

Abstract
The high frequency of RAS mutations in human cancers (33%) has stimulated intense interest in the development of anti-Ras inhibitors for cancer therapy. Currently, the major focus of these efforts is centered on inhibitors of components involved in Ras downstream effector signaling. In particular, more than 40 inhibitors of the Raf-MEK-ERK mitogen-activated protein kinase cascade and phosphoinositide 3-kinase-AKT-mTOR effector signaling networks are currently under clinical evaluation. However, these efforts are complicated by the fact that Ras can utilize at least 9 additional functionally distinct effectors, with at least 3 additional effectors with validated roles in Ras-mediated oncogenesis. Of these, the guanine nucleotide exchange factors of the Ras-like (Ral) small GTPases (RalGEFs) have emerged as important effectors of mutant Ras in pancreatic, colon, and other cancers. In this review, we summarize the evidence for the importance of this effector pathway in cancer and discuss possible directions for therapeutic inhibition of aberrant Ral activation and signaling.
Keywords: exocyst complex, RalBP1/RLIP76, geranylgeranyltransferase-I inhibitor, Aurora A
Introduction
The frequent mutational activation of Ras in human cancers,1 in particular those in which there remains a dire need for new therapies (e.g., pancreatic, colon, lung, melanoma), has prompted intense research interest and pharmaceutical industry effort to develop anti-Ras inhibitors for cancer treatment.2 Because Ras itself has not, to date, been a tractable target for the development of direct inhibitors, much of these efforts have involved indirect approaches for blocking either Ras membrane association, critical for Ras function, or Ras downstream effector signaling. In light of the disappointing failure of farnesyltransferase inhibitors to block the membrane association of the Ras isoforms most commonly mutated in human cancers (K-Ras and N-Ras),3 much of the current effort is now focused on inhibitors of Ras effector signaling, in particular the Raf-MEK-ERK mitogen-activated protein kinase (MAPK) cascade and phosphoinositide 3-kinase (PI3K)– AKT-mTOR effector signaling networks. In this review, we focus on a lesser studied effector pathway, the RalGEF-Ral small GTPase signaling network. Although not mutated frequently in human cancer, the validation of the importance of this pathway in cancer continues to mount. We summarize the validation of Ral in cancer and discuss approaches for targeting Ral for cancer treatment.
Effectors of Ras
A diverse spectrum of extracellular stimuli activates Ras guanine nucleotide exchange factors (RasGEFs) (e.g., Sos, RasGRF), leading to transient formation of active GTP-bound Ras.4 Ras GTPase-activating proteins (RasGAPs) (e.g., neurofibromin) then stimulate GTP hydrolysis, returning Ras to its inactive GDP-bound state. In human cancers, Ras proteins harbor single amino acid substitutions, most commonly at residues 12, 13, and 61, that render Ras persistently GTP bound and active.2 Activated Ras-GTP binds preferentially to a spectrum of functionally diverse downstream effectors (Fig. 1). Most effectors are characterized by Ras binding (RBD) or Ras association (RA; also RalGDS/AF-6) domains that preferentially interact with Ras-GTP.5,6 The Raf serine/threonine kinases (Raf-1, A-Raf, and B-Raf) are the best characterized effectors of Ras.7,8 Ras-mediated Raf activation in turn activates the MEK1 and MEK2 dual-specificity kinases, which then activate the ERK1 and ERK2 MAPKs. The second best characterized effectors of Ras are the p110 (α, β, γ, and δ) catalytic subunits of class I PI3Ks.9-11 The critical role of these 2 effector classes in Ras-mediated oncogenesis is supported by the frequent mutational activation of the genes that encode B-Raf (BRAF) (13%, COSMIC [http://www.sanger.ac.uk/genetics/CGP/cosmic/]) and p110α (PIK3CA) (19%, COSMIC) in human cancers.
Figure 1.
