than 200 bp in length.
Conventional PCR amplicons were amplified on a MasterCycler using thermocycling conditions as defined in Table 3. Amplifications were completed in triplicate to ensure reproducibility. PCR reactions were performed using 1X manufacturer-supplied high-fidelity buffer, 1.5 mmol/L MgCl2, 0.2 mmol/L dNTPs, 0.2 μmol/L primers, 1.0X LCGreen+ dye (Idaho Technologies Inc., Salt Lake City, UT), 5 U/μL Phusion high fidelity polymerase, and 1 μL of a 200-fold diluted multiplex-PCR product. Amplified products were evaluated by electrophoresis on an agarose gel with ethidium-bromide staining to confirm robust amplification of the correct length.
To evaluate the sensitivity of our approach, and to screen for the presence of mutations, approximately 10 μL of each PCR product, with a 20 μL mineral oil overlay, was subjected to HRM analysis on the LightScanner HR96 system (Idaho Technologies Inc.). Melting curves were analyzed via the LightScanner software with Call-IT 2.0 (Idaho Technologies Inc.) to discern the presence of a mutation. Products amplified from the colorectal and glioblastoma tumor specimens were directly compared with each other and with several wild-type, genomic male DNA reference samples in parallel. Samples exhibiting aberrant melting profiles were subsequently subjected to Exonuclease I/shrimp alkaline phosphatase digestion and sequenced at the Dana-Farber Cancer Institute, Molecular Biology Core Facility. Sequence chromatograms were evaluated using BioEdit biological sequence alignment editor (http://www.mbio.ncsu.edu/BioEdit/BioEdit.html, May 2005).
COLD-PCR-HRM and Sequence Analysis
The same 14 regions were amplified in the serial dilution and tumor sample sets using COLD-PCR. To define the critical denaturation temperatures for COLD-PCR of a given amplicon, a melting curve was first evaluated for a wild-type sample amplified via conventional PCR, followed by melting curve analysis to identify the experimentally-derived amplicon melting temperature (Tm); the melting curve analysis was performed post-PCR on the SmartCycler (Cepheid Inc., Sunnyvale, CA) using LCGreen+ as a real-time intercalating dye. The critical denaturation temperatures (Tc) were defined following the general rule: Tc = Tm − 1°C. Defining the Tc in this manner resulted in robust PCR amplification and also demonstrated excellent mutation enrichment.
COLD-PCR was performed on a SmartCycler II (Cepheid Inc.). Because the critical denaturation temperature during COLD-PCR has to be controlled precisely (eg, to within ±0.2°C), it is important to use a thermocycler with high temperature precision. The Cepheid SmartCycler II enables temperature calibration of each individual well, thereby ensuring minimal well-to-well temperature variation and satisfactory reproducibility in the mutation enrichment obtained from replicate samples and across all wells. COLD-PCR thermocycling was performed as described in Table 3. A melting analysis was performed as the last step of the thermocycling on the Cepheid SmartCycler to ensure strong amplification before further HRM evaluation.
COLD-PCR reactions were performed using 1X manufacturer-supplied high-fidelity buffer, 1.5 mmol/L MgCl2, 0.2 mmol/L dNTPs, 0.2 μmol/L primers, 1.0X LCGreen+dye (Idaho Technologies Inc.) 5 U/μL Phusion (Thermo Scientific Inc.) high-fidelity polymerase, and 1 μL of a 200-fold diluted multiplex-PCR product. As with conventional PCR, 10 μL of each COLD-PCR product, with a 20 μL mineral oil overlay, was subjected to high-resolution melting on the LightScanner HR96 system (Idaho Technologies Inc.). Melting curves were analyzed via the LightScanner software with Call-IT 2.0 to discern the presence of a mutation. HRM melt profiles were directly compared with the wild-type reference genomic DNA. HRM mutation detection sensitivity was compared between conventional and COLD-PCR. Aberrant melting profiles were subsequently analyzed via Sanger sequencing to identify variants.
Independent Verification by Restriction Enzyme Analysis
In scenarios when both the HRM results and the sequence analysis reveal very low-abundance mutations, representing the borderline limit of the application, independent confirmation by an alternative approach should be performed via restriction enzyme digestion. After a specific nucleotide change is putatively identified via COLD-PCR-sequencing, a sequence-specific reaction can be customized and performed. In this investigation, we independently confirmed a G>T mutation (c.782 + 1G>T) in exon 7 of specimen CT11. A PspGI recognition site (CCWGG) is present in the wild-type sequence; however, the presence of a mutation in that recognition sequence prevents digestion of the mutant strands (eg, a “CCATG” sequence string characterizes the same region in CT11). As such, on digestion by the restriction enzyme, the wild-type sequences are digested and the mutant sequences remain intact. We thus used this PCR – restriction fragment length polymorphisms (PCR-RFLP) approach to independently verify the mutation in sample CT11.
In this evaluation, both genomic DNA and multiplex-amplified product were evaluated for the colon tumor specimen CT11 and wild-type DNA. The aforementioned 185-bp target region was PCR amplified using the primers defined in Table 2. The 185 bp amplicon (4 μL) was incubated with five units of the restriction enzyme PspGI (New England Biolabs, Ipswich, MA) and 1X restriction buffer #2 (New England Biolabs), at 75°C for 2 hours. Resulting digested product was used directly in a subsequent nested conventional PCR, as described above (forward primer 5′-CCTCATCTTGGGCCTGTGTTAT-3′; reverse primer 5′-TGTGCAGGGTGGCAAGT-3′; annealing temperature 57°C), to yield a 166-bp product. The 166 bp amplicon was subsequently Sanger sequenced, and the mutation was verified by visual analysis of the chromatogram.
Results
Multiplex-PCR and Conventional PCR Validation
Following multiplex-PCR, each of the 14 regions of interest, spanning the coding and splice regions of exons 2 through 11, were amplified via conventional PCR for validation. Amplicons were visualized via agarose gels with ethidium-bromide staining and robust amplification of the correct length was confirmed for each target region (see Supplemental Figure S1 at http://jmd.amjpathol.org). The amplification of the 14 TP53 regions of interest was robust in all cases.
Mutation Detection Sensitivity via Serial-Dilution Analysis
PCR amplicons (conventional PCR and COLD-PCR) of mutant serial dilutions for varying amounts of template DNA were analyzed by HRM to determine mutation detection sensitivity. The sensitivity was comparable for each of three starting amounts (30 ng, 10 ng, and 3 ng). As this approach is focused on the amplification of small amounts of DNA from precious samples, we proceeded with 3 ng of DNA in all subsequent analyses. Our results for the serial dilution analysis of 3 ng of DNA are presented below.
HRM analysis of 3 ng serial dilutions of mutation-containing human cancer cell lines diluted into wild-type human genomic DNA revealed increased mutation-detection sensitivity in products of COLD-PCR amplification over conventional PCR amplification. After PCR amplification and HRM analysis of human cancer cell-line T47D serial dilutions in wild-type DNA, fluorescence difference curve plots relative to wild-type DNA were generated using the LightScanner Call-IT 2.0 software (Idaho Technologies Inc.). Conventional PCR-HRM was performed in parallel for comparison with COLD-PCR-HRM. After conventional-PCR, the exon 6 (129-bp amplicon) T47D cell-line mutation (c.580C>T, p.L194F) remains clearly differentiated from the wild-type amplicons down to 2% to 3%. In comparison, after COLD-PCR, a 0.5% mutation-abundance of T47D diluted in wild-type DNA can be differentiated from the wild-type DNA melting curves. Thus, for the 129-bp exon 6 amplicon, HRM analysis of COLD-PCR products provides an approximate fourfold to sixfold improvement in mutation detection over conventional-PCR-HRM (see Supplemental Figure S2 at http://jmd.amjpathol.org). To determine the degree of mutant enrichment and the ability to identify the mutation through downstream applications, Sanger sequencing of the low-abundance mutation was performed. Despite the inherent sensitivity of HRM screening, a mutant concentration of 10% cannot be identified in the sequence chromatograms of the conventional PCR. In comparison, after COLD-PCR amplification and sequencing, the 5% T47D mutation-abundance can be identified (see Supplemental Figure S2 at http://jmd.amjpathol.org), and results in an approximate ∼35% mutation abundance by examining the sequencing chromatograms.
