

Abstract
CEBPA mutations are of prognostic relevance in acute myeloid leukemia (AML) and are currently detected using a combination of denaturing high-performance liquid chromatography (DHPLC), gene scan/fragment length analysis, and direct Sanger sequencing. Next-generation deep pyrosequencing, principally, allows for the highly sensitive detection of molecular mutations. However, standard 454 chemistry laboratory procedures lack efficient amplification of guanine-cytosine (GC)-rich amplicons during the emulsion PCR (emPCR) steps allowing direct massively parallel clonal amplification of PCR products. To solve this problem, we investigated six distinct emPCR conditions. The coding sequence of CEBPA was subdivided into four overlapping amplicons: GC content for amplicon 1, 74%; amplicon 2, 76%; amplicon 3, 77%; and amplicon 4, 69%. A new emPCR condition, improving the standard titanium assay, presents a robust solution to sequence amplicons with a GC content of up to 77%. Moreover, this assay was subsequently tested on a larger independent cohort of 23 AML patients. For each patient, a median of 737 reads was generated (coverage range, 397-fold to 1194-fold) and therefore allowed a robust detection of insertions, deletions, and point mutations. In conclusion, next-generation amplicon sequencing enables the highly sensitive detection of molecular mutations and is a feasible assay for routine assessment of GC-rich content amplicons.
CEBPA (CCAAT/enhancer binding protein α) encodes a protein member of the basic region leucine zipper (bZIP) transcription factor family that is essential for myeloid differentiation.1,2 CEBPA mutations occur predominantly in AML with a normal karyotype and have been reported to occur in a range of 13% to 19% of such patients.1,3,4 Importantly, CEBPA mutations have been associated with a relatively favorable outcome and have therefore gained interest as a biomarker with prognostic utility.5 CEBPA-mutant AMLs have recently been included into the World Health Organization 2008 classification as a provisional entity.6 It is therefore warranted to provide a robust, objective, and sensitive detection method for the two prototypical classes of mutations: N-terminal mutations are located between the major translational start codon and a second ATG in the same open reading frame. These mutations most frequently introduce a premature stop of translation of the p42 CEBPA protein while preserving translation of a p30 isoform that has been reported to inhibit the function of full-length protein. Second, mutations in the C-terminal basic leucine zipper (bZIP) region, in contrast, are usually in-frame and may impair DNA binding and/or homodimerization and heterodimerization. The remaining mutations observed are found between the N-terminus and the bZIP region.7,8 Most CEBPA mutated AMLs carry two mutations, which most frequently involves a combination of an N-terminal and a bZIP gene mutation, whereas single heterozygous mutations are less frequently seen.8,9 Today, the screening of CEBPA mutations in patients with AML is often performed applying a combination of fragment length analysis, DHPLC, and subsequent direct Sanger sequencing.10,11
The 454 deep-pyrosequencing method includes emulsion PCR (emPCR) steps that allow a direct, massively parallel clonal amplification of PCR products. In principle, amplicon sequencing allows for a highly sensitive detection of molecular mutations. CEBPA mutations have not yet been investigated using next-generation sequencing technology. Moreover, standard Titanium chemistry laboratory procedures lack efficient amplification of highly guanine-cytosine (GC)-rich amplicons.
We investigated six distinct emulsion-based clonal amplification conditions and present a robust solution for subsequent sequencing of PCR amplicons with a GC content of up to 77%. In this assay, the coding sequence of CEBPA was subdivided into four distinct amplicons. Subsequently, the performance of this optimized emPCR condition was tested on a larger cohort of 23 AML patients and allowed for the sensitive detection of insertions, deletions, and point mutations.
Materials and Methods
Patient Samples
This work included 25 AML cases sent to the MLL Munich Leukemia Laboratory for diagnostic assessment between June 2005 and October 2009. All samples in this study were obtained from untreated AML patients and were preselected according to their known CEBPA mutation status. Bone marrow or peripheral blood mononuclear cells were enriched for molecular analyses using a Ficoll density gradient. The study design adhered to the tenets of the Declaration of Helsinki and was approved by the institutional review board before its initiation.
Molecular Diagnostics
The CEBPA gene was first amplified in four PCR fragments and subsequently analyzed by DHPLC. All fragments that revealed an aberrant dissociation pattern by DHPLC analysis were subsequently analyzed by Sanger sequencing (according to standard laboratory protocols).
