

Abstract
Typical general transcription factors, such as TATA binding protein and TFII B, have not yet been identified in any member of the Trypanosomatidae family of parasitic protozoa. Interestingly, mRNA coding genes do not appear to have discrete transcriptional start sites, although in most cases they require an RNA polymerase that has the biochemical properties of eukaryotic RNA polymerase II. A discrete transcription initiation site may not be necessary for mRNA synthesis since the sequences upstream of each transcribed coding region are trimmed from the nascent transcript when a short m7G-capped RNA is added during mRNA maturation. This short 39 nt m7G-capped RNA, the spliced leader (SL) sequence, is expressed as an ∼100 nt long RNA from a set of reiterated, though independently transcribed, genes in the trypanosome genome. Punctuation of the 5′ end of mRNAs by a m7G cap-containing spliced leader is a developing theme in the lower eukaryotic world; organisms as diverse as Euglena and nematode worms, including Caenorhabditis elegans, utilize SL RNA in their mRNA maturation programs. Towards understanding the coordination of SL RNA and mRNA expression in trypanosomes, we have begun by characterizing SL RNA gene expression in the model trypanosome Leptomonas seymouri. Using a homologous in vitro transcription system, we demonstrate in this study that the SL RNA is transcribed by RNA polymerase II. During SL RNA transcription, accurate initiation is determined by an initiator element with a loose consensus of CYAC/AYR(+1). This element, as well as two additional basal promoter elements, is divergent in sequence from the basal transcription elements seen in other eukaryotic gene promoters. We show here that the in vitro transcription extract contains a binding activity that is specific for the initiator element and thus may participate in recruiting RNA polymerase II to the SL RNA gene promoter.
INTRODUCTION
The trypanosomatid family of parasitic protozoa has an unusual method of gene expression that consists of polycistronic transcription of precursor mRNAs (reviewed in 1). Polyadenylation and trans-splicing of the polycistronic RNAs lead to mature individual mRNAs. The addition of a 39 nt capped RNA through trans-splicing provides the mRNA with a cap structure that contains the m7G and modification of the first four bases (2), thus endowing all mRNAs with a cap4 structure. This capped RNA is part of an ∼100 nt RNA that is transcribed from the spliced leader (SL) RNA gene promoter. This promoter has been widely studied in various trypanosomatids including Trypanosoma brucei (3), various Leishmania species (4–6) and Leptomonas spp. (7–10). In Leptomonas seymouri, the promoter has a tripartite structure (11). At least two protein activities, PBP-1 and PBP-2 previously defined in our laboratory, are required for transcription from this promoter (12,13). In addition, a third element, the initiator element is also required for both accurate initiation and efficient transcription (11).
Studies to determine the identity of the RNA polymerase that transcribes the SL RNA genes have proven inconclusive. As in higher order Eukaryotes, three RNA polymerases have been biochemically identified in trypanosomatids (14–19). RNA polymerase (pol) I, in addition to transcribing ribosomal RNAs as in metazoans and yeast, also transcribes the structural genes, variant surface glycoprotein and procyclic acidic repetitive protein, of T.brucei (reviewed in 20). RNA pol III transcribes the small U RNA and tRNA genes, as expected (21). While RNA pol II transcribes most protein coding genes, precise transcription initiation start sites have not been mapped (22,23). Indeed, intergenic regions have long been used as a means of driving transcription of reporter genes. In Leishmania and Leptomonas, expression of a reporter gene requires a bona fide trypanosome 3′ splice acceptor site (SL RNA addition site), but no specific upstream sequence seems to be required for transcription initiation.
To understand trypanosome mRNA expression we need to understand how RNA pol II is recruited to DNA. Clearly, defining a discrete RNA pol II transcription unit is a first step in this process. The suggestion that the SL RNA gene promoter might be RNA pol II-dependent came from the identification of a 5′ m7G cap on the RNA (24) and from pharmacokinetic studies. Tagetitoxin, a potent inhibitor of RNA pol III activity, had no effect on SL RNA transcription (4, 25). However, α-amanitin, a low-dose inhibitor of RNA pol II, inhibited SL RNA transcription at doses higher than needed for inhibition of α-tubulin, a prototypic RNA pol II gene (3,4,26,27). The SL RNA transcript is generally not polyadenylated (28,29) and termination of SL RNA transcription requires an intact 3′ poly(T) tract, both hallmarks of RNA pol III-dependent genes (30). Since drug and structural studies have produced conflicting results, a more direct approach is required to determine the identity of the RNA polymerase that drives SL RNA expression. The power of an in vitro transcription system has enabled us to examine, in detail, the mechanism of SL RNA transcription in trypanosomes. With this system, we and others have defined promoter elements and potential transcription factors (5,6,10,31,32). Now we have used this system to identify the RNA polymerase recruited by these factors to the SL RNA gene promoter.
To test the hypothesis that RNA pol II transcribes the SL RNA gene, we cloned the L.seymouri RNA pol II largest subunit and prepared histidine-tagged recombinant protein from the C-terminal domain (CTD) portion of the protein. Anti-CTD polyclonal antibodies, which specifically recognized the CTD, were generated in rabbits immunized with the recombinant protein. The antibodies functioned to deplete RNA pol II from a transcriptionally competent nuclear extract. These nuclear extracts were used in in vitro transcription reactions and loss of specific transcription was seen in anti-CTD depleted extracts but not in pre-immune depleted extracts. Transcription of the RNA pol III-dependent U6 small nuclear (sn)RNA gene was unaltered. The dependence of SL RNA transcription upon RNA pol II directly defines for the first time a discrete RNA pol II-dependent transcription unit in trypanosomes.
MATERIALS AND METHODS
PCR
Two sets of degenerate primers were designed based on the sequence of the T.brucei RNA pol II largest subunit gene (14). One primer set, corresponding to T.brucei amino acids 488–493 and 871–876, was used in a PCR reaction with L.seymouri genomic DNA as template. A second set of primers, corresponding to amino acids 789–793 and 818–823, was then used in a second PCR reaction, using the first PCR reaction products as template. The nested PCR yielded a product of the expected length, 102 bp, which was cloned into pBlueScript II SK+ (Stratagene, La Jolla, CA) to produce pSKIIa1. The sequence of the 102 bp insert displayed regions of homology with RNA pol II largest subunit genes from other genera in a BLASTX database search. This insert was then used in screening the genomic library and in subsequent Southern analyses.
Genomic library screening and Southern blot analysis
A genomic library was constructed by partial digestion of L.seymouri genomic DNA with the enzyme Sau3A and ligation into the BamH1 site of the λ phage EMBL3 (Promega, Madison, WI) (A.Das, personal communication). Plaques were screened by hybridization using the radiolabeled insert from pSKIIa1. Two initial positives, obtained in the primary screen, were used in a secondary screen. The secondary screen identified one positive clone, which was further plaque-purified. Phage DNA was purified using the Qiagen (Valencia, CA) λ phage mini prep kit.