Similarly, for the exon 7 (185 bp) amplicon, after conventional-PCR, the human cell-line HCC2157 mutation (c.742C>T, p.R248W) can be differentiated from the wild-type amplicons down to 3%. In comparison, after COLD-PCR, a 0.5% mutation-abundance of HCC2157 diluted in wild-type DNA remains clearly differentiated from the wild-type DNA melting curves. Thus, for the 185 bp exon 7 amplicon, COLD-PCR-HRM results in an approximate sixfold improvement in mutation detection over conventional-PCR-HRM (Figure 1). In Sanger sequence chromatograms, a mutant concentration of 5% is not discernable; however, after COLD-PCR amplification and sequencing, the 3% HCC2157 mutation abundance can be clearly identified (Figure 1), and results in an approximately 50% mutation abundance after COLD-PCR enrichment.
Figure 1.
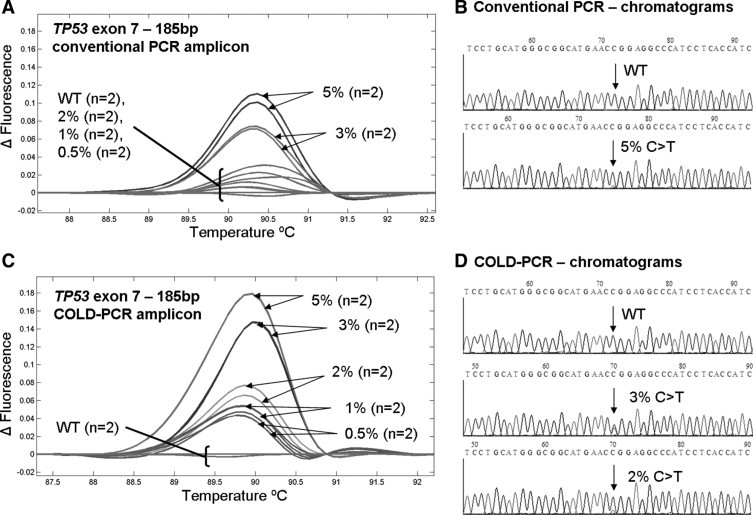
A 185-bp amplicon of TP53 exon 7 was analyzed via HRM after conventional PCR (A) and COLD-PCR (C). Amplicons were subsequently sequenced (Sanger, sense strand). While a 5% mutant abundance of the HCC2157 cell line (C>T) is unreliable in the chromatogram of conventional PCR amplicons (B), a 3% mutant abundance is easily visible in the chromatogram of COLD-PCR (D). Wild-type (WT) sequences are presented for comparison.
Analysis of the exon 8−128 bp amplicon demonstrates that HRM screening of the cell line MD-MBA-435 (mutation c.797G>A, p.G266E) after conventional PCR can reliably detect a mutation above 5%. In comparison, after COLD-PCR, a 2% mutation abundance of MD-MBA-435 diluted in wild-type DNA can be differentiated from wild-type DNA melting curves. Thus, for the exon 8−128 bp amplicon, COLD-PCR-HRM analysis provides an approximate threefold improvement in detection sensitivity over conventional-PCR-HRM (see Supplemental Figure S3 at http://jmd.amjpathol.org). In Sanger sequence chromatograms, a mutant concentration of 5% is not discernable; however, after COLD-PCR amplification and sequencing, the 5% MD-MBA-435 mutation abundance can be clearly identified (see Supplemental Figure S3 at http://jmd.amjpathol.org).
Analysis of the exon 8−155 bp amplicon reveals that HRM screening of the cell-line HCC2218 mutation (c.847C>T, p.R283C) after conventional-PCR can detect a mutation abundance of 5%. However, after COLD-PCR amplification, a 1% mutation abundance of HCC2218 diluted in wild-type DNA can be clearly differentiated from wild-type DNA melting curves. Thus, for the 155-bp exon 8 amplicon, COLD-PCR-HRM analysis results in an approximate fivefold improvement in detection sensitivity over HRM analysis of conventional PCR amplicons (see Supplemental Figure S4 at http://jmd.amjpathol.org). In Sanger sequence chromatograms, a mutant concentration of 5% is not reliably detected; however, after COLD-PCR amplification and sequencing, the 5% HCC2218 mutation-abundance can be clearly identified (see Supplemental Figure S4 at http://jmd.amjpathol.org).
Tumor Mutation Scanning
Mutations and single-nucleotide polymorphisms (SNPs) were detected by HRM analysis of both conventional and COLD-PCR amplicons (Table 4) in all coding exons excluding exons 3, 10, and 11. Additionally, mutations and SNPs were detected in the intronic splice sites between exons 7-8, exons 8-9, and exons 9-10. Mutations were detected in 75% and 42% of the colorectal and glioblastoma specimens examined, respectively; 25% and 33% of the colorectal and glioblastoma specimens examined contained more than one mutation in TP53. Twenty-four mutations were identified (Table 4): 14 (58%) were missense mutations, three (12.5%) were deletions resulting in frameshifts, two (8.3%) were found in splice regions, and five were intronic (21%).
Table 4.
Mutational Status of CT and CMK-T Specimens in Exons 2–11 of TP53
| Tumor specimen | TP53 exon | Variant (nt) | Protein change | Result | MPLX + HRM- conv. PCR | MPLX + HRM-COLD-PCR | Chromatogram conv. PCR | Chromatogram COLD-PCR | Matched normal conv./COLD-PCR | Chromatogram genomic DNA conv. PCR | Chromatogram genomic DNA COLD-PCR |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CMK-T7 | 2 | c.40del2 | p.L14del2 | Frameshift (novel) | Y | Y | Y | Y | |||
| CT12 | 4 | c.108G>A | p.P36P | SNP- synonymous | Y | Y | Y | Y | Y | ||
| CMK-T3 | 4 | c.108G>A | p.P36P | SNP- synonymous | Y | Y | Y | Y | |||
| CMK-T12 | 4 | c.108G>A | p.P36P | SNP- synonymous | Y | Y | Y | Y | |||
| CMK-T13 | 4 | c.108G>A | p.P36P | SNP- synonymous | Y | Y | Y | Y | |||
| CMK-T14 | 4 | c.108G>A | p.P36P | SNP- synonymous | Y | Y | Y | Y | |||
| CMK-T1 | 5 | c.403del3 | p.C135del3 | Frameshift (novel) | Y | Y | Y | Y | |||
| CT6 | 5 | c.476C>T | p.A159V | Missense | Y | Y | Y | ||||
| CT2 | 5 | c.523C>A | p.R175S | Missense | Undetectable | Y | Undetectable | Y | WT/WT | Undetectable | Y |
| CT10 | 5 | c.524G>T | p.R175K | Missense | Y | Y | Y | ||||
| CT17 | 5 | c.524G>A | p.R175H | Missense | Y | Y | Y | ||||
| CT20 | 5 | c.527G>T | p.C176F | Missense | Y | Y | Y | Y | WT/MUTANT | Y | Y |
| CMK-T3 | 6 | c.568C>T | p.P190L | Missense | Y | Y | Y | ||||
| CMK-T5 | 6 | c.610del3 | p.E204del3 | Frameshift | Y | Y | Y | ||||
| CMK-T12 | 6 | c.639A>G | p.R213R | SNP- synonymous | Y | Y | Y | ||||
| CMK-T16 | 7 | c.712T>G | p.C238W | Missense | Y | Y | Y | ||||
| CT2 | 7 | c.739A>T | p.N247I | Missense | Y | Y | Y | ||||
| CT11 | 7-intronic | c.782 + 1G>T | Splice | Undetectable | Y | Undetectable | Y | WT/WT | Undetectable | Y | |
| CT10 | 8 | c.817C>T | p.R273C | Missense | Y | Y | Y | ||||
| CMK-T7 | 8 | c.817C>T | p.R273C | Missense | Y | Y | Y | ||||
| CT20 | 8 | c.818G>A | p.R273H | Missense | Undetectable | Y | Undetectable | Y | WT/WT | Undetectable | Y |
| CT5 | 8 | c.853G>A | p.E285K | Missense | Y | Y | Y | ||||
| CMK-T16 | 8 | c.916C>T | p.R306X | Nonsense | Y | Y | Y | ||||
| CT20 | 9 | c.925C>T | p.P309S | Missense | Undetectable | Y | Undetectable | Y | WT/WT | Undetectable | Y |
| CT12 | 9-intronic | c.993 + 12T>C | Intronic | Y | Y | Y | Y | ||||
| CMK-T3 | 9-intronic | c.993 + 12T>C | Intronic | Y | Y | Y | Y | ||||
| CMK-T12 | 9-intronic | c.993 + 12T>C | Intronic | Y | Y | Y | Y | ||||
| CMK-T13 | 9-intronic | c.993 + 12T>C | Intronic | Y | Y | Y | Y | ||||
| CMK-T14 | 9-intronic | c.993 + 12T>C | Intronic | Y | Y | Y | Y | ||||
| CT4 | 9-intronic | c.994-1G>C | Splice | Y | Y | Y |
Mid- to high-abundance mutants and SNPs could be detected in HRM analysis of both conventional (conv.) and COLD-PCR amplicons (denoted “Y”) amplified from the multiplex-PCR product; however, four low-abundance mutations (in bold) were not detected by HRM analysis of conventional PCR (denoted “undetectable”), though could be detected in COLD-PCR amplicons (denoted “Y”). Sanger sequence analysis was used to identify the mutations in the product amplified from the multiplex-PCR. Furthermore, low-abundance mutations, and additional mutations of interest, were secondarily sequenced from amplified genomic DNA.