Next-Generation Sequencing
NGS was performed using 454 Titanium amplicon chemistry (Roche Applied Science, Penzberg, Germany).12 For each of the CEBPA patients, four overlapping amplicons were processed. Table 1 lists the corresponding primer sequences and depicts the GC content for the four amplicons. The GC-RICH PCR System (Roche Applied Science) was used to amplify CEBPA fragments from genomic patient DNA according to the manufacturer's recommendations. The PCR conditions were as follows: 95°C for 3 minutes; 45 cycles of 95°C for 60 seconds, 60°C for 60 seconds, and 72°C for 60 seconds; 72°C for 10 minutes; and 12°C hold. PCR products were subsequently purified using AMPure XP beads (Beckman Coulter, Krefeld, Germany) and quantified using the Quant-iT PicoGreen kit (Invitrogen, Carlsbad, CA). For each patient, 100 ng each of the four CEBPA amplicons were pooled in an equimolar ratio to generate one single patient-specific CEBPA library. According to the manufacturer's recommendations, the multiplexed amplicon pools were subsequently diluted to 4 × 106 molecules/μL. This working dilution was used as starting concentration for the GS FLX Titanium small volume (SV) emPCR Kit (Lib-A) (Roche Applied Science), processing forward (A) and reverse (B) reactions in separate amplification mixes. The emPCR thermocycler conditions were as follows: 94°C for 4 minutes; 50 cycles of 94°C for 30 seconds, 58°C for 4.5 minutes, 68°C for 30 seconds; and 10°C hold (Table 2). Following the emPCR amplification, clonally amplified beads were enriched for 454 next-generation sequencing following the manufacturer's recommendations, processing forward (A) and reverse (B) beads separately until combining them at the step of loading the respective PicoTiterPlate (PTP) lanes. Two molecular barcodes were used, ie, standard 454 Life Sciences 10-base sequences MID1 (ACGAGTGCGT) and MID2 (ACGCTCGACA), incorporated in the PCR primer sequences to allow multiplexing of distinct patient libraries per sequencing lane on the 454 PTP.
Table 1.
Primers Used for Amplification of CEBPA Fragments and Their GC Content
| Primer set | Forward | Temp °C | Reverse | Tm °C | Length (including sequence-specific primer) | Region (AA) | G | C | GC content |
|---|---|---|---|---|---|---|---|---|---|
| CEBPA Set A1 | 5′-GCCATGCCGGGAGAACT-3′ | 62 | 5′-CCCGGGTAGTCAAAGTCG-3′ | 59 | 357 bp | 1–103 | 116 | 147 | 74% |
| CEBPA Set A2 | 5′-CCTTCAACGACGAGTTCCTG-3′ | 61 | 5′-CGGCTGGTAAGGGAAGAGG-3′ | 62 | 335 bp | 79–176 | 130 | 126 | 76% |
| CEBPA Set A3 | 5′-GAGGAGGATGAAGCCAAGC-3′ | 60 | 5′-CTCGTTGCTGTTCTTGTCCA-3′ | 60 | 357 bp | 173–277 | 113 | 163 | 77% |
| CEBPA Set A4 | 5′-TGGCAGCGCGCTCAAG-3′ | 65 | 5′-CCAGGGCGGTCCCACA-3′ | 65 | 357 bp | 255–359 | 136 | 110 | 69% |
CEBPA, CCAAT/enhancer binding protein α; G, guanine; C, cytosine.
Table 2.
Standard emPCR Recommendations (454) in 100 μL Reaction Volume and Optimized emPCR Protocol in 50 μL Reaction Volume
| Component | Standard emPCR recommendations |
Optimized emPCR protocol |
||
|---|---|---|---|---|
| 16 × (μL) | 1 × (μL) | 16 × (μL) | 1 × (μL) | |
| Molecular biology grade water | 1200 | 75 | — | — |
| emPCR additive | 1500 | 93.8 | 2700 | 168.8 |
| 5 × amplification mix | 780 | 48.8 | 780 | 48.8 |
| Amplification primer A or B | 230 | 14.4 | 230 | 14.4 |
| emPCR enzyme mix | 200 | 12.5 | 200 | 12.5 |
| PPiase enzyme | 5 | 0.3 | 5 | 0.3 |
emPCR, emulsion PCR.
Data Generation and Analysis
The 454 sequencing data were generated during several runs on a Genome Sequencer FLX instrument using the GS FLX Titanium Sequencing Kit XLR70 (Roche Applied Science). The PTP was divided into eight distinct lanes. After image generation using the GS Sequencer software version 2.3 and processing of the data using the GS Image and Signal Processing software version 2.3, the obtained reads for each amplicon were aligned against the reference sequences of CEBPA (Ensemble ID: ENSG00000245848) using the GS Amplicon Variant Analyzer software version 2.3 (Roche Applied Science). Mapping results and detected variants were exported to R/Bioconductor13 for further analysis and visualization by diagnostic plots shown in this article. Wilcoxon's matched pairs rank test was used to study the influence of the molecular barcodes MID1 and MID2 on the total number of reads generated per sample across all four PCR amplicons. Two samples with MID1 and MID2, respectively, located on the same PTP and lane were considered as paired. Because of the experimental design and loading of the PTP lanes, five of 23 test samples had to be removed from the statistical analysis due to a missing appropriate corresponding MID pair in the respective run.