For Southern analysis, phage DNA and genomic DNA were digested with SalI, electrophoresed on a 0.8% agarose gel and transferred by capillary action overnight to a Nytran membrane. The membrane was hybridized at 37°C in 50% formamide with the radiolabeled insert for 48 h, ultimately washed in 0.1× SSC/0.1% SDS at room temperature for 10 min and exposed to a phosphorimager screen.
Plasmids
pHL-U6, containing the 296 nt internally tagged L.seymouri U6 snRNA gene, was described previously (11). pJM-1 was constructed by cloning a PCR product, which contained 100 nt upstream of the SL transcription initiation site and 100 nt downstream of this site, into the Invitrogen (Carlsbad, CA) vector pCR2.1TOPO. pHisCTD was constructed by PCR amplification of the λ phage clone 7-1-2 using two primers, one complementary to the 5′ end of the CTD with an Ndel site at the 5′ end, and one complementary to the 3′ end of the CTD with a BamHI site at the 3′ end. This product was digested with NdeI and BamHI and ligated to a similarly digested Novagen (Madison, WI) vector pET15b. Transcription by T7 RNA polymerase produced a fusion protein containing six histidine residues at the N-terminal end followed, in frame, by the L.seymouri CTD sequence.
Purification of histidine-tagged (His)CTD and antibody production
pHisCTD was transformed into the Escherichia coli strain, pBL21(DE3)LysS. Cells were grown at 30°C for 3 h, induced with 0.2 mM IPTG for 3 h and harvested by three cycles of freeze–thaw. The debris was removed by centrifugation and the supernatant was passed through a 0.45 filter and applied to a NiSO4 column. The purified HisCTD was eluted with 1 M imidazole, desalted using a P-10 column and rechromatographed. The final eluate was desalted into phosphate-buffered saline (PBS) and lyophilized. The lyophilizate (2 mg) was sent to Research Genetics (Mobile, AL) for injection into rabbits. All immunoprecipitations and western blots were performed using anti-serum 10 weeks post-immunization (p.i.).
Nuclear extracts
Leptomonas seymouri extracts were made as described previously (9,11). Briefly, L.seymouri were grown to a density of 107/ml, harvested by centrifugation and lysed by douncing in hypotonic buffer. Nuclear proteins were collected by 55% NH4SO4 precipitation of supernatant from the high-speed centrifugation. The NH4SO4 precipitated material was resuspended in transcription buffer (10 mM HEPES–KOH pH 7.9, 150 mM sucrose, 20 mM potassium glutamate, 2.5 mM MgCl2, 2.5 mM DTT, 1 mM EDTA, 0.1 mM PMSF, 1 mM pepstatin and 1 mM leupeptin) and dialyzed against this buffer. The extract was briefly microcentrifuged and frozen in aliquots at –70°C.
Immunoprecipitations
The extract was pre-cleared by incubating 50 µl of protein A–Sepharose in PBS with 5 µl of rabbit IgG and 330 µl of nuclear extract for 35 min at 4°C, and then centrifuging at 2000 g for 2 min. This step removed non-specific protein–immunoglobulin interactions. At the same time, 25 µl of rabbit anti-CTD antibody or 25 µl of pre-immune serum was incubated with 50 µl of protein A–Sepharose for 30 min at 4°C, washed three times with PBS and then with transcription buffer to remove contaminants. After removal of 70 µl of the pre-cleared extract for subsequent analysis, the remaining extract was incubated with antibody-treated or pre-immune serum-treated protein A–Sepharose for 90 min at 4°C, after which the sample was centrifuged for 2 min at 2000 g. The supernatant was used directly in transcription reactions and for western blot analysis. The pellet was washed in transcription buffer and then resuspended in SDS sample buffer for western blot analysis.
In vitro transcriptions
In vitro transcriptions were performed as described previously (11) with the following exceptions: 0.14 µg of plasmid, either JM-1 or HL-U6, was used to program each reaction. pHL-U6 and extract were pre-incubated for 10 min before the addition of pJM-1 and rNTPs. RNAs were resuspended in primer extension buffer and half of the RNA was annealed to [γ-32P]ATP-labeled primer VB207gi by incubation at 75°C for 5 min, and then slowly cooled to 45°C. Superscript reverse transcriptase (Gibco-BRL, Rockville, MD) was added and the extension proceeded for 30 min at 42°C. The products were denatured in 90% formamide and then electrophoresed on a 10% polyacrylamide–7 M urea gel. Dried gels were exposed to phosphorimager screens.
Western assays
Denatured extract (10 µl) was electrophoresed through a 7% SDS–polyacrylamide gel, then transferred overnight onto PVDF membrane. The membrane was blocked for 1 h in 0.1% PBS–Tween with 5% non-fat milk and then incubated for 1 h in 0.1% PBS–Tween, 5% non-fat milk containing a 1/500 dilution of the CTD antibody. The membrane was washed three times in 0.1% PBS–Tween and then incubated for 1 h in 0.1% PBS–Tween, 5% non-fat milk and a 1/10 000 dilution of horseradish peroxidase anti-rabbit antibody. The membrane was washed three times in 0.1% PBS–Tween and the antibody-bound proteins were visualized using the ECL kit from Amersham Pharmacia (Piscataway, NJ). In experiments where competing peptides were used, 100 ng of either HisCTD or an irrelevant His fusion protein (HisORF50; 33) was incubated with a 1/500 dilution of antibody for 30 min at 37°C and then the antibody was used in western blotting as described above.
Electrophoretic mobility shift analysis (EMSA)
PBP-1 and PBP-2 gel shifts were performed as described previously (13). For initiator protein detection, 10 000 c.p.m. of DNA probe, (containing the –1/–20 bp of the SL RNA gene promoter, flanked by pBlueScript II SK+ sequence; see Fig. 7), was incubated in 10% glycerol, 100 mM KCl, 10 mM HEPES pH 7.6, 5 mM DTT, 0.1 mM PMSF and ∼5 µg of nuclear extract, at 30°C for 15 min. The protein–DNA complexes were resolved on a 5% polyacrylamide, 5% glycerol gel electrophoresed in 0.5× TBE at 140 V for 3 h (34). The gel was dried and placed on a phosphorimager screen for visualization.
Figure 7.
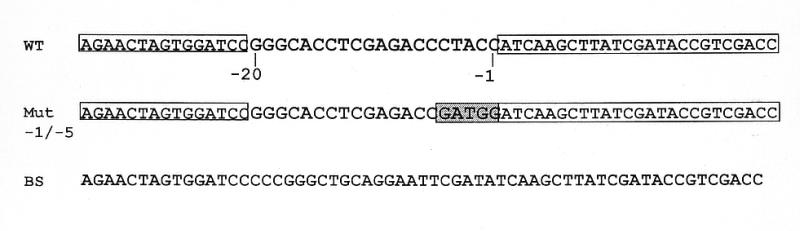
Diagram of DNA competitor fragments used in EMSAs. The wild-type (WT) DNA sequence used in Inrt competitions is indicated in the first line of the figure. SL RNA promoter sequences from –1 to –20 bp are flanked by sequences from the pBlueScript II SK+ (indicated by boxes). The names of the competitor fragments are indicated to the left of the sequences. The mutated sequences in Mut –1/–5 are indicated by the shaded region.