CT, colorectal tumor; CMK-T, glioblastoma tumor.
Seven and eight moderate abundance mutations were detected in the conventional amplicons of the glioblastoma and colorectal tumor specimens, respectively. Although no low-abundance mutations were detected after COLD-PCR amplification of glioblastoma specimens, four low-abundance mutations were detected after COLD-PCR amplification of the colorectal specimens. COLD-PCR increased the mutation detection of HRM by 33% in the colorectal specimens. Low-abundance mutations were confirmed in COLD-PCR amplifications from the genomic DNA; evaluation of the matched normal genomic DNA was performed simultaneously.
Four low-abundance mutations were detected in exons 5, 8, and 9 of colorectal tumor specimens. Colorectal tumor specimen CT20 possessed two low-abundance mutations (exons 8 and 9) in addition to another heterozygous mutation in exon 5. Exon 8 exhibited a G>A mutation at the commonly mutated codon 273; and results in a missense protein change from arginine to histidine. This low-abundance mutation was detectable only in COLD-PCR amplicons (Figure 2). Sequence chromatograms of the COLD-PCR amplicons from both the multiplex-PCR product as well as the genomic DNA were used to identify and confirm the mutation; the mutation was not present in the matched normal amplicon from genomic DNA. Similarly a second low-abundance mutation was detected by HRM for CT20 in exon 9 in the COLD-PCR amplicons only (Figure 3, sequence of antisense strand presented). This CT20 mutation is a C>T mutation at codon 309 and results in a missense protein change from proline to serine; the mutation was not present in the matched normal amplicon from genomic DNA. The exon 5 mutation for CT20 is, in contrast, a heterozygous G>T mutation (missense p.C176F), and can be detected in both conventional and COLD-PCR amplicons (Figure 4). However, this mutation is unique in that it is detected in the matched normal sample when amplified by COLD-PCR; the mutation was not observed in sequence chromatograms from the genomic male control sample amplified in parallel (Figure 4). Although located close to a mutational hotspot associated with germline Li-Fraumeni syndrome,38 this mutation (c.527G>T, missense p.C176F) is not documented as a common germline mutation in the IARC TP53 database (http://www-p53.iarc.fr/, December 2009). A potential explanation for the occurrence of this mutation is that the matched normal tissue may not be purely normal (due to the macrodissection); this tissue may contain tissue from the tumor margin.
Figure 2.
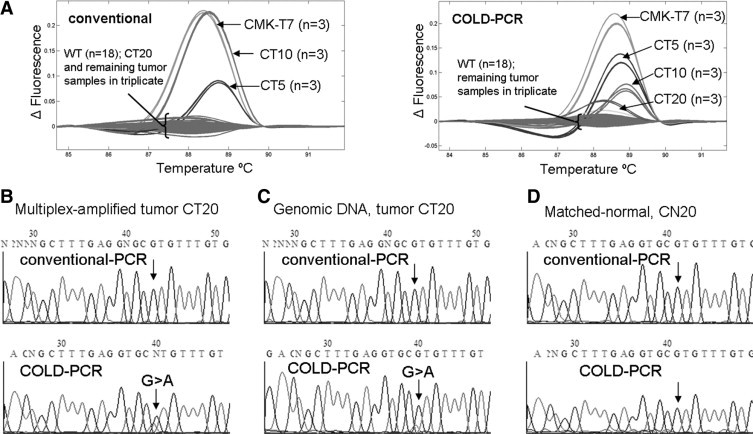
In the exon 8 (128 bp) amplicon, a low-abundance mutation in a colorectal tumor sample (CT20) was detected after analyzing COLD-PCR amplicons with HRM (A); however, this mutation was not detected in conventional PCR amplicons. An additional three samples possessed mid- to high-abundance mutations detectable by HRM screening of both conventional and COLD-PCR amplicons. For the exon 8 (128 bp) amplicon, Sanger sequencing analysis (sense strand) of the multiplexed-PCR product for CT20 revealed a G>A mutation in the sequence chromatograms of COLD-PCR (B); however, this mutation was not detected in conventional PCR amplicons. The mutation was confirmed in subsequent analysis of the COLD-PCR-amplified genomic DNA (C); however, the mutation was not observed in either the conventional PCR amplicon or the matched normal sample (D).
Figure 3.
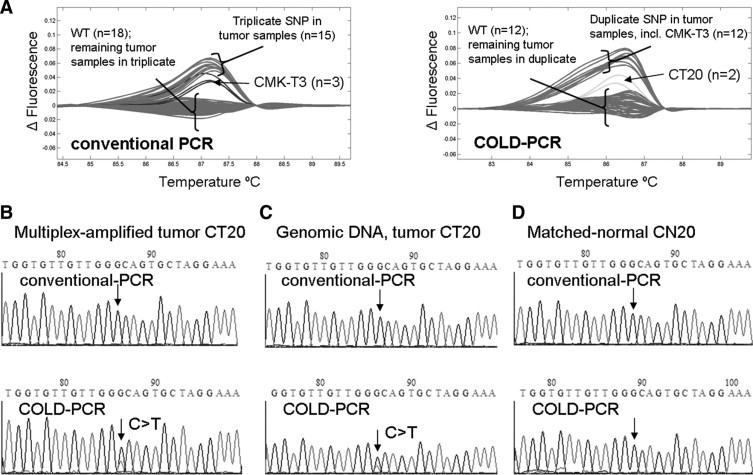
In the exon 9 amplicon, a low abundance mutation in a colorectal tumor sample (CT20) was detected after analyzing COLD-PCR amplicons with HRM (A); however, this mutation was not detectable in conventional PCR amplicons. An additional six samples possessed mid- to high-abundance SNPs detectable by HRM screening of both conventional and COLD-PCR amplicons. For the exon 9 amplicon, Sanger sequencing analysis of the multiplexed-PCR product for CT20 revealed a C>T mutation in the sequence chromatograms of COLD-PCR (B, anti-sense strand sequencing presented); however, this mutation was not detectable in conventional PCR amplicons or the matched normal sample (D). The mutation was confirmed in subsequent analysis of the COLD-PCR amplified genomic DNA (C).
Figure 4.
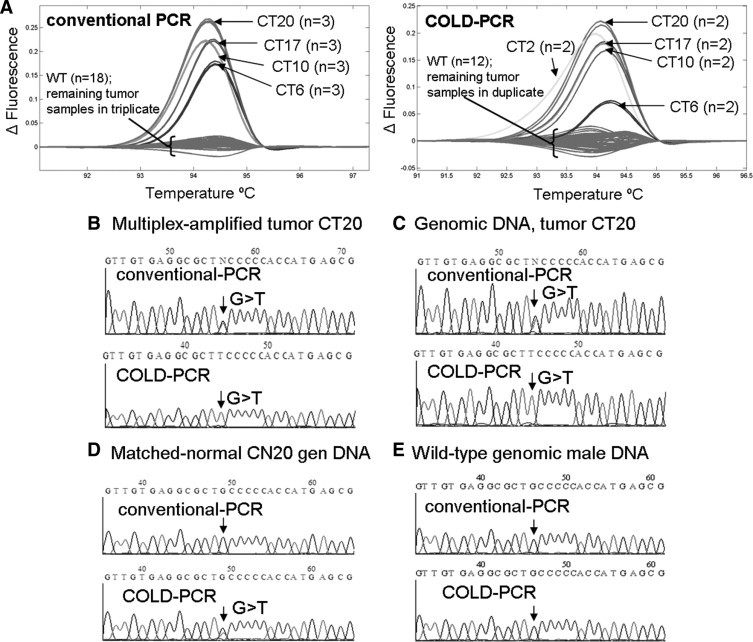
In the exon 5 (148bp) amplicon, several mid- to high- abundance mutations were detected in the tumor samples, including colorectal tumor sample CT20. Additionally, after analyzing COLD-PCR amplicons with HRM (A), a low-abundance mutation was detected in CT2; however, this mutation was not detectable in conventional PCR amplicons. A mid- to high-abundance mutation was detected in CT20 by HRM for both PCR amplicons of the exon 5 148bp amplicon (in addition to the previously discussed low-abundance mutations in exons 8 and 9). Sanger sequencing analysis (sense strand) of the multiplexed-PCR product (B) and genomic DNA (C) for CT20 revealed a heterozygous G>T mutation in conventional PCR amplicons; the T allele was enriched during COLD-PCR. The mutation was also detected in the COLD-PCR amplification of the matched normal tissue (D), suggesting that the putatively normal sample may contain tissue from the tumor margin. This G>T is unlikely to be artifact of COLD-PCR as it is not observed in the wild-type (genomic human male) amplicons (E).