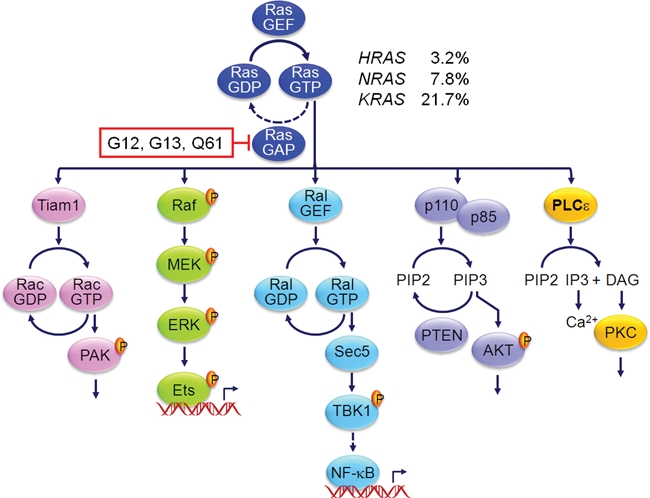
Effectors implicated in Ras-mediated oncogenesis. Missense mutations at Ras residues, primarily at Ras residues G12, G13, and Q61, impair GAP stimulation of intrinsic Ras GTP hydrolysis activity. Thus, mutant Ras is persistently GTP bound and active. In addition to the Raf, PI3K, and RalGEF effector families, the Tiam1 Rho family GEF and phospholipase Cϵ have also been identified as effectors important in Ras-mediated tumorigenesis. RAS mutation frequencies are compiled from COSMIC (http://www.sanger.ac.uk/genetics/CGP/cosmic/). PIP3 = phosphatidylinositol 3,4,5-bisphosphate; PIP2 = phosphatidylinositol 4,5-bisphosphate; IP3 = inositol 1,4,5-trisphosphate; DAG = diacyglycerol.
Despite the strong experimental evidence validating key roles for Raf and PI3K in Ras-mediated oncogenesis, there is substantial evidence that additional effectors must also contribute critical functions for mutant Ras in cancer growth.3 There are at least 9 additional functionally distinct effector classes identified for Ras, and of these, the validated roles of 3 additional effectors in Ras-mediated oncogenesis have been established.2 Of these, substantial and rapidly accumulating evidence validates a key role for the RalGEF-Ral effector pathway in a diversity of human cancers,12 in particular KRAS mutant pancreatic and colorectal cancer. In this review, we first provide an overview of RalGEF-Ral effector regulation and effector signaling. We then summarize the data supporting essential roles for Ral GTPases in various human cancers. Finally, we discuss possible therapeutic approaches for blocking RalGEF-Ral signaling for cancer treatment.
Discovery of the RalGEF-Ral Effector Pathway
Ral (Ras-like) GTPases (46%-51% identity with human Ras) were identified initially in a search for RAS-related genes.13 Using oligonucleotide probes corresponding to a sequence of 7 amino acids strictly conserved in Ras, RhoA, and the yeast YPT Rab protein (Ras residues 57-63; DTAGQE/D), a screen of a cDNA library derived from B95-8 Epstein-Barr virus, immortalized simian B lymphocytes identified RALA, which encodes a 206 amino acid protein with approximately 50% sequence identity with Ras. This initial source of Ral isolation accounts for why the approved RAL gene name is “v-ral simian leukemia viral oncogene homolog” (HUGO). The simian RALA sequence was then used to isolate human RALA, and additionally, the related RALB gene, from a human pheochromocytoma library.14 As described below, although the 2 human RAL genes encode highly related proteins (82% sequence identity) (Fig. 2A and 2B), they exhibit very distinct functional roles in cancer cell biology. RAL genes are expressed ubiquitously and conserved in evolution, with related genes found in Caenorhabditis elegans and Drosophila melanogaster that encode highly related Ral GTPases (Fig. 2B).
Figure 2.