RESULTS
The L.seymouri RNA pol II largest subunit gene contains a divergent CTD
To determine the identity of the polymerase that transcribes the SL RNA gene, we began by cloning and characterizing the gene for the largest subunit of the multi-subunit L.seymouri RNA pol II enzyme. To amplify a portion of this gene we used two sets of degenerate primers based on the T.brucei RNA pol II largest subunit gene sequence (14) in a PCR reaction with genomic DNA from L.seymouri. We obtained a 102 bp DNA fragment using a nested PCR procedure as described in Materials and Methods. This 102 bp DNA fragment identified a recombinant phage clone, 7-1-2, which contained a portion of the RNA pol II largest subunit gene. Rescreening of the genomic library produced a second recombinant phage, which contained the entire RNA pol II largest subunit gene (data not shown). Southern blot analysis showed that the 102 bp fragment (representing amino acids 762–796 within the coding region) hybridized to a single 5 kb band in a SalI-digestion of Leptomonas genomic DNA. These data suggest that the RNA pol II largest subunit is a single copy gene (Fig. 1).
Figure 1.
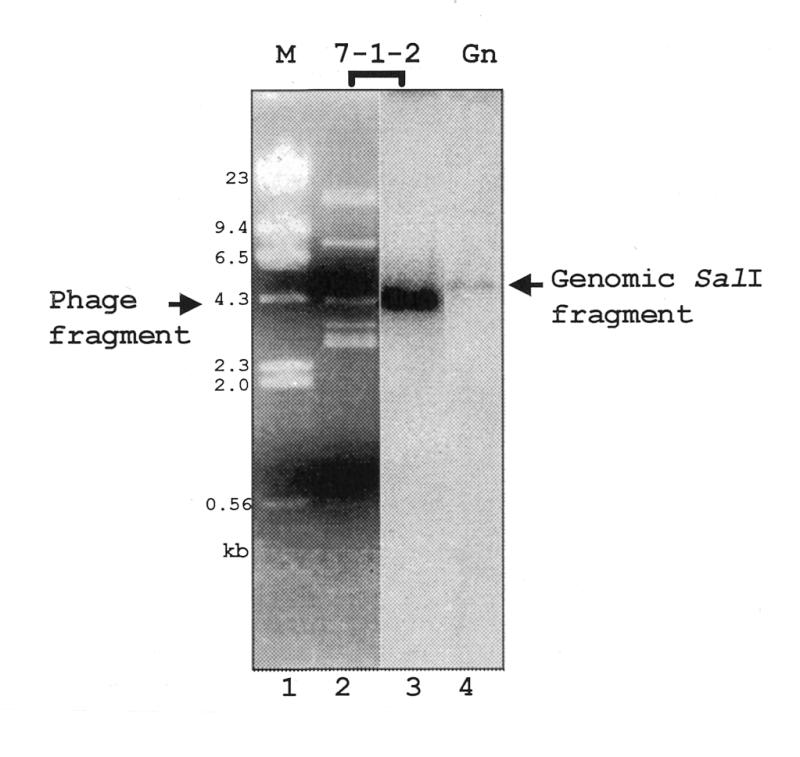
Identification of the L.seymouri RNA pol II largest subunit gene. SalI-digested recombinant λ phage 7-1-2 was size-separated on a 0.8% agarose gel and stained with SYBR Green (FMC Bioproducts, Chicago, IL) (lanes 1 and 2). The 4.3 kb phage DNA fragment in lane 2 is indicated. Lane 3 is the hybridization result when the DNA in lane 2 was transferred to Nytran and probed with radiolabeled pSKPIIa1. Lane 4 contains SalI-digested L.seymouri genomic DNA also probed with radiolabeled oligonucleotide SKPIIa1. The genomic DNA-hybridized fragment is indicated. Markers are λ phage DNA digested with HindIII.
The L.seymouri largest subunit of RNA pol II shares significant similarity (71–90% identity and 83–92% similarity) with the homologous protein from other trypanosomatids, as expected (NCBI accession number AF338253). The seven domains (domains A–G), which are requisite components of all eukaryotic largest subunits of RNA polymerase proteins, are present in the L.seymouri polypeptide (data not shown) (35). Analysis of the A–G domain region of the Leptomonas protein showed 40% identity and 57% similarity to the human (NP_000928) and Drosophila (AAF89203) largest subunit of RNA pol II. These data confirm the similarity of the Leptomonas RNA pol II largest subunit protein to the homologous proteins in other trypansomatids and eukaryotes. The amino acids following domain G and beginning at amino acid 1454 constitute an acidic (pI 4.5) CTD (Fig. 2). Of note, there is a complete absence of the heptapeptide repeat found in metazoans and yeast. A comparison of this CTD among four trypanosomatid genera, Crithidia, Leishmania, Trypanosoma and Leptomonas, indicates significant divergence in primary sequence. In particular, L.seymouri diverges almost completely from T.brucei except in a region (L.seymouri amino acids 1573–1582) containing a di-serine motif (boxed in Fig. 2). Di-serines in the CTD are a common theme in all the trypanosomatids. Serines are potential phosphorylation sites, and phosphorylation of RNA pol II has been reported in trypanosomes (36). It is not yet clear whether phosphorylation of the largest subunit occurs in the CTD.
Figure 2.

Protein sequence comparison of the L.seymouri RNA pol II subunit CTD and other trypanosomatid species. T.brucei (18), Leishmania major (67) and Crithidia fasiculata (68) CTD sequences were compared with L.seymouri. Leptomonas seymouri amino acid numbers, on the right, are based on the entire RNA pol II largest subunit protein sequence. Amino acid identity with L.seymouri is represented by a dash. Gaps in sequences (represented by dots) were inserted to improve alignment. The di-serine motifs are underlined. The largest region of identity among the trypanosomatids is boxed. Sequences were aligned using the Gapped BLAST algorithm.
Antiserum generated against the HisCTD fusion protein recognizes both the fusion protein and the L.seymouri RNA pol II largest subunit protein
To produce anti-CTD polyclonal antiserum, we generated a recombinant CTD protein. Six histidine residues were placed at the N-terminal end of the CTD sequence of the RNA pol II largest subunit gene using the vector pET15b. The bacterial expression plasmid, pHisCTD, was transformed into pBL21(DE3)LysS and recombinant protein was induced and purified. Coomassie blue-stained protein gels indicated that the fusion protein was highly purified (Fig. 3A). Polyclonal rabbit antibodies were generated against the HisCTD fusion protein. The specificity of the antiserum was tested by western analysis (Fig. 3B). The data indicate that the anti-CTD antiserum recognizes the recombinant 28 kDa fusion protein produced in bacteria. The specific antiserum recognized a single polypeptide of the anticipated size, 182 kDa, which was not observed in immunoblots probed with the pre-immune serum (Fig. 3C). To confirm antibody specificity, the anti-CTD antibody was reacted with either the HisCTD fusion protein or an irrelevant His-fusion protein (33) prior to its use in immunoblotting. The HisCTD protein specifically inhibited anti-CTD binding to the 182 kDa endogenous protein present in L.seymouri nuclear extract, whereas the His ORF50 protein did not block antibody binding (Fig. 3D).