A low-abundance mutation was detected by HRM for CT2 in the COLD-PCR amplicons of exon 5 (148 bp amplicon) (see Supplemental Figure S5 at http://jmd.amjpathol.org). This CT2 mutation (c.523C>A, p.R175S) results in a missense protein change from arginine to serine; the mutation was not present in the matched normal amplicon from genomic DNA. CT2 contains another mutation in exon 7 which is a heterozygous A>T mutation (c.739A>T, p.N247I missense mutation), and can be detected in both conventional and COLD-PCR amplicons (see Supplemental Figure S5 at http://jmd.amjpathol.org).
The last low-abundance mutation was detected in the intronic spice region between exons 7 and 8 in colorectal tumor specimen CT11. After COLD-PCR amplification, a G>T mutation (c.782 + 1G>T) was detected by HRM via the Call-IT 2.0 software (in triplicate) and observed in sequence chromatograms of both the multiplex-PCR product and the independent analysis of the genomic DNA (see Supplemental Figure S6 at http://jmd.amjpathol.org); the mutation was not observed in either the conventional PCR amplicons or the matched normal sample. However, due to the very low abundance of the mutation observed via both HRM and Sanger sequencing, the CT11 mutation is close to the sensitivity limits of this method. The HRM differentiation was quite small, and the enriched mutant fraction via COLD-PCR was quite low. We performed an independent verification of this approach using a restriction enzyme (PspGI) to digest the wild-type amplicons and effectively enrich the mutant fraction following a first conventional PCR (PCR-RFLP). A second, nested PCR and sequence analysis of the digested product was then performed in parallel on both the multiplex-amplified CT11 DNA and the genomic CT11 DNA, as well as both multiplex-amplified and genomic wild-type DNA. The analysis revealed clearly the G>T mutation (c.782 + 1G>T) at an appreciably enriched level in CT11, while the wild-type sequence remained without evidence of a mutation (see Supplemental Figure S7 at http://jmd.amjpathol.org).
Two novel mutations, both heterozygous deletions, were detected in two glioblastoma tumor specimens. They were detected in both the conventional and COLD-PCR amplicons by HRM, and identified via Sanger sequencing. CMK-T7 exhibits a 2-bp deletion in exon 2 (p.L14del2, c.40del2) and the CMK-T1 3-bp deletion is in exon 5 (p.C135del3, c.403del3). Sequence chromatograms are presented in Supplemental Figure S8 (http://jmd.amjpathol.org) for products amplified from both the multiplex-PCR and the original genomic DNA. These mutations are not documented in either the IARC TP53 database (http://www-p53.iarc.fr/, December 2009), the TP53–free database (http://p53.free.fr/Database/p53_database.html, November 2008), or the COSMIC database (http://www.sanger.ac.uk/genetics/CGP/cosmic/, May 2010).
Lastly, a panel of 12 FFPE glioblastoma tissue sections were evaluated and compared with the matched panel of DNA specimens prepared from frozen specimens. For each of the 14 amplicons, amplified by both conventional PCR and COLD-PCR, there was 100% concordance in the HRM mutation detection analysis. A representative result demonstrating this concordance is represented in Supplemental Figure S9 (see http://jmd.amjpathol.org).
Discussion
Many molecular applications and their respective depth of analysis can be limited by low-yield specimen samples. The application of a targeted multiplex-PCR on low-yield precious specimen samples generates high quantities of template that can be used in downstream applications, allowing for an in-depth analysis of mutation spectra. Primers used in multiplex-PCR can be selected for particular regions of interest. For example, Fredriksson et al37 amplified the entire coding sequence of ten human cancer genes in one assay, generating an extensive template panel for biomarker screening. Here we demonstrate a multiplex-PCR tailored for the amplification of exons 2-11 of TP53 that can be performed on as little as 3 ng of DNA. The merging of multiplex-PCR with downstream COLD-PCR reactions not only increases the number of downstream assays that can be performed, but simultaneously allows for mutation and minority allele enrichment from as little as 1000 genome equivalents. Subsequent amplicon analysis with HRM creates a high-throughput, efficient, and inexpensive screening method to detect mutant-containing amplicons.
The use of multiplex-PCR as a pre-amplification step, as opposed to whole-genome pre-amplification approaches,1–6 enables better mutation enrichment during subsequent COLD-PCR amplification. Thus, enriching the target(s) of interest during the multiplex-PCR pre-amplification simplifies the target and allows COLD-PCR to be initiated after only five cycles of the conventional PCR mode, thereby enabling more cycles in COLD-PCR mode, and subsequently increasing the enrichment potential. However, it is important to limit the number of PCR cycles that are performed to exclude the possibility for polymerase-introduced errors. Though PCR errors were not observed in this investigation, such errors in principle can occur if an excessive number of PCR cycles are performed. To reduce the probability of errors, we use the highest-fidelity polymerase commercially available (Phusion DNA Polymerase; Thermo Scientific Inc.), and we analyze multiple wild-type control samples within all evaluations.
For this particular study and method development, we elected to use fast-COLD-PCR29,30,39,40 to enrich mutations. Fast-COLD PCR will enrich only those mutations that result in decreasing the melting temperature (Tm) of a particular amplicon. We chose to use the fast-COLD approach for several reasons. The majority of human TP53 mutations are Tm-reducing,41 a bias which putatively reflects the methylation of 5′-CpG-3′ dinucleotides.42,43 Fast-COLD-PCR assays can be run in less time, and products yield robust enrichment. Full-COLD PCR is another COLD-PCR format that can be used in place of fast-COLD-PCR to enrich all mutation types; however, in its original format, full-COLD-PCR requires much longer hybridization times and the achievable enrichment is typically lower than fast-COLD-PCR. Nevertheless, improved full-COLD-PCR protocols that generate better enrichment are currently under development in our laboratory. These will allow faster full-COLD-PCR reactions that can be used in the present approach to identify all possible mutation types.
Twenty-four clinical tumor specimens were evaluated in this study; 12 colorectal tumor and 12 glioblastoma tumor specimens. The 12 glioblastoma samples were evaluated using matched DNA isolated from frozen tissue as well as FFPE histological slide sections. FFPE preserved tumor tissues are widely available and used for large-scale clinical investigations; however, it is well known that the FFPE preservation method may result in high levels of DNA damage and induced errors. As such, the analysis of low-abundance mutations in such low-quality DNA has the potential to yield false-positive results. In this evaluation, we compared mutation detection in frozen and FFPE glioblastoma sections, and recovered 100% concordance across all 14 targets amplified by conventional and COLD-PCR.
Although these FFPE-based results are promising, one must keep in mind that COLD-PCR is designed to enrich low-abundance variants. As such, an error induced by FFPE preservation may be enriched in the same manner as a true mutation. Therefore, the methodology presented herein provides an efficient and sensitive approach for large-scale screening. However, after an aberrant HRM melting curve is detected, it is important to analyze FFPE-preserved specimens with a secondary independent method to prevent false-positive results.
Within the glioblastoma specimens analyzed, seven mutations and nine SNPs were detected via conventional PCR-HRM screening; however, no additional low-abundance mutations were detected after amplification with COLD-PCR. Although still unresolved,44 it was reported that TP53 mutation is an early event in the progression of gliomas.45–48 Similarly, it has been observed that the total number and the frequency of nucleic mutations and variants are substantially reduced in gliomas when compared against other cancers such as colorectal, breast, and pancreatic.49 In reference to an early study,50 Parsons et al49 attribute the fact that fewer cell generations occur in glial cells before neoplasia inception as a potential explanation for a relatively small number of genetic aberrations in gliomas. As such, one might anticipate finding fewer low-abundance mutations in the glioma tumor specimens compared to the colorectal tumor specimens.