Results
Amplification of CEBPA Fragments Using the Standard emPCR Condition
For 454 deep-sequencing analyses, the CEBPA gene was covered by four overlapping PCR fragments and processed using the small-volume emulsion-based clonal amplification Lib-A assay for PCR amplicons. In this first analysis, two patients (T13 and T15) were investigated using GS FLX Titanium Sequencing Chemistry standard emPCR amplification condition as recommended by the manufacturer. The live amplification mix was prepared for forward and reverse reactions, as given in Table 2, using the recommended total reaction volume of 100 μL per emPCR reaction in a 96-well plate format.
As demonstrated in Figure 1, for two patients, T13 and T15, the next-generation massively parallel pyrosequencing assay revealed mutations in the N- and C-terminal regions for both patients. All mutations were known from a previous DHPLC analysis at routine diagnostic work-up including subsequent Sanger sequencing. In detail, as given in Figure 1A, in patient T13 amplicon 1 was represented by 516 reads (228 forward and 288 reverse). The respective mutations were observed with the following percentages: H18Q (51%) and G53AfsX107 (47%). Amplicon 4 was represented by 580 reads (368 forward and 212 reverse) and contained the dupT310_R325 in 24%. In patient T15, amplicon 1 was represented by 263 reads (74 forward and 189 reverse) and contained the Q83SfsX76 mutation in 53% of obtained reads (Figure 1B). Amplicon 4 was represented by 870 reads (480 forward and 390 reverse) and contained the A295V mutation in 47% of sequencing reads (Figure 1C).
Figure 1.
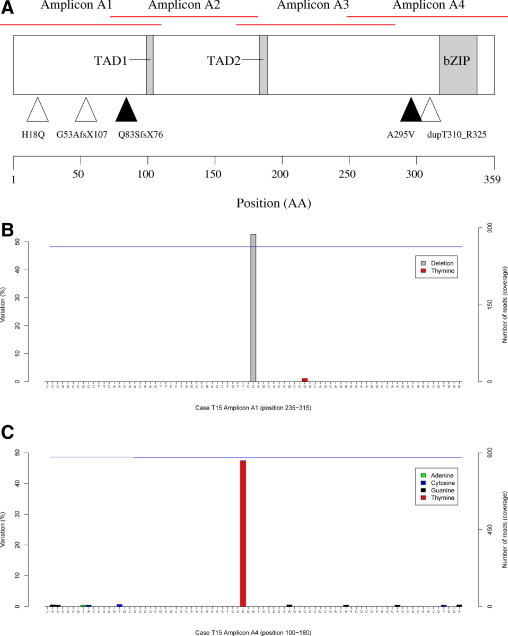
Next-generation sequencing of two AML cases. For both patients T13 and T15 mutations observed by next-generation amplicon sequencing are highlighted. A: Location of the mutations of sample T13 (open triangles) and T15 (filled triangles), corresponding to the functional protein domains. The CEBPA protein structure is given according to coding amino acids (lower part). Upper panel visualizes the overlapping amplicon design covering the complete coding sequence of CEBPA. B: Example of the Q83SfsX76 mutation in patient T15. As given on the right y axis for this amplicon, a 263-fold coverage was generated (blue line). The single-base deletion leading to the frameshift was detected in 139 (53%) of reads. C: Example of the A295V variant in patient T15. A substitution (884C>T) was observed in 412 (47%) of reads (left y axis). The overall coverage of 870 reads is highlighted by the blue line.
On investigation of the corresponding next-generation sequencing reads, the highest coverage was observed for amplicon 4 with a GC content of 69% (Figure 2). Although a lower coverage was observed for amplicon 1, amplicons 2 and 3 failed completely to amplify when the recommended standard GS FLX Titanium SV emPCR condition was applied. Therefore, amplicons with a GC content of >74% did not amplify during the emPCR (Figure 2).
Figure 2.