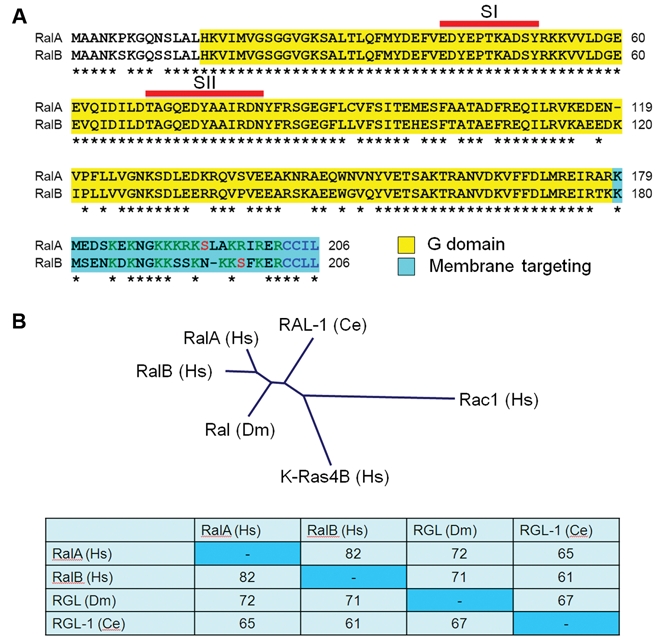
Ral small GTPases. (A) Sequence alignment of human (Hs) RalA and RalB. There is 100% identity in the switch I (SI: 41-51) and switch II (SII: 69-81) sequences that change conformation in the GDP- and GTP-bound states and are involved in effector binding. The greatest sequence divergence is in the C-terminal membrane-targeting sequences, with polybasic (green), phosphorylation site (red), and CAAX motif (blue) residues indicated. G domain = residues 15-178/9 (corresponding to Ras residues 4-166). Asterisks indicate sequence identity. (B) Conservation of Ral GTPases in evolution. Drosophila melanogaster (Dm) and Caenorhabditis elegans (Ce) possess a single Ral GTPase ortholog with strong sequence identity to human Ral. The dendrogram was generated by Clustal/W multiple sequence alignment of the indicated Ral proteins, human K-Ras4B and Rac1 (Rho family).
The first RalGEF, RalGDS (Ral guanine nucleotide dissociation stimulator), was identified in a PCR-based screen of a mouse cDNA library for genes with sequence identity to yeast RasGEFs15 (Fig. 3A). RalGDS was found to possess sequence homology with the REM (Ras exchange motif) and CDC25 RasGEF catalytic domains characteristic of Ras-GEFs (e.g., Sos, RasGRP, RasGRF). However, Ral-GDS did not display exchange activity for Ras and instead was selective for RalA and RalB.
Figure 3.
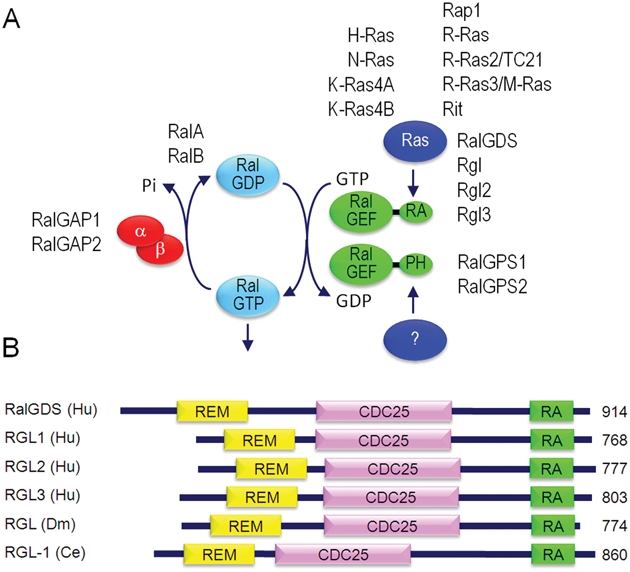
Regulators and effectors of Ral. (A) Regulators of the Ral GDP-GTP cycle. The 2 classes of RalGEFs are defined by the presence of an RA or PH domain. (B) RalGEF domain structure and evolutionary conservation. REM = Ras exchange motif; CDC25 homology domain = RalGEF catalytic domain; RA = Ras association domain.