Figure 3.

Purification of HisCTD using a nickel column and western analysis of recombinant protein. (A) Bacterial extract and nickel column eluates were electrophoresed on a 10% SDS–polyacrylamide gel, fixed in 10% acetic acid/10% methanol and stained with Coomassie blue. Samples are as follows: bacterial extract (lane 1), flow-through fraction (lane 2), wash fraction (lane 3), 1 M imidazole eluate (lane 4). The recombinant HisCTD protein band is indicated. (B) Recombinant HisCTD was electrophoresed as in (A), transferred to PVDF membrane and probed with either pre-immune serum (left) or anti-CTD antibody (right). The recombinant protein band is indicated. (C) Nuclear extract was electrophoresed on a 7% SDS–polyacrylamide gel transferred and probed with either the pre-immune serum (right) or anti-CTD antibody (left). Lanes 1 and 2 contain two independent extracts. (D) The blot in (C) was stripped and reprobed with anti-CTD antibody that had been blocked with either the HisORF 50 protein (lane 1) or the HisCTD protein (lane 2).
Nuclear extract depleted of RNA pol II is unable to transcribe the SL RNA gene but retains the ability to transcribe the U6 snRNA gene
To determine whether RNA pol II is required for transcription of the SL RNA, we used immunoprecipitation to deplete a nuclear extract of this polymerase. The extract was incubated with anti-CTD antibodies or pre-immune serum in the presence of protein A–Sepharose beads. After incubation, the immune complexes were pelleted and the supernatant collected. Samples of both supernatant and pellet were loaded onto 7% SDS–polyacrylamide gels, and processed for western analysis (Fig. 4). The anti-CTD antibody recognized protein in the starting extract, the pre-cleared extract and the pellet from immunoprecipitations performed with the anti-CTD antiserum (lanes 1, 2 and 5). The RNA pol II largest subunit was completely removed from the extract treated with the anti-CTD antiserum (lane 3). The protein remained in the supernatant from pre-immune immunoprecipitations, as was expected (lane 4). Therefore, the supernatant from the immunoprecipitation with the anti-CTD antibody was depleted of RNA pol II largest subunit to the limits of detection using western analysis.
Figure 4.
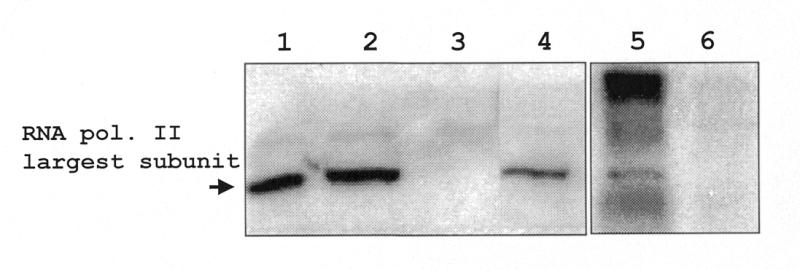
Western blot analysis of immunoprecipitated (IP) extracts. IP complexes and supernatants were electrophoresed on a 7% SDS–polyacrylamide gel, transferred overnight and probed with anti-CTD antibody (10 weeks p.i.). The extracts used were as follows: nuclear extract (lane 1), pre-cleared extract (lane 2), anti-CTD IP supernatant (lane 3), pre-immune IP supernatant (lane 4), anti-CTD IP pellet (lane 5), pre-immune IP pellet (lane 6). The band representing the RNA pol II largest subunit is indicated.
The extracts were then used in in vitro transcription reactions programmed with two templates, pJM-1, which contains the SL RNA gene, and pHL-U6, which contains the RNA pol III-dependent U6 snRNA gene. When pre-cleared extract was used in in vitro transcription/primer extension reactions, bands of 69 and 99 nt appear, corresponding to the two reverse transcribed products of SL RNA and U6 snRNA transcripts, respectively (Fig. 5A, lane 1). If anti-CTD-depleted extract was used to program the reaction, the level of the SL RNA transcript was significantly diminished (lane 2). U6 snRNA levels remained unchanged. Quantitation of SL RNA transcript levels, relative to U6 snRNA, indicated that transcription of the SL RNA gene is reduced by ∼25-fold (Fig. 5B). In the presence of pre-immune depleted extract there is no change in intensities of either U6 snRNA or SL RNA transcripts (lane 3). Therefore, depletion of RNA pol II specifically blocks SL RNA gene transcription.
Figure 5.

In vitro transcription with immunodepleted extracts. (A) In vitro transcription products were primer extended with a 32P-labeled primer (VB207gi) specific to a 19 nt tag present in both SL and U6 snRNA genes. The extension products were electrophoresed on a 10% polyacrylamide–7 M urea denaturing gel and exposed to a phosphorimager screen. Specific SL RNA and U6 RNA primer extended products are indicated. The extract used in each reaction was: preclear (lane 1), anti-CTD depleted (lane 2) and pre-immune depleted (lane 3). (B) Quantitation of the primer extended products. RNA activity is represented as arbitrary units of phosphorescence in a ratio of SL RNA activity divided by U6 snRNA activity. The P-values with respect to the pre-clear are 0.016 and 0.18 for anti-CTD antibody and pre-immune treated extracts, respectively.
The RNA pol II-depleted extract retains the protein activities, PBP-1/-2 as is evident by the presence of shifted bands in an EMSA
As reported previously by our laboratory, two protein activities, PBP-1 and PBP-2, interact with their cognate sites in the SL RNA promoter and facilitate transcription initiation (11,13). The interaction of PBP-2 with the promoter requires the interaction of the –60 to –70 bp upstream region of the promoter with PBP-1. To determine whether the removal of RNA pol II from nuclear extract affected PBP-1/-2 activities, we performed DNA EMSAs using immuno-depleted extracts. The gel shift data presented in Figure 6 demonstrate that both PBP-1 and PBP-2 proteins remained in anti-CTD-depleted extract. Therefore, the loss of transcription activity seen in anti-CTD-depleted extract is not due to the removal of necessary transcription factors.
Figure 6.