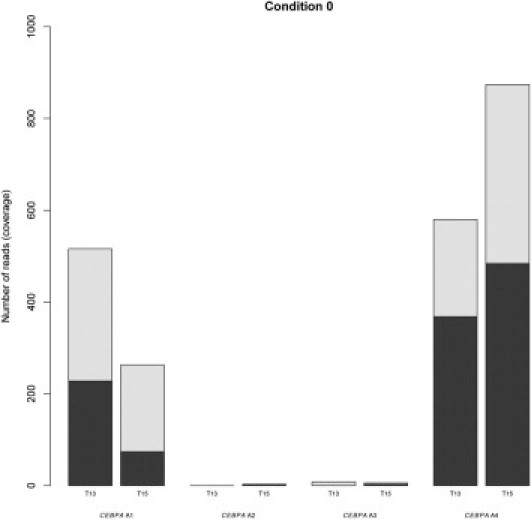
Sequencing coverage of the four CEBPA amplicons using standard GS FLX Titanium Sequencing emPCR conditions. For both patients T13 and T15, the number of obtained sequencing reads using the procedure as recommended by the manufacturer is given on the y axis (referred to as Condition 0). For each amplicon A1 to A4, the bar is colored according to the distribution of forward reads (dark area) and reverse reads (light area).
Amplification of CEBPA Fragments Using Four Different emPCR Conditions
In a second sequencing run, four different emPCR conditions were analyzed to improve the amplification of GC-rich template amplicons during the emPCR: Condition A, GS FLX Titanium Sequencing standard emPCR amplification conditions but 50-μL reaction volume instead of 100 μL; Condition B, GS FLX Titanium Sequencing emPCR master mix containing only Additive reagent instead of H2O and 50-μL reaction volume; Condition C, GS FLX Titanium Sequencing emPCR master mix containing only Additive reagent instead of H2O in a 100-μL reaction volume; and Condition D, GS FLX Titanium Sequencing emPCR master mix in a 100-μL reaction volume containing only 50% of H2O compared with standard conditions but 50% increased volume of Additive reagent.
As seen in Figure 3, condition A still failed to yield reads for amplicons 2 and 3. In contrast, condition B homogeneously amplified fragments 1, 2, and 4. Moreover, condition B also amplified the fragment 3 with the highest GC content of 77%. A substantially lower coverage for amplicon 3 was observed for conditions C and D. The latter two conditions C and D revealed a substantially more heterogeneous distribution of reads than condition B and were not selected for the next round of optimization. Overall, for each of the two cases (patients T13 and T15), a similar trend was observed across all distinct emPCR conditions and fragments. In conclusion, of this series of four experiments, condition B was chosen for further investigation.
Figure 3.
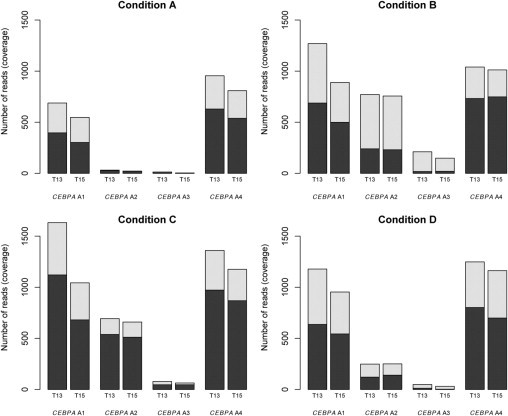
Sequencing coverage of the four CEBPA amplicons using various emPCR conditions. For both patients T13 and T15, four distinct GS FLX Titanium Sequencing emPCR conditions were tested (A–D). The number of obtained sequencing reads is given on the y axis. For each amplicon A1 to A4, the bar is colored according to the distribution of forward reads (dark area) and reverse reads (light area).
Amplification of CEBPA Fragments Using Condition B and Diverse Amounts of PCR Products and Forward (A) and Reverse (B) Beads
Next, to further improve on the coverage of each amplicon, a third sequencing run was performed. This series of experiments was based on amplicon pools that included twice the amount of input PCR product for amplicon 2 and a fourfold input amount of PCR product for amplicon 3. As seen in Figure 4, condition B.1 homogeneously amplified fragments 1, 2, and 4. Fragment 3 also produced a significant number of sequencing reads that was improved over the previous condition B. A final iteration tested a variation of forward (A) and reverse (B) beads (ie, a 1.5-fold input of A beads was used). As observed for the condition B.2, a slightly higher percentage of forward reads was obtained for fragment 3; however, for other amplicons, the number of forward and reverse reads was less homogeneously distributed (Figure 4). Therefore, condition B.1 and not B.2 was selected as the final optimized condition for the emPCR amplification reaction and was selected for being tested on a larger cohort of AML patients.
Figure 4.
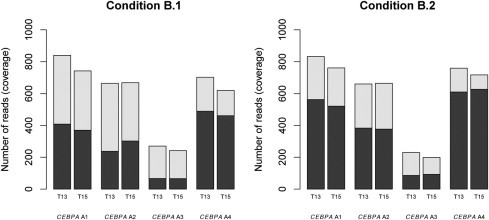
Sequencing coverage of the four CEBPA amplicons using the optimized emPCR conditions B. For patients T13 and T15, two distinct variations of condition B (B.1 and B.2) were tested. The number of obtained sequencing reads is given on the y axis. For each amplicon A1 to A4, the bar is colored according to the distribution of forward reads (dark area) and reverse reads (light area).