Eight mutations were detected in the conventional PCR-HRM screening of the colorectal tumor specimens compared to 12 mutations via COLD-PCR-HRM screening, allowing us to detect an additional four mutations and demonstrating a 33% increase in the mutation detection of colorectal tumor mutations. For each of the four low-abundance mutations detected in COLD-PCR products amplified from the multiplex-PCR-template, we confirmed the presence of the mutation in the genomic DNA. Previous analysis30 of TP53 exon 8 in the specimen CT20 has validated the low-abundance mutation by independent diagnostic approaches such as restriction fragment length polymorphism analysis.
Unlike the glioma tumor specimens, the percent tumor content of the colorectal tumor specimens was not confirmed by histopathology before genomic DNA extraction, so we cannot rule out the possibility that stromal contamination may reduce the relative mutation concentration. However, the data indicate that stromal contamination is at most a minor contribution in these specimens as other high-abundance mutations were detected in these same specimens that contain also the low-level mutations. For example, in two of the samples within which low-abundance mutations were detected (CT20 and CT2), a high-abundance mutation was also detected in exons 5 and 7, respectively. A likely explanation for the observed variability in the colorectal tumor specimens is that TP53 mutational events occur throughout the course of cancer progression, resulting in intratumoral heterogeneity. Another explanation would be that the mutation does not possess a strong selective advantage and, therefore, it is present in only a low-abundance relative to the other alleles. Recent investigations51–54 of colorectal adenocarcinoma specimens have reported intratumoral heterogeneity in mutations of KRAS, BRAF, P1K3CA, transforming growth factor b type II receptor (TGFBRII), insulin-like growth factor II receptor (IGFIIR), BAX, MSH3, and MSH6, as well as TP53. Supporting our observations, Losi et al52 and Giaretti et al54 observed TP53 intratumoral heterogeneity in up to 59% of the colorectal adenocarcinoma specimens they evaluated, and they report that TP53 were late events in the progression of colorectal adenocarcinoma.
Using this approach, we demonstrate that low-abundance mutations that may be missed by conventional assays can be detected for several genetic regions of interest without the normal limitations presented by small sample size or low-concentration specimens. The clinical significance of low-abundance mutations remains largely undetermined at this time, and will be highly variable depending on tumor type or stage. It is likely that some mutations will remain insignificant for tumor development and treatment (passenger mutations), whereas others will demonstrate a strong selective advantage throughout the course of disease progression and/or treatment (driver mutation).55–59 We are particularly interested in evaluating low-abundance mutations that fall into the driver mutation category because they hold the potential to influence and determine patient response to treatment, disease progression and evolution, and ultimately survival.
We elected to evaluate TP53 because the mutational type and position often define their prognostic value.60 Certain TP53 mutations have been associated with poor prognosis for several cancers60,61 and loss of TP53-function correlates with the onset of multidrug resistance.62 By using multiplex PCR as a preliminary template construction tool, COLD-PCR for mutation enrichment, and HRM as a scanning method, only those amplicons with aberrant HRM profiles require downstream processing and analysis (such as Sanger sequencing). The downstream analysis load is reduced appreciably while a great number of targets can be rapidly and efficiently analyzed. The present multiplex COLD-PCR-HRM approach provides a convenient and low-cost method for the detection and identification of low-level somatic mutations in tumors.
Conclusions
Using a multiplex-PCR approach37 to amplify from small amounts (eg, 3 ng, 1000 genome equivalents) of DNA provides a large quantity of DNA template for downstream applications. The multiplex PCR can be tailored to specific regions of interest (ie, tumor suppressor and oncogenic gene panels) for the analysis of precious DNA such as fine needle aspirations, circulating-DNA, or tumor cells where extensive molecular testing is difficult due to the small amount of starting material. The use of downstream COLD-PCR before HRM increases the mutation scanning sensitivity and allows low-abundance mutations to be enriched to levels that can be easily identified in Sanger sequence chromatograms. We demonstrate that the use of COLD-PCR increased the mutations detected in colorectal adenocarcinoma specimens by 33% and increased the sensitivity of HRM by approximately threefold to sixfold.
Acknowledgments
We thank Dr. Jin Li for his insight and critique throughout the course of this project.
Footnotes
Supported by grants T32-CA009078 from the NCI (C.A.M.), and NIH grants CA-138280 and CA-111994.
The contents of this manuscript are the responsibility of the authors and do not necessarily represent the official views of the NIH.
Supplemental material for this article can be found at http://jmd.amjpathol.org or at doi:10.1016/j.jmoldx.2010.10.008.
Supplementary data
Representative amplicons generated for each of the 14 regions of interest following the initial TP53 multiplex PCR.
A 129 base pair amplicon of TP53 exon 6 was analyzed via HRM after conventional PCR (A) and COLD-PCR(C). Conventional-PCR amplicons of the 2% mutant mixture were scored as “unknown” by the Call-IT software, indicating a potential presence of variants that differ from wild-type. Amplicons were subsequently sequenced (Sanger; anti-sense strand presented). While a 10% abundance of the T47D mutant cell line (c.580C>T, p.L194F) cannot be observed in the chromatogram of conventional-PCR amplicons (B), a 5% mutant abundance is easily visible in the chromatogram of COLD-PCR (D). Wild-type (WT) sequences are presented for comparison.
A 128 base pair amplicon of TP53 exon 8 was analyzed via HRM after conventional PCR (A) and COLD-PCR (C). Amplicons were subsequently sequenced (Sanger; anti-sense strand presented). While a 5% mutant abundance of the MDMBA-435 cell-line (c.797G>A, p.G266E) cannot be detected in the chromatogram of conventional-PCR amplicons (B), a 5% mutant abundance is easily visible in the chromatogram of COLD-PCR (D). Wild-type (WT) sequences are presented for comparison.
A 155 base pair amplicon of TP53 exon 8 was analyzed via HRM after conventional PCR (A) and COLD-PCR (C). Amplicons were subsequently sequenced (Sanger; anti-sense strand presented). While a 5% mutant abundance of the HCC2218 cell-line (c.847 C>T, p.R283C) is unreliable in the chromatogram of conventional-PCR amplicons (B), a 3% mutant abundance is easily visible in the chromatogram of COLD-PCR (D). Wild-type (WT) sequences are presented for comparison.
For the exon 5 (148bp) amplicon, Sanger sequencing analysis (sense strand) of the multiplexed-PCR product for CT2 revealed a low-abundance C>A mutation (c.523C>A, p.R175S) in the sequence chromatograms of COLD-PCR (A); however, this mutation could not be detected in conventional PCR amplicons by either HRM or sequencing. The mutation was confirmed in subsequent analysis of the COLD-PCR amplified genomic DNA (B); however, the mutation was not observed in the matched normal sample (C). A second mutation A>T (c.739A>T, p.N247I) was detected by HRM in CT2 in the exon 7 conventional PCR amplicon and identified via Sanger sequencing.
In the exon 7 amplicon, a low abundance mutation in a colorectal tumor sample (CT11) was detected after HRM analysis of COLD-PCR amplicons (A); however, this mutation was not detected in conventional PCR amplicons. An additional two samples possessed mid- to high-abundance mutations detectable by HRM screening of both conventional and COLD-PCR amplicons. For the exon 7 amplicon, Sanger sequencing analysis (sense strand) of the multiplexed-PCR product for CT11 revealed a low-abundance G>T mutation (c.782+1G>T) in the sequence chromatograms of COLD-PCR (B); however, this mutation could not be detected in conventional PCR amplicons by either HRM or sequencing. The mutation was confirmed in subsequent analysis of the COLD-PCR amplified genomic DNA (C); however, the mutation was not observed in the matched normal sample (D).
Genomic DNA and multiplex-amplified template, of specimen CT11 and wild-type DNA, were PCR amplified (TP53 exon 7, 185 bp) and subjected to PspGI digestion (sequence region containing the recognition site, CCWGG, is underlined). Digested product was subsequently re-amplified (TP53 exon 7, 166 bp) and analyzed via Sanger sequencing (sense strand). The G>T mutation is clearly enriched in both the CT11 genomic DNA (A) and multiplex-amplified product (B); yet remains absent in the respective wild-type control samples (C and D).