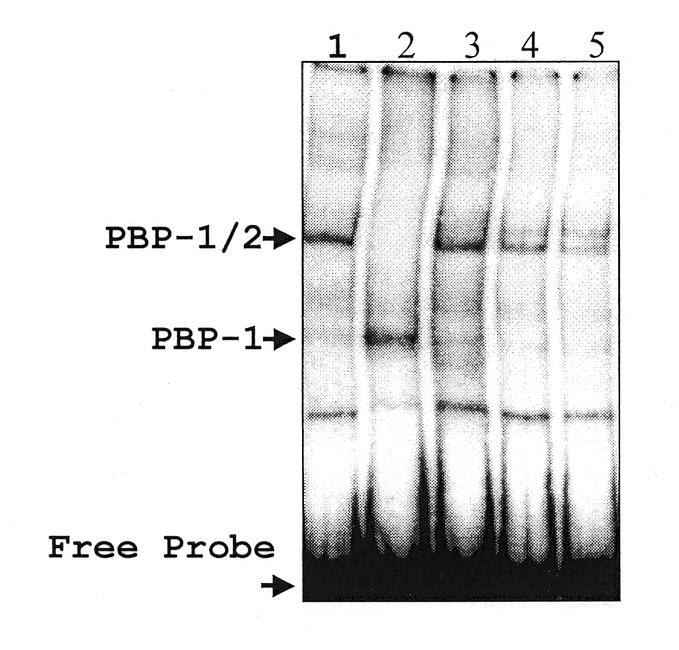
PBP-1 and PBP-2 EMSAs. IP extracts were used in an EMSA reaction along with partially purified PBP-1 and PBP-1/-2. Extracts were incubated with a 95 bp 32P-labeled probe containing the promoter elements, PBP-1 and PBP-2, and electrophoresed on a native 4% polyacrylamide gel. The reactions contain the following proteins: affinity purified PBP-1/-2 (lane 1), PBP-1 purified from a non-specific double-stranded DNA column (lane 2), pre-cleared extract (lane 3), anti-CTD IP supernatant (lane 4), pre-immune IP supernatant (lane 5).
An initiator binding factor that recognizes the Inrt of the SL RNA gene promoter does not co-immunoprecipitate with RNA pol II
In vitro, as well as in vivo, SL RNA transcription requires a consensus sequence CYAC/AYR(+1) immediately upstream of the transcription start site, which is defined as the Inrt (11,12). To determine whether specific transcription factors bind Inrt, we performed EMSAs. For these assays we used a DNA fragment that contained the –1 to –20 bp region of the SL RNA promoter, including the Inrt sequence, flanked by vector sequence (Fig. 7). In the DNA binding assay, factors from nuclear extract specifically recognized this sequence (WT) as evidenced by the appearance of two shifted bands (Fig. 8A). These bands were competed away by WT but not by a mutated –1 to –5 bp competitor. This result agrees with in vitro transcription results reported by our laboratory in which the most severe transcriptional abrogation occurred by alteration of the –1 to –5 bp promoter sequences (11). When RNA pol II-depleted extracts were used in the Inr gel shift assay, Inr activity was seen (Fig. 8B). Therefore, depletion of RNA pol II does not affect formation of protein–DNA complexes at the Inrt of the SL RNA promoter.
Figure 8.

EMSA identifies Inrt binding activities within Leptomonas nuclear extracts. (A) Nuclear extracts were incubated with an Inrt-containing probe (as described in Materials and Methods) alone or in the presence of DNA competitors. The competitor amounts used in competitions were 5, 10 and 20 ng. The competitors used were: WT, the wild-type sequence; Mut –1/–5, which contains mutated sequences in the –1 to –5 bp region of the Inrt; and BS, BlueScript sequence. (B) Nuclear extract alone (lane 1), depleted with anti-CTD antibody (lane 2) or depleted with pre-immune serum (lane 3) was incubated with the Inrt-containing probe used in (A). The arrows indicate the Inrt-specific complexes.
DISCUSSION
In this paper we have presented, for the first time, conclusive evidence that the SL RNA gene is transcribed by RNA pol II. We were able to deplete a transcriptionally active nuclear extract of the RNA pol II as evidenced by western analysis. This depleted extract was unable to support SL RNA transcription in vitro, yet was able to support U6 snRNA transcription. U6 snRNA is transcribed by RNA pol III (21), indicating that we have not removed this enzyme or any tightly associated factors. The necessary transcription factors for SL RNA gene transcription, PBP-1 and PBP-2, are present in the depleted extract, indicating that the disruption of transcription is not due to loss of SL RNA specific transcription factors. Thus, it is clear that SL RNA transcription requires RNA pol II.
One striking feature of trypanosome pol II-dependent mRNA synthesis is that in permeabilized cells it is 100-fold more resistant to α-amanitin than is mRNA synthesis in permeabilized mammalian cells. The different levels of drug sensitivities could reflect differences in RNA pol II affinity for α-amanitin. α-Amanitin binding sites have been indirectly mapped through the identification of mutations that generate α-amanitin resistance phenotypes in mouse (37–39), Drosophila (40–42) and Caenorhabditis elegans (43–45). The largest number of amino acid mutations was found in mice (38). Here, mutation of any of four different amino acid residues in domain E of the largest subunit of the enzyme could independently confer drug resistance (murine amino acid numbers 745, 749, 779, 792). Accordingly, we analyzed similarly positioned amino acids in the largest subunit gene from L.seymouri and from Trichomonas vaginalis, a primitive organism with an RNA pol II that is somewhat resistant to α-amanitin (Fig. 9) (46). The T.vaginalis RNA pol II largest subunit diverged at three of the four amino acid residues associated with α-amanitin sensitivity. The L.seymouri protein, on the other hand, exhibits identity at three of the four amino acid residues; the fourth change is a conservative one (leucine to isoleucine). Other trypanosomatid RNA pol II largest subunit proteins are identical to the L.seymouri protein at these amino acids. Therefore, the unusually high α-amanitin resistance of the trypanosomal RNA pol II is not due to alterations of the amino acids deemed relevant for α-amanitin sensitivity in mice but could be a result of differences in amino acid residues yet to be associated with α-amanitin binding. In addition, amino acid variations in the trypanosome enzyme might result in a tertiary protein structure that would effect drug sensitivity. Alternatively, differences in drug response could be explained by the presence, in permeabilized trypanosomatids, of an activity affecting the stability of α-amanitin.
Figure 9.

Comparison of the α-amanitin resistance coding region of the RNA pol II largest subunit. Mouse (Mus musculus) (35) and Trichomonas (T.vaginalis) (46) sequences from domain E of the RNA pol II largest subunit are compared with L.seymouri amino acid sequences 685–760. Boxed residues indicate amino acid residues found to be individually mutated in four independently isolated α-amanitin-resistant mouse mutants (38). The sequences were aligned using the Gapped BLAST algorithm. Gaps in sequences (represented by dots) were inserted to improve alignment. Dashes represent identity with L.seymouri sequences.
The ambiguous results of drug studies have made it extremely difficult to determine the identity of the RNA polymerase responsible for transcription of the SL RNA gene. In permeabilized trypanosomes, mRNA genes (notably α-tubulin) were downregulated by 5–10 µg/ml α-amanitin and rRNA genes required substantially more (500–1000 µg/ml) (26,47). RNA pol III-dependent small RNAs, including many of the U snRNAs, tRNA and 5S RNA, had the expected intermediate α-amanitin sensitivity; 100–200 µg/ml blocked transcription. Interestingly, the α-amanitin inhibition profile of SL transcription did not precisely coincide with any of the three polymerase profiles. However, biochemical and genomic analyses indicated that only three types of DNA-dependent RNA polymerases exist in trypanosomes. Therefore, it remains unexplained why two classes of RNA pol II-dependent genes, namely the SL RNA genes and mRNA-encoding genes, have different α-amanitin sensitivities. This may be due to the fact that the SL RNA gene copy number is much higher, and transcript size much shorter, than gene copy numbers and transcription unit lengths for protein-coding genes. It is possible that at any given moment the number of polymerases actively elongating the polycistronic transcripts may be large compared to the number of polymerases at the site of transcription initiation. Just the opposite condition may exist within the SL RNA gene. Since α-amanitin binds to elongating polymerase, transcription of the protein-coding genes would be most sensitive to the drug. Inhibition of SL RNA transcription would be more refractory to the drug since the ratio of initiating polymerases to elongating polymerases would be high.