Characterization of Patients Using Optimized emPCR Amplification Condition
The optimized emPCR amplification protocol B.1 was tested on an independent cohort of 23 AML patients. All patients were fully characterized according to the World Health Organization 2008 classification. As depicted in Table 3, a median coverage of 737-fold was obtained per patient across all amplicons for these 23 cases. In all cases, condition B.1 proved to robustly yield sufficient reads for all four CEBPA fragments. This is depicted in Figure 5 for all 23 AML patients, both for the overall amplicon coverage (Figure 5A) and for the respective representation of forward and reverse reads (Figure 5B). Overall, the cases processed with molecular barcode MID1 produced slightly more sequencing reads than MID2. Table 4 lists the corresponding median values and range for each amplicon according to the MID used (P = 0.01). Therefore, distinct performances according to the 10-base molecular barcode were observed and would benefit from further investigation. In this independent cohort of 23 patients, after excluding silent mutations and polymorphisms, we observed 35 mutations (Table 3). Of those mutations that were previously known from the time point of routine assessment (n = 31), NGS analysis was able to detect and confirm all of them. However, it is important to note that in four of 23 cases (17%), NGS ultra-deep sequencing detected mutations not observed by the previous routine assessment. In detail, these mutations ranged from 10% (M15V) to 49% (R297P). The latter mutation was not detected in the WAVE assay because of technical limitations and thus was not further processed by Sanger sequencing.
Table 3.
Characteristics and Sequencing Results of CEBPA Mutations (Amplification Protocol B.1)
| Sample | Sex | Age (years) | Diagnosis | Cytogenetics | Median coverage | NGS results |
|---|---|---|---|---|---|---|
| T13 | F | 25 | AML M2 | 46,XX [20] | 683 | H18Q (58%), G53AfsX107 (58%), T310_R325dup (15%) |
| T15 | F | 48 | AML | 46,XX [20] | 644 | Q83SfsX77 (48%), A295V (51%) |
| T266 | F | 65 | AML | 46,XX [15] | 860 | No mutation found |
| T267 | M | 68 | AML M1 | 46,XY [20] | 1194 | dupE309_E316 (30%) |
| T268 | M | 25 | AML M1 | 46,XY [25] | 618 | P22LfsX118 (64%), dupL315 (50%) |
| T270 | F | 26 | AML M1 | 46,XX [20] | 865 | A84PfsX112 (39%), V308GinsR (35%) |
| T271 | M | 36 | AML M1 | 46,XY [20] | 1153 | F106LfsX54 (27%), dupK302 (41%) |
| T273 | F | 51 | AML M1 | 46,XX [20] | 778 | H24AfsX136 (22%), R297P (49%) |
| T274 | F | 53 | AML | 46,XX [20] | 702 | No mutation found |
| T275 | F | 71 | AML M1 | 46,XX [20] | 851 | Q87X (37%) |
| T276 | F | 79 | AML | 46,XX [20] | 658 | R264GfsX55 (42%), L338HfsX22 (37%) |
| T277 | M | 43 | AML M6 | 46,XY [35] | 607 | G116RfsX54 (42%), dupK302_K313 (24%) |
| T278 | F | 87 | AML M2 | 46,XX [20] | 403 | Q41SfsX117 (46%), F82SfsX78 (37%) |
| T279 | F | 60 | AML M1 | 46,XX [17] | 440 | D80GfsX28 (37%) |
| T280 | F | 85 | AML M2 | 46,XX [20] | 440 | M15V (10%), C133WfsX17 (10%) |
| T281 | F | 71 | t-AML M1 | 46,XX [20] | 397 | G110RfsX212 (39%) |
| T282 | M | 83 | s-AML | 46,XY [20] | 737 | G54EfsX106 (70%) |
| T283 | M | 75 | AML | 48,XY,+8,+18 [19] | 729 | A152T (48%) |
| T284 | F | 63 | AML M2 | 46,XX [20] | 1047 | Y181LfsX140 (27%) |
| T285 | M | 76 | MDS | 47,XY,+15 [19] | 808 | P23QfsX81 (34%) |
| T286 | M | 72 | s-AML | 46,XY [20] | 879 | Y285C (83%) |
| T287 | F | 78 | AML M1 | 46,XX [20] | 763 | H24AfsX84 (33%), dupQ312 (44%) |
| T288 | M | 63 | s-AML | 46,XY [21] | 633 | No mutation found |
| T289 | F | 87 | AML M2 | 47,XX,+13 [14] | 1101 | P187_P189del (57%) |
| T290 | M | 73 | s-AML | Complex aberrant karyotype including abnormalities on chromosomes 11 and 13 [9] | 656 | I68V (15%), P128RfsX32 (18%) |
F, female; M, male.