Two novel deletions were detected in the glioma tumor samples. (A) For the exon 2 amplicon, Sanger sequencing analysis (sense strand) of the multiplexed-PCR and genomic DNA amplicons for CMK-T7 revealed a previously undocumented two base pair deletion (c.40del2, p.L14del2). The deletion (CT) was located in codon 14, and results in a frameshift. (B) For the exon 5 (120bp) amplicon, Sanger sequencing analysis (anti-sense strand) of the multiplexed-PCR and genomic DNA amplicons for CMK-T1 revealed a previously undocumented three base pair deletion (c.403del3, p.C135del3). The deletion (TTG) was located at codon 135, and results in a frameshift.
Comparative analysis of the frozen (panels A and B) and FFPE (panels C and D) specimens revealed concordance. Representative results are presented for TP53 exon 8, 155 bp amplicon. HRM difference plots demonstrate the CMK-16 nonsense mutation (p.R306X, c.916C>T), represented by the red melting curve as amplified by both conventional (panels A and C) and COLD-PCR (panels B and D) relative to wild-type melting curves (gray).
References
- 1.Hosono S., Faruqi A.F., Dean F.B., Du Y., Sun Z., Wu X., Du J., Kingsmore S.F., Egholm M., Lasken R.S. Unbiased whole-genome amplification directly from clinical samples. Genome Res. 2003;13:954–964. doi: 10.1101/gr.816903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dean F.B., Hosono S., Fang L., Wu X., Faruqi A.F., Bray-Ward P., Sun Z., Zong Q., Du Y., Du J., Driscoll M., Song W., Kingsmore S.F., Egholm M., Lasken R.S. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci USA. 2002;99:5261–5266. doi: 10.1073/pnas.082089499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hughes S., Arneson N., Done S., Squire J. The use of whole genome amplification in the study of human disease. Prog Biophys Mol Biol. 2005;88:173–189. doi: 10.1016/j.pbiomolbio.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Wang G., Maher E., Brennan C., Chin L., Leo C., Kaur M., Zhu P., Rook M., Wolfe J.L., Makrigiorgos G.M. DNA amplification method tolerant to sample degradation. Genome Res. 2004;14:2357–2366. doi: 10.1101/gr.2813404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li J., Harris L., Mamon H., Kulke M., Liu W., Zhu P., Makrigiorgos G.M. Whole genome amplification of plasma-circulating DNA enables expanded screening for allelic imbalance in plasma. J Mol Diagn. 2006;8:22–30. doi: 10.2353/jmoldx.2006.050074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang G., Brennan C., Rook M., Wolfe J.L., Leo C., Chin L., Pan H., Liu W.H., Price B., Makrigiorgos G.M. Balanced-PCR amplification allows unbiased identification of genomic copy changes in minute cell and tissue samples. Nucleic Acids Res. 2004;32:e76. doi: 10.1093/nar/gnh070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim E.H., Zhang S.-L., Li J.-L., Yap W.-S., Howe T.-C., Tan B.-P., Lee Y.-S., Wong D., Khoo K.-L., Seto K.-Y., Tan L., Agasthian T., Koong H.-N., Tam J., Tan C., Caleb M., Chang A., Ng A., Tan P. Using whole genome amplification (WGA) of low-volume biopsies to assess the prognostic role of EGFR, KRAS, p53, and CMET mutations in advanced-stage non-small cell lung cancer (NSCLC) J Thorac Oncol. 2009;4:12–21. doi: 10.1097/JTO.0b013e3181913e28. [DOI] [PubMed] [Google Scholar]
- 8.Wittwer C.T., Reed G.H., Gundry C.N., Vandersteen J.G., Pryor R.J. High-resolution genotyping by amplicon melting analysis using LCGreen. Clin Chem. 2003;49:853–860. doi: 10.1373/49.6.853. [DOI] [PubMed] [Google Scholar]
- 9.Reed G.H., Wittwer C.T. Sensitivity and specificity of single-nucleotide polymorphism scanning by high-resolution melting analysis. Clin Chem. 2004;50:1748–1754. doi: 10.1373/clinchem.2003.029751. [DOI] [PubMed] [Google Scholar]
- 10.Vaughn C.P., Elenitoba-Johnson K.S.J. High-resolution melting analysis for detection of internal tandem duplications. J Mol Diagn. 2004;6:211–216. doi: 10.1016/S1525-1578(10)60512-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou L., Wang L., Palais R., Pryor R., Wittwer C.T. High-resolution DNA melting analysis for simultaneous mutation scanning and genotyping in solution. Clin Chem. 2005;51:1770–1777. doi: 10.1373/clinchem.2005.054924. [DOI] [PubMed] [Google Scholar]
- 12.Herrmann M.G., Durtschi J.D., Bromley L.K., Wittwer C.T., Voelkerding K.V. Amplicon DNA melting analysis for mutation scanning and genotyping: cross-platform comparison of instruments and dyes. Clin Chem. 2006;52:494–503. doi: 10.1373/clinchem.2005.063438. [DOI] [PubMed] [Google Scholar]
- 13.De Leeneer K., Coene I., Poppe B., De Paepe A., Claes K. Rapid and sensitive detection of BRCA1/2 mutations in a diagnostic setting: comparison of two high-resolution melting platforms. Clin Chem. 2008;54:982–989. doi: 10.1373/clinchem.2007.098764. [DOI] [PubMed] [Google Scholar]
- 14.Polakova K.M., Lopotova T., Klamova H., Moravcova J. High-resolution melt curve analysis: initial screening for mutations in BCR-ABL kinase domain. Leuk Res. 2008;32:1236–1243. doi: 10.1016/j.leukres.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 15.Seipp M.T., Pattison D., Durtschi J.D., Jama M., Voelkerding K.V., Wittwer C.T. Quadruplex genotyping of F5. F2, and MTHFR variants in a single closed tube by high-resolution amplicon melting. Clin Chem. 2008;54:108–115. doi: 10.1373/clinchem.2007.097121. [DOI] [PubMed] [Google Scholar]
- 16.Smith G.D., Chadwick B.E., Willmore-Payne C., Bentz J.S. Detection of epidermal growth factor receptor gene mutations in cytology specimens from patients with non-small cell lung cancer utilising high-resolution melting amplicon analysis. J Clin Path. 2008;61:487–493. doi: 10.1136/jcp.2007.051425. [DOI] [PubMed] [Google Scholar]
- 17.Vandersteen J.G., Bayrak-Toydemir P., Palais R.A., Wittwer C.T. Identifying common genetic variants by high-resolution melting. Clin Chem. 2007;53:1191–1198. doi: 10.1373/clinchem.2007.085407. [DOI] [PubMed] [Google Scholar]
- 18.Takano T., Ohe Y., Tsuta K., Fukui T., Sakamoto H., Yoshida T., Tateishi U., Nokihara H., Yamamoto N., Sekine I., Kunitoh H., Matsuno Y., Furuta K., Tamura T. Epidermal growth factor receptor mutation detection using high-resolution melting analysis predicts outcomes in patients with advanced non small cell lung cancer treated with gefitinib. Clin Cancer Res. 2007;13:5385–5390. doi: 10.1158/1078-0432.CCR-07-0627. [DOI] [PubMed] [Google Scholar]
- 19.Monis P.T., Giglio S., Saint C.P. Comparison of SYTO9 and SYBR Green I for real-time polymerase chain reaction and investigation of the effect of dye concentration on amplification and DNA melting curve analysis. Anal Biochem. 2005;340:24–34. doi: 10.1016/j.ab.2005.01.046. [DOI] [PubMed] [Google Scholar]
- 20.Gudnason H., Dufva M., Bang D.D., Wolff A. Comparison of multiple DNA dyes for real-time PCR: effects of dye concentration and sequence composition on DNA amplification and melting temperature. Nucl Acids Res. 2007;35:e127. doi: 10.1093/nar/gkm671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erali M., Voelkerding K.V., Wittwer C.T. High resolution melting applications for clinical laboratory medicine. Exp Mol Pathol. 2008;85:50–58. doi: 10.1016/j.yexmp.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vossen R., H A.M., Aten E., Roos A., den Dunnen J.T. High-Resolution Melting Analysis (HRMA) - more than just sequence variant screening. Hum Mutat. 2009;30:860–866. doi: 10.1002/humu.21019. [DOI] [PubMed] [Google Scholar]
- 23.Bastien R., Lewis T.B., Hawkes J.E., Quackenbush J.F., Robbins T.C., Palazzo J., Perou C.M., Bernard P.S. High-throughput amplicon scanning of the TP53 gene in breast cancer using high-resolution fluorescent melting curve analyses and automatic mutation calling. Hum Mutat. 2008;29:757–764. doi: 10.1002/humu.20726. [DOI] [PubMed] [Google Scholar]