In metazoans and yeast, the CTD is capable of interacting with a variety of factors involved in transcription, including initiation, splicing, capping and polyadenylation factors (48–54). Typically, the CTD in pre-initiation complexes is hypophosphorylated, but actively elongating enzyme contains hyperphosphorylated CTD (reviewed in 55). A variety of kinases have been identified that phosphorylate the CTD, including CDK7 and CDK9/PITALRE (reviewed in 56), thus suggesting the potential for differential phosphorylation of RNA pol II CTDs. Moreover, studies in yeast have shown that subpopulations of genes are transcribed by different phosphorylated forms of RNA pol II largest subunit CTD (57). Our work now establishes that there are clearly two classes of RNA pol II-dependent genes in trypanosomes. A correlation of enzyme phosphorylation states with both pre-initiation complex formation and elongation during transcription of SL RNA and mRNA genes will determine whether these two classes of RNA pol II-dependent genes are initiated and elongated by differentially phosphorylated forms of the RNA pol II largest subunit CTD.
RNA pol II is probably brought to the SL gene promoter to form the pre-initiation complex via interactions with DNA binding proteins. We have previously characterized two transcription factors, PBP-1 and PBP-2, that bind this tripartite promoter (11,13). In this current study, we identified a sequence-specific binding activity that recognizes the Inrt. Initiator elements have been described in metazoans (for review, see 58) and in an evolutionarily ancient protozoan, T.vaginalis (59). A variety of factors have been identified that interact with these elements (60–66). Considering that T.vaginalis and trypanosome species do not appear to contain the TATA binding protein TBP, and do not contain a heptapeptide repeated CTD, it is possible that there was a coevolution of RNA pol II and basal transcription factors. Hopefully, trypanosomes will provide us with a unique perspective on understanding the complex interactions of the CTD with factors involved in transcription.
Acknowledgments
ACKNOWLEDGEMENTS
The authors thank A.Das for the genomic library, A.Matkin for PBP-1/-2 preparations, J.Milone for the plasmid JM-1, R.Donnelly for all DNA sequencing, and D.Lukac for providing the HisORF 50 protein. V.B. is a Burroughs Wellcome New Investigator in Molecular Parasitology. This work was supported by NIH-NIAID grant AI29478 to V.B.
References
- 1.Vanhamme L. and Pays,E. (1995) Control of gene expression in trypanosomes. Microbiol. Rev., 59, 223–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bangs J.D., Crain,P.F., Hashizume,T., McCloskey,J.A. and Boothroyd,J.C. (1992) Mass spectrometry of mRNA cap 4 from trypanosomatids reveals two novel nucleosides. J. Biol. Chem., 267, 9805–9815. [PubMed] [Google Scholar]
- 3.Günzl A., Ullu,E., Dörner,M., Fragoso,S., Hoffmann,K., Milner,J., Morita,Y., Nguu,E., Vanacova,S., Wünsch,S., Dare,A., Kwon,H. and Tschudi,C. (1997) Transcription of the Trypanosoma brucei spliced leader RNA gene is dependent only on the presence of upstream regulatory elements. Mol. Biochem. Parasitol., 85, 67–76. [DOI] [PubMed] [Google Scholar]
- 4.Saito R.M., Elgort,M.G. and Campbell,D.A. (1994) A conserved upstream element is essential for transcription of the Leishmania tarentolae mini-exon. EMBO J., 13, 5460–5469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agami R., Aly,R., Halman,S. and Shapira,M. (1994) Functional analysis of cis-acting DNA elements required for expression of the SL RNA gene in the parasitic protozoan Leishmania amazonensis. Nucleic Acids Res., 22, 1959–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu M.C., Sturm,N., Saito,R., Roberts,T. and Campbell,D. (1998) Single nucleotide resolution of promoter activity and protein binding for the Leishmania tarentolae spliced leader RNA gene. Mol. Biochem. Parasitol., 94, 265–281. [DOI] [PubMed] [Google Scholar]
- 7.Bellofatto V. and Cross,G.A.M. (1989) Expression of a bacterial gene in a trypanosomatid protozoan. Science, 244, 1167–1169. [DOI] [PubMed] [Google Scholar]
- 8.Bellofatto V., Torres,J. and Cross,G.A.M. (1991) Stable transformation of Leptomonas seymouri by circular extrachromosomal elements. Proc. Natl Acad. Sci. USA, 88, 6711–6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huie J., He,P. and Bellofatto,V. (1997) In vitro transcription of the Leptomonas seymouri SL RNA gene using homologous cell extracts. Mol. Biochem. Parasitol., 90, 183–192. [DOI] [PubMed] [Google Scholar]
- 10.Goldring A., Karchi,M. and Michaeli,S. (1995) The spliced leader RNA gene of Leptomonas collosoma. Exp. Parasitol., 80, 333–338. [DOI] [PubMed] [Google Scholar]
- 11.Luo H., Gilinger,G., Mukherjee,D. and Bellofatto,V. (1999) Transcription initiation at the TATA-less spliced leader RNA gene promoter requires at least two DNA-binding proteins and a tripartite architecture that includes an initiator element. J. Biol. Chem., 274, 31947–31954. [DOI] [PubMed] [Google Scholar]
- 12.Hartree D. and Bellofatto,V. (1995) Essential components of the mini-exon promoter in the trypanosomatid Leptomonas seymouri. Mol. Biochem. Parasitol., 71, 27–39. [DOI] [PubMed] [Google Scholar]
- 13.Luo H. and Bellofatto,V. (1997) Characterization of two protein activities that interact at the promoter of the trypanosomatid Spliced Leader RNA. J. Biol. Chem., 272, 33344–33352. [DOI] [PubMed] [Google Scholar]
- 14.Evers R., Hammer,A., Kock,J., Jess,W., Borst,P., Memet,S. and Cornelissen,A.W. (1989) Trypanosoma brucei contains two RNA polymerase II largest subunit genes with an altered C-terminal domain. Cell, 56, 585–597. [DOI] [PubMed] [Google Scholar]
- 15.Croan D., Morrison,D. and Ellis,J. (1997) Evolution of the genus Leishmania revealed by comparison of DNA and RNA polymerase gene sequences. Mol. Biochem. Parasitol., 89, 149–159. [DOI] [PubMed] [Google Scholar]
- 16.Croan D.G. and Ellis,J. (2000) The Leishmania major RNA polymerase II largest subunit lacks a carboxy-terminus heptad repeat structure and its encoding gene is linked with the calreticulin gene. Protist, 151, 57–68. [DOI] [PubMed] [Google Scholar]