Bold type incidates mutations that were not detectable with conventional routine assays because of technical limitations of the WAVE assay.
Note: Subsequent Sanger sequencing confirmed the R297P (49%) and H24AfsX84 (33%) mutations.
Figure 5.
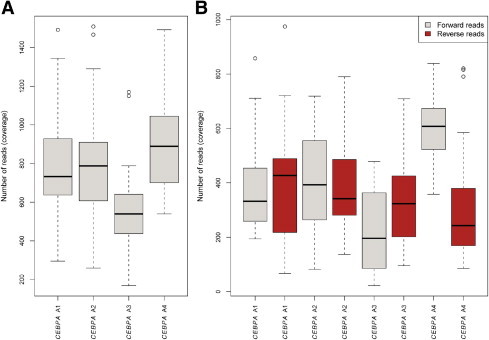
Independent testing cohort of 23 AML patients using the optimized emPCR condition B.1. The number of obtained sequencing reads is given on the y axis. A: Box-and-whisker plots depict the overall sequencing coverage of the four CEBPA amplicons A1 to A4. B: Box-and-whisker plots depict the overall sequencing coverage of the four CEBPA amplicons represented as forward and reverse reads.
Table 4.
Performance of the Distinct Molecular Barcodes MID1 and MID2
| Amplicon | Median MID1 (range) | Median MID2 (range) |
|---|---|---|
| CEBPA A1 | 830 (621–998) | 658 (295–1492) |
| CEBPA A2 | 865 (723–1136) | 655 (259–1509) |
| CEBPA A3 | 596 (463–1170) | 499 (390–1150) |
| CEBPA A4 | 1047 (802–1492) | 708 (539–1052) |
Discussion
CEBPA (CCAAT/enhancer binding protein α) encodes a protein member of the basic region leucine zipper (bZIP) transcription factor family that is essential for myeloid differentiation.1,2 According to current standard laboratory procedures, CEBPA mutations are currently detected using a combination of DHPLC, gene scan/fragment length analysis, and direct Sanger sequencing. Fragment length analysis cannot detect base substitutions, and therefore it would not be technically possible to detect eight of 35 mutations (23%) as observed in our cohort. Moreover, DHPLC analyses can miss rare mutations (eg, if these are located at the very end of the respective amplicon). In addition, DHPLC can miss changes in AT- or GC-rich regions, particularly if it is a base duplication (eg, A>AA, T>TT, G>GG or C>CC). Finally, conventional Sanger sequencing allows an immediate and detailed analysis of PCR-amplified DNA fragments,10 but has a currently accepted lower cut-off value of 20% diagnostic sensitivity.
As presented in this study, the NGS analysis would have outperformed the current sequencing-based workflow. Because of the high GC content of two CEBPA fragments, the emPCR, as part of the 454 assay, was not sufficiently amplifying these two PCR products. Therefore, we have optimized the conditions for CEBPA analysis on a 454 next-generation sequencing platform. In total, 23 patients with known CEBPA mutation status were analyzed in a second step for independent validation of this modified emPCR assay. Most CEBPA-mutant AMLs carry two mutations, which frequently involves a combination of an N-terminal and a bZIP gene mutation.8 The data for this selected cohort showed that in 12 samples the patients harbored more than one mutation. In three cases, these mutations were detected in the same respective amplicons, and in nine cases the mutations occurred in separate amplicons.
Moreover, in those three cases with double mutations within the same respective amplicons, 454 deep-sequencing allowed for the prediction of the presence or absence of distinct subclones harboring different mutations. In patient T13, we detected two distinct mutations in the same amplicon occurring in the same sequencing reads, and for patients T276 and T278 we were able to detect separate subclones because the mutated codons were not found concomitantly within the same sequencing read but were separated across distinct individual reads.
It has been shown that biallelic disruption of the N and C terminus of CEBPA is required for the favorable clinical outcome.9 As such, it is expected that with a future increase in read length of next-generation sequencing assays it will be possible to clearly correlate the reads of all double mutations to a monoallelic or biallelic status of the mutation, thereby underlining the clinical utility of such a diagnostic assay.