- 24.Nomoto K., Tsuta K., Takano T., Fukui T., Yokozawa K., Sakamoto H., Yoshida T., Maeshima A.M., Shibata T., Furuta K., Ohe Y., Matsuno Y. Detection of EGFR mutations in archived cytologic specimens of non-small cell lung cancer using high-resolution melting analysis. Am J Clin Pathol. 2006;126:608–615. doi: 10.1309/N5PQNGW2QKMX09X7. [DOI] [PubMed] [Google Scholar]
- 25.Krypuy M., Newnham G.M., Thomas D.M., Conron M., Dobrovic A. High resolution melting analysis for the rapid and sensitive detection of mutations in clinical samples: KRAS codon 12 and 13 mutations in non-small cell lung cancer. BMC Cancer. 2006;6:295. doi: 10.1186/1471-2407-6-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Do H., Krypuy M., Mitchell P.L., Fox S.B., Dobrovic A. High resolution melting analysis for rapid and sensitive EGFR and KRAS mutation detection in formalin fixed paraffin embedded biopsies. BMC Cancer. 2008;8:142. doi: 10.1186/1471-2407-8-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krypuy M., Ahmed A.A., Etemadmoghadam D., Hyland S.J., deFazio A., Fox S.B., Brenton J.D., Bowtell D.D., Dobrovic A. High resolution melting for mutation scanning of TP53 exons 5-8. BMC Cancer. 2007;7:168. doi: 10.1186/1471-2407-7-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fukui T., Ohe Y., Tsuta K., Furuta K., Sakamoto H., Takano T., Nokihara H., Yamamoto N., Sekine I., Kunitoh H., Asamura H., Tsuchida T., Kaneko M., Kusumoto M., Yamamoto S., Yoshida T., Tamura T. Prospective study of the accuracy of EGFR mutational analysis by high-resolution melting analysis in small samples obtained from patients with non-small cell lung cancer. Clin Cancer Res. 2008;14:4751–4757. doi: 10.1158/1078-0432.CCR-07-5207. [DOI] [PubMed] [Google Scholar]
- 29.Milbury C.A., Li J., Makrigiorgos G.M. COLD-PCR-enhanced high-resolution melting enables rapid and selective identification of low-level unknown mutations. Clin Chem. 2009;55:2130–2143. doi: 10.1373/clinchem.2009.131029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li J., Wang L., Mamon H., Kulke M.H., Berbeco R., Makrigiorgos G.M. Replacing PCR with COLD-PCR enriches variant DNA sequences and redefines the sensitivity of genetic testing. Nat Med. 2008;14:579–584. doi: 10.1038/nm1708. [DOI] [PubMed] [Google Scholar]
- 31.Li J., Milbury C.A., Li C., Makrigiorgos G.M. Two-round coamplification at lower denaturation temperature-PCR (COLD-PCR)-based sanger sequencing identifies a novel spectrum of low-level mutations in lung adenocarcinoma. Hum Mutat. 2009;30:1583–1590. doi: 10.1002/humu.21112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zuo Z., Soape M., Doan S., Chandra P., Galbincea J., Barkoh B., Luthra R. Application of COLD-PCR for improved detection of KRAS mutations in clinical samples. Mod Pathol. 2009;22:1773. doi: 10.1038/modpathol.2009.59. [DOI] [PubMed] [Google Scholar]
- 33.Li J., Wang L., Janne P.A., Makrigiorgos G.M. Coamplification at lower denaturation temperature-PCR increases mutation-detection selectivity of TaqMan-based real-time PCR. Clin Chem. 2009;55:748–756. doi: 10.1373/clinchem.2008.113381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maekawa M., Taniguchi T., Hamada E., Takeshita A. Efficiency of COLD-PCR for enrichment of K-ras mutation; a proof by use of SSCP analysis. Clin Chem. 2009;55 A219-A219. [Google Scholar]
- 35.Delaney D., Diss T.C., Presneau N., Hing S., Berisha F., Idowu B.D., O'Donnell P., Skinner J.A., Tirabosco R., Flanagan A.M. GNAS1 mutations occur more commonly than previously thought in intramuscular myxoma. Mod Pathol. 2009;22:718–724. doi: 10.1038/modpathol.2009.32. [DOI] [PubMed] [Google Scholar]
- 36.Luthra R., Zuo Z. COLD-PCR finds hot application in mutation analysis. Clin Chem. 2009;55:2077–2078. doi: 10.1373/clinchem.2009.136143. [DOI] [PubMed] [Google Scholar]
- 37.Fredriksson S., Baner J., Dahl F., Chu A., Ji H., Welch K., Davis R.W. Multiplex amplification of all coding sequences within 10 cancer genes by Gene-Collector. Nucl Acids Res. 2007;35:e47. doi: 10.1093/nar/gkm078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Varley J.M. Germline TP53 mutations and Li-Fraumeni syndrome. Hum Mutat. 2003;21:313–320. doi: 10.1002/humu.10185. [DOI] [PubMed] [Google Scholar]
- 39.Milbury C.A., Li J., Makrigiorgos G.M. PCR-based methods for the enrichment of minority alleles and mutations. Clin Chem. 2009;55:632–640. doi: 10.1373/clinchem.2008.113035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J., Makrigiorgos G.M. COLD-PCR: a new platform for highly improved mutation detection in cancer and genetic testing. Biochemical Society Transactions. 2009;37:427–432. doi: 10.1042/BST0370427. [DOI] [PubMed] [Google Scholar]
- 41.Olivier M., Eeles R., Hollstein M., Khan M.A., Harris C.C., Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat. 2002;19:607–614. doi: 10.1002/humu.10081. [DOI] [PubMed] [Google Scholar]
- 42.Sjoblom T., Jones S., Wood L.D., Parsons D.W., Lin J., Barber T.D., Mandelker D., Leary R.J., Ptak J., Silliman N., Szabo S., Buckhaults P., Farrell C., Meeh P., Markowitz S.D., Willis J., Dawson D., Willson J.K.V., Gazdar A.F., Hartigan J., Wu L., Liu C., Parmigiani G., Park B.H., Bachman K.E., Papadopoulos N., Vogelstein B., Kinzler K.W., Velculescu V.E. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 43.Soussi T., Lozano G. p53 mutation heterogeneity in cancer. Biochem Biophys Res Comm. 2005;331:834–842. doi: 10.1016/j.bbrc.2005.03.190. [DOI] [PubMed] [Google Scholar]
- 44.Mawrin C. Molecular genetic alterations in gliomatosis cerebri: what can we learn about the origin and course of the disease? Acta Neuropathol. 2005;110:527–536. doi: 10.1007/s00401-005-1083-8. [DOI] [PubMed] [Google Scholar]
- 45.Ohgaki H., Dessen P., Jourde B., Horstmann S., Nishikawa T., Di Patre P.-L., Burkhard C., Schuler D., Probst-Hensch N.M., Maiorka P.C., Baeza N., Pisani P., Yonekawa Y., Yasargil M.G., Lutolf U.M., Kleihues P. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004;64:6892–6899. doi: 10.1158/0008-5472.CAN-04-1337. [DOI] [PubMed] [Google Scholar]
- 46.Ohgaki H. Genetic pathways to glioblastomas. Neuropathology. 2005;25:1–7. doi: 10.1111/j.1440-1789.2004.00600.x. [DOI] [PubMed] [Google Scholar]
- 47.Ohgaki H., Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170:1445–1453. doi: 10.2353/ajpath.2007.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Watanabe K., Sato K., Biernat W., Tachibana O., von Ammon K., Ogata N., Yonekawa Y., Kleihues P., Ohgaki H. Incidence and timing of p53 mutations during astrocytoma progression in patients with multiple biopsies. Clin Cancer Res. 1997;3:523–530. [PubMed] [Google Scholar]
- 49.Parsons D.W., Jones S., Zhang X., Lin J.C.-H., Leary R.J., Angenendt P., Mankoo P., Carter H., Siu I.M., Gallia G.L., Olivi A., McLendon R., Rasheed B.A., Keir S., Nikolskaya T., Nikolsky Y., Busam D.A., Tekleab H., Diaz L.A., Jr., Hartigan J., Smith D.R., Strausberg R.L., Marie S.K.N., Shinjo S.M.O., Yan H., Riggins G.J., Bigner D.D., Karchin R., Papadopoulos N., Parmigiani G., Vogelstein B., Velculescu V.E., Kinzler K.W. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kraus-Ruppert R., Laissue J., Bürki H., Odartchenko N. Proliferation and turnover of glial cells in the forebrain of young adult mice as studied by repeated injections of H-thymidine over a prolonged period of time. J Compar Neurol. 1973;148:211–216. doi: 10.1002/cne.901480206. [DOI] [PubMed] [Google Scholar]
- 51.Baldus S.E., Schaefer K.-L., Engers R., Hartleb D., Stoecklein N.H., Gabbert H.E. Prevalence and heterogeneity of KRAS: BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin Cancer Res. 2010;16:790–799. doi: 10.1158/1078-0432.CCR-09-2446. [DOI] [PubMed] [Google Scholar]