- 17.Kock J., Evers,R. and Cornelissen,A.W.C.A. (1988) Structure and sequence of the gene for the largest subunit of trypanosomal RNA polymerase III. Nucleic Acids Res., 16, 8753–8772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith J.L., Levin,J.R., Ingles,C.J. and Agabian,N. (1989) In trypanosomes the homolog of the largest subunit of RNA polymerase II is encoded by two genes and has a highly unusual C-terminal domain structure. Cell, 56, 815–827. [DOI] [PubMed] [Google Scholar]
- 19.Smith J.L., Chapman,A.B. and Agabian,N. (1993) Trypanosoma vivax: evidence for only one RNA polymerase II largest subunit gene in a trypanosome which undergoes antigenic variation. Exp. Parasitol., 76, 242–246. [DOI] [PubMed] [Google Scholar]
- 20.Lee M.G. and Van der Ploeg,L.H. (1997) Transcription of protein-coding genes in trypanosomes by RNA polymerase I. Annu. Rev. Microbiol., 51, 463–489. [DOI] [PubMed] [Google Scholar]
- 21.Fantoni A., Dare,A.O. and Tschudi,C. (1994) RNA polymerase III-mediated transcription of the trypanosome U2 small nuclear RNA gene is controlled by both intragenic and extragenic regulatory elements. Mol. Cell. Biol., 14, 2021–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee M.G. (1996) An RNA polymerase II promoter in the hsp70 locus of Trypanosoma brucei. Mol. Cell. Biol., 16, 1220–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McAndrew M., Graham,S., Hartmann,C. and Clayton,C. (1998) Testing promoter activity in the trypanosome genome: isolation of a metacyclic-type VSG promoter and unexpected insights into RNA polymerase II transcription. Exp. Parasitol., 90, 65–76. [DOI] [PubMed] [Google Scholar]
- 24.Laird P.W., Kooter,J.M., Loosbroek,N. and Borst,P. (1985) Mature mRNAs of Trypanosoma brucei possess a 5′ cap acquired by discontinuous RNA synthesis. Nucleic Acids Res., 13, 4253–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grondal E.J.M., Evers,R., Kosubek,K. and Cornelissen,A.W.C.A. (1989) Characterization of the RNA polymerases of Trypanosoma brucei: trypanosomal mRNAs are composed of transcripts derived from both RNA polymerase II and III. EMBO J., 8, 3383–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crenshaw-Williams K. and Bellofatto,V. (1999) In vivo transcriptional analysis of the spliced leader RNA gene in the trypanosomatid Leptomonas seymouri. Parasitol. Res., 85, 700–706. [DOI] [PubMed] [Google Scholar]
- 27.Rudenko G. and Van der Ploeg,L.H.T. (1992) The PARP and VSG genes of Trypanosoma brucei do not resemble RNA polymerase II transcription units in sensitivity to Sarkosyl in nuclear run-on assays. Nucleic Acids Res., 20, 303–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agabian N. (1990) Trans splicing of nuclear pre-mRNAs. Cell, 61, 1157–1160. [DOI] [PubMed] [Google Scholar]
- 29.Lamontagne J. and Papadopoulou,B. (1999) Developmental regulation of spliced leader RNA gene in Leishmania donovani amastigotes is mediated by specific polyadenylation. J. Biol. Chem., 274, 6602–6609. [DOI] [PubMed] [Google Scholar]
- 30.Sturm N.R., Yu,M.C. and Campbell,D.A. (1999)Transcription termination and 3′-end processing of the spliced leader RNA in kinetoplastids. Mol. Cell. Biol., 19, 1595–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wen L.M., Xu,P., Benegal,G., Carvalho,M.R. and Buck,G.A. (2000) PPB1, a putative spliced leader RNA gene transcription factor in Trypanosoma cruzi. Mol. Biochem. Parasitol., 110, 207–221. [DOI] [PubMed] [Google Scholar]
- 32.Gunzl A., Ullu,E., Dorner,M., Fragoso,S.P., Hoffmann,K.F., Milner,J.D., Morita,Y., Nguu,E.K., Vanacova,S., Wunsch,S., Dare,A.O., Kwon,H. and Tschudi,C. (1997) Transcription of the Trypanosoma brucei spliced leader RNA gene is dependent only on the presence of upstream regulatory elements. Mol. Biochem. Parasitol., 85, 67–76. [DOI] [PubMed] [Google Scholar]
- 33.Lukac D.M., Renne,R., Kirshner,J.R. and Ganem,D. (1998) Reactivation of Kaposi’s sarcoma-associated herpes virus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology, 252, 304–312. [DOI] [PubMed] [Google Scholar]
- 34.Roy A.L., Meisterernst,M., Pognonec,P. and Roeder,R.G. (1991) Cooperative interaction of an initiator-binding transcription initiation factor and the helix–loop–helix activator USF. Nature, 354, 245–248. [DOI] [PubMed] [Google Scholar]
- 35.Ahearn J.M.,Jr, Bartolomei,M.S., West,M.L., Cisek,L.J. and Corden,J.L. (1987) Cloning and sequence analysis of the mouse genomic locus encoding the largest subunit of RNA polymerase II. J. Biol. Chem., 262, 10695–10705. [PubMed] [Google Scholar]
- 36.Chapman A.B. and Agabian,N. (1994) Trypanosoma brucei RNA polymerase II is phosphorylated in the absence of carboxyl-terminal domain heptapeptide repeats. J. Biol. Chem., 269, 4754–4760. [PubMed] [Google Scholar]
- 37.Bartolomei M.S. and Corden,J.L. (1987) Localization of an α-amanitin resistance mutation in the gene encoding the largest subunit of mouse RNA polymerase II. Mol. Cell Biol., 7, 586–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bartolomei M.S. and Corden,J.L. (1995) Clustered α-amanitin resistance mutations in mouse. Mol. Gen. Genet., 246, 778–782. [DOI] [PubMed] [Google Scholar]
- 39.Bryant R.E., Adelberg,E.A. and Magee,P.T. (1977) Properties of an altered RNA polymerase II activity from an α-amanitin-resistant mouse cell line. Biochemistry, 16, 4237–4244. [DOI] [PubMed] [Google Scholar]
- 40.Coulter D.E. and Greenleaf,A.L. (1982) Properties of mutationally altered RNA polymerases II of Drosophila. J. Biol. Chem., 257, 1945–1952. [PubMed] [Google Scholar]
- 41.Greenleaf A.L., Borsett,L.M., Jiamachello,P.F. and Coulter,D.E. (1979) α-Amanitin-resistant D. melanogaster with an altered RNA polymerase II. Cell, 18, 613–622. [DOI] [PubMed] [Google Scholar]
- 42.Greenleaf A.L., Weeks,J.R., Voelker,R.A., Ohnishi,S. and Dickson,B. (1980) Genetic and biochemical characterization of mutants at an RNA polymerase II locus in D. melanogaster. Cell, 21, 785–792. [DOI] [PubMed] [Google Scholar]