In conclusion, our results indicate that 454 next-generation amplicon sequencing is a sensitive assay suitable for routine assessment of the important parameter of CEBPA mutations in AML. We were able to show that this procedure allowed for the detection of all CEBPA mutations as known from current routine assays. Moreover, some CEBPA mutations that were not identifiable using the current standard workflow were detected and therefore highlight the utility of this method for a total comprehensive screening of CEBPA mutations. Of note, we observed in our cohort two distinct mutations with a mutational burden of <15%, underlining the sensitivity of amplicon-based deep-sequencing. Yet, further research will be necessary to fully understand the clinical relevance of these small subclones, as amplicon deep-sequencing not only provides a technical means to detect these subclones at diagnosis but also allows for the quantitative monitoring of these aberrations during a course of treatment. Furthermore, NGS also allowed new insights into the distinction of separate subclones with mutations derived from the same PCR amplicon. Because double mutations of CEBPA have been proposed to represent a distinct molecular subtype of AML with a normal karyotype, this method may add to future risk-adapted therapeutic strategies and improve the outcome of AML.8 Although this study was designed to address the methodology of NGS and certain amplification considerations using a single gene, it is further anticipated that NGS will evolve as a suitable platform to cover the needs of providing data on multiple molecular mutations in a high-level throughput and accuracy, not only in hematological malignancies, and as such will rapidly advance into the field of standard molecular diagnostics. Recently, such a study was presented in which 43 amplicons covering seven candidate genes (RUNX1, TET2, CBL, JAK2, MPL, NRAS, and KRAS) were investigated in a cohort of 81 chronic myelomonocytic leukemia patients, generating novel molecular insights into this poorly characterized category of myeloid malignancy.14 As for AML, amplicon deep-sequencing not only can be applied to cover mutational hotspot regions of established markers (NPM1, FLT3) but also enables the broader in-depth investigation of novel candidates, such as TET2,15 EZH2,16 and ASXL1,17 or the characterization of even larger genes, such as NF1.18
Footnotes
Disclosures: Su.S., W.K., T.H., and C.H. are partial owners of the MLL Munich Leukemia Laboratory GmbH. V.G., So.S., C.E., and A.K. are employed by MLL Munich Leukemia Laboratory GmbH. The other authors declare no conflict of interest.
References
- 1.Marcucci G., Maharry K., Radmacher M.D., Mrozek K., Vukosavljevic T., Paschka P., Whitman S.P., Langer C., Baldus C.D., Liu C.G., Ruppert A.S., Powell B.L., Carroll A.J., Caligiuri M.A., Kolitz J.E., Larson R.A., Bloomfield C.D. Prognostic significance of, and gene and microRNA expression signatures associated with. CEBPA mutations in cytogenetically normal acute myeloid leukemia with high-risk molecular features: a Cancer and Leukemia Group B Study. J Clin Oncol. 2008;26:5078–5087. doi: 10.1200/JCO.2008.17.5554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nerlov C. C/EBPalpha mutations in acute myeloid leukaemias. Nat Rev Cancer. 2004;4:394–400. doi: 10.1038/nrc1363. [DOI] [PubMed] [Google Scholar]
- 3.Frohling S., Schlenk R.F., Stolze I., Bihlmayr J., Benner A., Kreitmeier S., Tobis K., Dohner H., Dohner K. CEBPA mutations in younger adults with acute myeloid leukemia and normal cytogenetics: prognostic relevance and analysis of cooperating mutations. J Clin Oncol. 2004;22:624–633. doi: 10.1200/JCO.2004.06.060. [DOI] [PubMed] [Google Scholar]
- 4.Pabst T., Mueller B.U., Zhang P., Radomska H.S., Narravula S., Schnittger S., Behre G., Hiddemann W., Tenen D.G. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet. 2001;27:263–270. doi: 10.1038/85820. [DOI] [PubMed] [Google Scholar]
- 5.Schlenk R.F., Dohner K., Krauter J., Frohling S., Corbacioglu A., Bullinger L., Habdank M., Spath D., Morgan M., Benner A., Schlegelberger B., Heil G., Ganser A., Dohner H. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909–1918. doi: 10.1056/NEJMoa074306. [DOI] [PubMed] [Google Scholar]
- 6.Swerdlow S.H., Campo E., Harris N.L., Jaffe E.S., Pileri S.A., Stein H., Thiele J., Vardiman J.W., 4th . International Agency for Research on Cancer (IARC); Lyon: 2008. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. [Google Scholar]