- 52.Losi L., Baisse B., Bouzourene H., Benhattar J. Evolution of intratumoral genetic heterogeneity during colorectal cancer progression. Carcinogenesis. 2005;26:916–922. doi: 10.1093/carcin/bgi044. [DOI] [PubMed] [Google Scholar]
- 53.Barnetson R., Jass J., Tse R., Eckstein R., Robinson B., Schnitzler M. Mutations associated with microsatellite unstable colorectal carcinomas exhibit widespread intratumoral heterogeneity. Genes Chromosomes Cancer. 2000;29:130–136. doi: 10.1002/1098-2264(200010)29:2<130::aid-gcc1023>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 54.Giaretti W., Macciocu B., Geido E., Hermsen M.A.J.A., Postma C., Baak J.P.A., Williams R.A., Meijer G.A. Intratumor heterogeneity of k-ras and p53 mutations among human colorectal adenomas containing early cancer. Analyt Cellular Pathol. 2000;21:49–57. doi: 10.1155/2000/747524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Greenman C., Stephens P., Smith R., Dalgliesh G.L., Hunter C., Bignell G., Davies H., Teague J., Butler A., Stevens C., Edkins S., Meara S., Vastrik I., Schmidt E.E., Avis T., Barthorpe S., Bhamra G., Buck G., Choudhury B., Clements J. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Campbell P.J., Pleasance E.D., Stephens P.J., Dicks E., Rance R., Goodhead I., Follows G.A., Green A.R., Futreal P.A., Stratton M.R. Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing. Proc Natl Acad Sci. 2008;105:13081–13086. doi: 10.1073/pnas.0801523105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jones S., Chen W.-d., Parmigiani G., Diehl F., Beerenwinkel N., Antal T., Traulsen A., Nowak M.A., Siegel C., Velculescu V.E., Kinzler K.W., Vogelstein B., Willis J., Markowitz S.D. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci. 2008;105:4283–4288. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Loeb L.A., Bielas J.H., Beckman R.A., Bodmer I.W. Cancers exhibit a mutator phenotype: clinical implications. Cancer Res. 2008;68:3551–3557. doi: 10.1158/0008-5472.CAN-07-5835. [DOI] [PubMed] [Google Scholar]
- 59.Torkamani A., Verkhivker G., Schork N.J. Cancer driver mutations in protein kinase genes. Cancer Lett. 2009;281:117–127. doi: 10.1016/j.canlet.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang C., Taki T., Adachi M., Konishi T., Higashiyama M., Miyake M. Mutations in exon 7 and 8 of p53 as poor prognostic factors in patients with non-small cell lung cancer. Oncogene. 1998;16:2469–2477. doi: 10.1038/sj.onc.1201776. [DOI] [PubMed] [Google Scholar]
- 61.Ahrendt S.A., Hu Y., Buta M., McDermott M.P., Benoit N., Yang S.C., Wu L., Sidransky D. p53 mutations and survival in stage I non-small-cell lung cancer: results of a prospective study. J Natl Cancer Inst. 2003;95:961–970. doi: 10.1093/jnci/95.13.961. [DOI] [PubMed] [Google Scholar]
- 62.Wallace-Brodeur R.R., Lowe S.W. Clinical implications of p53 mutations. Cell Mol Life Sci. 1999;55:64–75. doi: 10.1007/s000180050270. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative amplicons generated for each of the 14 regions of interest following the initial TP53 multiplex PCR.
A 129 base pair amplicon of TP53 exon 6 was analyzed via HRM after conventional PCR (A) and COLD-PCR(C). Conventional-PCR amplicons of the 2% mutant mixture were scored as “unknown” by the Call-IT software, indicating a potential presence of variants that differ from wild-type. Amplicons were subsequently sequenced (Sanger; anti-sense strand presented). While a 10% abundance of the T47D mutant cell line (c.580C>T, p.L194F) cannot be observed in the chromatogram of conventional-PCR amplicons (B), a 5% mutant abundance is easily visible in the chromatogram of COLD-PCR (D). Wild-type (WT) sequences are presented for comparison.
A 128 base pair amplicon of TP53 exon 8 was analyzed via HRM after conventional PCR (A) and COLD-PCR (C). Amplicons were subsequently sequenced (Sanger; anti-sense strand presented). While a 5% mutant abundance of the MDMBA-435 cell-line (c.797G>A, p.G266E) cannot be detected in the chromatogram of conventional-PCR amplicons (B), a 5% mutant abundance is easily visible in the chromatogram of COLD-PCR (D). Wild-type (WT) sequences are presented for comparison.
A 155 base pair amplicon of TP53 exon 8 was analyzed via HRM after conventional PCR (A) and COLD-PCR (C). Amplicons were subsequently sequenced (Sanger; anti-sense strand presented). While a 5% mutant abundance of the HCC2218 cell-line (c.847 C>T, p.R283C) is unreliable in the chromatogram of conventional-PCR amplicons (B), a 3% mutant abundance is easily visible in the chromatogram of COLD-PCR (D). Wild-type (WT) sequences are presented for comparison.
For the exon 5 (148bp) amplicon, Sanger sequencing analysis (sense strand) of the multiplexed-PCR product for CT2 revealed a low-abundance C>A mutation (c.523C>A, p.R175S) in the sequence chromatograms of COLD-PCR (A); however, this mutation could not be detected in conventional PCR amplicons by either HRM or sequencing. The mutation was confirmed in subsequent analysis of the COLD-PCR amplified genomic DNA (B); however, the mutation was not observed in the matched normal sample (C). A second mutation A>T (c.739A>T, p.N247I) was detected by HRM in CT2 in the exon 7 conventional PCR amplicon and identified via Sanger sequencing.
In the exon 7 amplicon, a low abundance mutation in a colorectal tumor sample (CT11) was detected after HRM analysis of COLD-PCR amplicons (A); however, this mutation was not detected in conventional PCR amplicons. An additional two samples possessed mid- to high-abundance mutations detectable by HRM screening of both conventional and COLD-PCR amplicons. For the exon 7 amplicon, Sanger sequencing analysis (sense strand) of the multiplexed-PCR product for CT11 revealed a low-abundance G>T mutation (c.782+1G>T) in the sequence chromatograms of COLD-PCR (B); however, this mutation could not be detected in conventional PCR amplicons by either HRM or sequencing. The mutation was confirmed in subsequent analysis of the COLD-PCR amplified genomic DNA (C); however, the mutation was not observed in the matched normal sample (D).
Genomic DNA and multiplex-amplified template, of specimen CT11 and wild-type DNA, were PCR amplified (TP53 exon 7, 185 bp) and subjected to PspGI digestion (sequence region containing the recognition site, CCWGG, is underlined). Digested product was subsequently re-amplified (TP53 exon 7, 166 bp) and analyzed via Sanger sequencing (sense strand). The G>T mutation is clearly enriched in both the CT11 genomic DNA (A) and multiplex-amplified product (B); yet remains absent in the respective wild-type control samples (C and D).
Two novel deletions were detected in the glioma tumor samples. (A) For the exon 2 amplicon, Sanger sequencing analysis (sense strand) of the multiplexed-PCR and genomic DNA amplicons for CMK-T7 revealed a previously undocumented two base pair deletion (c.40del2, p.L14del2). The deletion (CT) was located in codon 14, and results in a frameshift. (B) For the exon 5 (120bp) amplicon, Sanger sequencing analysis (anti-sense strand) of the multiplexed-PCR and genomic DNA amplicons for CMK-T1 revealed a previously undocumented three base pair deletion (c.403del3, p.C135del3). The deletion (TTG) was located at codon 135, and results in a frameshift.
Comparative analysis of the frozen (panels A and B) and FFPE (panels C and D) specimens revealed concordance. Representative results are presented for TP53 exon 8, 155 bp amplicon. HRM difference plots demonstrate the CMK-16 nonsense mutation (p.R306X, c.916C>T), represented by the red melting curve as amplified by both conventional (panels A and C) and COLD-PCR (panels B and D) relative to wild-type melting curves (gray).