- 43.Bullerjahn A.M. and Riddle,D.L. (1988) Fine-structure genetics of ama-1, an essential gene encoding the amanitin-binding subunit of RNA polymerase II in Caenorhabditis elegans. Genetics, 120, 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rogalski T.M., Bullerjahn,A.M. and Riddle,D.L. (1988) Lethal and amanitin-resistance mutations in the Caenorhabditis elegans ama-1 and ama-2 genes. Genetics, 120, 409–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanford T., Golomb,M. and Riddle,D.L. (1983) RNA polymerase II from wild type and α-amanitin-resistant strains of Caenorhabditis elegans. J. Biol. Chem., 258, 12804–12809. [PubMed] [Google Scholar]
- 46.Quon D.V.K., Delgadillo,M.G. and Johnson,P.J. (1996) Transcription in the early diverging eukaryote Trichomonas vaginalis: an unusual RNA polymerase II and α-amanitin-resistant transcription of protein-coding genes. J. Mol. Evol., 43, 253–262. [DOI] [PubMed] [Google Scholar]
- 47.Campbell D.A., Sturm,N.R. and Yu,M.C. (2000) Transcription of the kinetoplastid spliced leader RNA gene. Parasitol. Today, 16, 78–82. [DOI] [PubMed] [Google Scholar]
- 48.McCracken S., Fong,N., Yankulov,K., Ballantyne,S., Pan,G., Greenblatt,J., Patterson,S.D., Wickens,M. and Bentley,D.L. (1997) The C-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature, 385, 357–361. [DOI] [PubMed] [Google Scholar]
- 49.McCracken S., Fong,N., Rosonina,E., Yankulov,K., Brothers,G., Siderovski,D., Hessel,A., Foster,S., Shuman,S. and Bentley,D.L. (1997) 5′-Capping enzymes are targeted to pre-mRNA by binding to the phosphorylated carboxy-terminal domain of RNA polymerase II. Genes Dev., 11, 3306–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Usheva A., Maldonado,E., Goldring,A., Lu,H., Houbavi,C., Reinberg,D. and Aloni,Y. (1992) Specific interaction between the nonphosphorylated form of RNA polymerase II and the TATA-binding protein. Cell, 69, 871–881. [DOI] [PubMed] [Google Scholar]
- 51.Kang M.E. and Dahmus,M.E. (1995) The photoactivated cross-linking of recombinant C-terminal domain to proteins in a HeLa cell transcription extract that comigrate with transcription factors IIE and IIF. J. Biol. Chem., 270, 23390–23397. [DOI] [PubMed] [Google Scholar]
- 52.Shuman S. (1997) Origins of mRNA identity: capping enzymes bind to the phosphorylated C-terminal domain of RNA polymerase II. Proc. Natl Acad. Sci. USA, 94, 12758–12760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cho E.J., Rodriguez,C.R., Takagi,T. and Buratowski,S. (1998) Allosteric interactions between capping enzyme subunits and the RNA polymerase II carboxy-terminal domain. Genes Dev., 12, 3482–3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ho C.K., Sriskanda,V., McCracken,S., Bentley,D., Schwer,B. and Shuman,S. (1998) The guanylyltransferase domain of mammalian mRNA capping enzyme binds to the phosphorylated carboxyl-terminal domain of RNA polymerase II. J. Biol. Chem., 273, 9577–9585. [DOI] [PubMed] [Google Scholar]
- 55.Shatkin A.J. and Manley,J.L. (2000) The ends of the affair: capping and polyadenylation. Nat. Struct. Biol., 7, 838–842. [DOI] [PubMed] [Google Scholar]
- 56.Bensaude O., Bonnet,F., Casse,C., Dubois,M.-F., Nguyen,V.T. and Palancade,B. (1999) Regulated phosphorylation of the RNA polymerase II C-terminal domain (CTD). Biochem. Cell Biol., 77, 249–255. [PubMed] [Google Scholar]
- 57.Patturajan M., Schulte,R.J., Sefton,B.M., Berezney,R., Vincent,M., Bensaude,O., Warren,S.L. and Corden,J.L. (1998) Growth-related changes in phosphorylation of yeast RNA polymerase II. J. Biol. Chem., 273, 4689–4694. [DOI] [PubMed] [Google Scholar]
- 58.Smale S.T., Jain,A., Kaufmann,J., Emami,K.H., Lo,K. and Garraway,I.P. (1998) The initiator element: a paradigm for core promoter heterogeneity within metazoan protein-coding genes. Cold Spring Harb. Symp. Quant. Biol., 63, 21–31. [DOI] [PubMed] [Google Scholar]
- 59.Liston D.R. and Johnson,P.J. (1999) Analysis of a ubiquitous promoter element in a primitive eukaryote: early evolution of the initiator element. Mol. Cell. Biol., 19, 2380–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carcamo J., Buckbinder,L. and Reinberg,D. (1991) The initiator directs the assembly of a transcription factor IID-dependent transcription complex. Proc. Natl Acad. Sci. USA, 88, 8052–8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aso T., Conaway,J.W. and Conaway,R.C. (1994) Role of core promoter structure in assembly of the RNA polymerase II preinitiation complex. A common pathway for formation of preinitiation intermediates at many TATA and TATA-less promoters. J. Biol. Chem., 269, 26575–26583. [PubMed] [Google Scholar]
- 62.Means A.L., Slansky,J.E., McMahon,S.L., Knuth,M.W. and Farnham,P.J. (1992) The HIP1 binding site is required for growth regulation of the dihydrofolate reductase gene promoter. Mol. Cell Biol., 12, 1054–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Usheva A. and Shenk,T. (1994) TATA-binding protein-independent initiation: YY1, TFIIB and RNA polymerase II direct basal transcription on supercoiled template DNA. Cell, 76, 1115–1121. [DOI] [PubMed] [Google Scholar]
- 64.Kaufmann J., Verrijzer,C.P., Shao,J. and Smale,S.T. (1996) CIF, an essential cofactor for TFIID-dependent initiator function. Genes Dev., 10, 873–886. [DOI] [PubMed] [Google Scholar]
- 65.Roy A.L., Malik,S., Meisterernst,M. and Roeder,R.G. (1993) An alternative pathway for transcription initiation involving TFII-I. Nature, 365, 355–359. [DOI] [PubMed] [Google Scholar]
- 66.Roy A.L., Du,H., Gregor,P.D., Novina,C.D., Martinez,E. and Roeder,R.G. (1997) Cloning of an inr- and E-box-binding protein, TFII-I, that interacts physically and functionally with USF1. EMBO J., 16, 7091–7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Croan D. and Ellis,J. (1996) Phylogenetic relationships between Leishmania, Viannia and Sauroleishmania inferred from comparison of a variable domain within the RNA polymerase II largest subunit gene. Mol. Biochem. Parasitol., 79, 97–102. [DOI] [PubMed] [Google Scholar]
- 68.Evers R., Hammer,A. and Cornelissen,A.W.C.A. (1989) Unusual C-terminal domain of the largest subunit of RNA polymerase II of Crithidia fasciculata. Nucleic Acids Res., 17, 3403–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]