- 7.Pabst T., Mueller B.U. Transcriptional dysregulation during myeloid transformation in AML. Oncogene. 2007;26:6829–6837. doi: 10.1038/sj.onc.1210765. [DOI] [PubMed] [Google Scholar]
- 8.Wouters B.J., Lowenberg B., Erpelinck-Verschueren C.A., van Putten W.L., Valk P.J., Delwel R. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood. 2009;113:3088–3091. doi: 10.1182/blood-2008-09-179895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dufour A., Schneider F., Metzeler K.H., Hoster E., Schneider S., Zellmeier E., Benthaus T., Sauerland M.C., Berdel W.E., Buchner T., Wormann B., Braess J., Hiddemann W., Bohlander S.K., Spiekermann K. Acute myeloid leukemia with biallelic CEBPA gene mutations and normal karyotype represents a distinct genetic entity associated with a favorable clinical outcome. J Clin Oncol. 2010;28:570–577. doi: 10.1200/JCO.2008.21.6010. [DOI] [PubMed] [Google Scholar]
- 10.Ahn J.Y., Seo K., Weinberg O., Boyd S.D., Arber D.A. A comparison of two methods for screening CEBPA mutations in patients with acute myeloid leukemia. J Mol Diagn. 2009;11:319–323. doi: 10.2353/jmoldx.2009.080121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Green C.L., Koo K.K., Hills R.K., Burnett A.K., Linch D.C., Gale R.E. Prognostic significance of CEBPA mutations in a large cohort of younger adult patients with acute myeloid leukemia: impact of double CEBPA mutations and the interaction with FLT3 and NPM1 mutations. J Clin Oncol. 2010;28:2739–2747. doi: 10.1200/JCO.2009.26.2501. [DOI] [PubMed] [Google Scholar]
- 12.Margulies M., Egholm M., Altman W.E., Attiya S., Bader J.S., Bemben L.A., Berka J., Braverman M.S., Chen Y.J., Chen Z., Dewell S.B., Du L., Fierro J.M., Gomes X.V., Godwin B.C., He W., Helgesen S., Ho C.H., Irzyk G.P., Jando S.C., Alenquer M.L., Jarvie T.P., Jirage K.B., Kim J.B., Knight J.R., Lanza J.R., Leamon J.H., Lefkowitz S.M., Lei M., Li J., Lohman K.L., Lu H., Makhijani V.B., McDade K.E., McKenna M.P., Myers E.W., Nickerson E., Nobile J.R., Plant R., Puc B.P., Ronan M.T., Roth G.T., Sarkis G.J., Simons J.F., Simpson J.W., Srinivasan M., Tartaro K.R., Tomasz A., Vogt K.A., Volkmer G.A., Wang S.H., Wang Y., Weiner M.P., Yu P., Begley R.F., Rothberg J.M. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., Hornik K., Hothorn T., Huber W., Iacus S., Irizarry R., Leisch F., Li C., Maechler M., Rossini A.J., Sawitzki G., Smith C., Smyth G., Tierney L., Yang J.Y., Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kohlmann A., Grossmann V., Klein H.U., Schindela S., Weiss T., Kazak B., Dicker F., Schnittger S., Dugas M., Kern W., Haferlach C., Haferlach T. Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2. CBL, RAS, and RUNX1. J Clin Oncol. 2010;28:3858–3865. doi: 10.1200/JCO.2009.27.1361. [DOI] [PubMed] [Google Scholar]
- 15.Delhommeau F., Dupont S., Della V., James C., Trannoy S., Masse A., Kosmider O., Le Couedic J.P., Robert F., Alberdi A., Lecluse Y., Plo I., Dreyfus F.J., Marzac C., Casadevall N., Lacombe C., Romana S.P., Dessen P., Soulier J., Viguie F., Fontenay M., Vainchenker W., Bernard O.A. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289–2301. doi: 10.1056/NEJMoa0810069. V. [DOI] [PubMed] [Google Scholar]
- 16.Ernst T., Chase A.J., Score J., Hidalgo-Curtis C.E., Bryant C., Jones A.V., Waghorn K., Zoi K., Ross F.M., Reiter A., Hochhaus A., Drexler H.G., Duncombe A., Cervantes F., Oscier D., Boultwood J., Grand F.H., Cross N.C. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42:722–726. doi: 10.1038/ng.621. [DOI] [PubMed] [Google Scholar]
- 17.Gelsi-Boyer V., Trouplin V., Adelaide J., Bonansea J., Cervera N., Carbuccia N., Lagarde A., Prebet T., Nezri M., Sainty D., Olschwang S., Xerri L., Chaffanet M., Mozziconacci M.J., Vey N., Birnbaum D. Mutations of polycomb–associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 2009;145:788–800. doi: 10.1111/j.1365-2141.2009.07697.x. [DOI] [PubMed] [Google Scholar]
- 18.Parkin B., Ouillette P., Wang Y., Liu Y., Wright W., Roulston D., Purkayastha A., Dressel A., Karp J., Bockenstedt P., Al-Zoubi A., Talpaz M., Kujawski L., Liu Y., Shedden K., Shakhan S., Li C., Erba H., Malek S.N. NF1 inactivation in adult acute myelogenous leukemia. Clin Cancer Res. 2010;16:4135–4147. doi: 10.1158/1078-0432.CCR-09-2639. [DOI] [PMC free article] [PubMed] [Google Scholar]