Stimulated by the success of yeast 2-hybrid library screening that identified Raf as a Ras effector,16 a flurry of similar studies established RalGEFs as Ras effectors. Independently, human RalGDS was identified in yeast 2-hybrid screens for H-Ras-GTP,17 R-Ras-GTP,18 or R-Ras2/TC21-GTP19 binding proteins. A mouse RalGDS-like (Rgl) protein was identified in a yeast 2-hybrid screen for H-Ras-GTP–interacting proteins.20 Subsequently, mouse (RalGDS-like factor [Rlf]) and human Rgl2 were identified as Rap 1A– or Rap 1B–GTP binding proteins, respectively.21,22 Yeast 2-hybrid library screening for Rit small GTPase binding proteins identified mouse Rgl3 (Ral GEF-like 3) and additionally RalGDS, Rgl, and Rgl2.23 Independently, Rgl3 was identified in a yeast 2-hybrid screen for R-Ras3/M-Ras24 or Rap1-interacting24 proteins. Both studies also established H-Ras, as well as R-Ras and Rap1, association with Rgl3. The 4 human RalGEFs that can serve as Ras effectors share a common domain structure: an N-terminal REM followed by a CDC25 homology RalGEF catalytic domain and a C-terminal RA domain (Fig. 3B).
All 4 RalGEFs can interact with Ras and the closely related (~50% identity) Rap and R-Ras small GTPases (Fig. 3A). Although ectopic overexpression studies show that RalGEFs can be activated by R-Ras and Rap family small GTPases,25 whether endogenous activation of these Ras family proteins can activate Ral is not clear. For example, it was found that mutationally activated H-Ras, but not R-Ras2/TC21, transformation was dependent on Ral activation.26 Whether the different RalGEFs are regulated by different Ras family GTPases and whether they differentially regulate RalA and RalB are issues that remain unresolved.27
The RA domain–containing RalGEFs are conserved in evolution, with highly homologous orthologs in D. melanogaster (RGL) and C. elegans (RGL1)28,29 (Fig. 3B). Finally, 2 other Ral-specific GEFs (Fig. 3A), RalGPS1/RalGEF2 and Ral-GPS2 (Ral GEFs with PH domain and SH3 binding motif), have been identified that lack RA domains and instead contain pleckstrin homology domains.30-33 Because RalGPS proteins lack RA or RBD domains, they are not activated directly by Ras; limited evidence suggests activation by the Grb2 adaptor or phospholipids. Another RalGEF, Rgr, was identified originally as a transforming protein and can also activate other small GTPases.34,35 Ral activation in RAS wild-type cancer cells may be mediated by these additional RalGEFs.
Recently, 2 RalGAPs have been identified, designated RalGAP1 and Ral-GAP236 (Fig. 3A). Both are large heterodimeric complexes, each consisting of a catalytic α1 or α2 subunit and a common β subunit. Rather than sharing sequence similarity to RasGAPs, the RalGAP complexes share structural and catalytic similarities with the tuberous sclerosis tumor suppressor Tsc1/Tsc2 complex, which acts as a GAP for the Rheb branch of the Ras family small GTPases. Mutational loss of function of the neurofibromin RasGAP and the Tsc1/2 RhebGAP has been identified in cancer.37 Whether a similar loss of function of RalGAPs may promote oncogenesis has not been addressed.
Ral Effector Signaling
Similar to Ras,2 activated Ral-GTP interacts with multiple, functionally divergent downstream effectors (Fig. 4). The first effector was identified in a yeast 2-hybrid library or cDNA expression library screens using RalA, leading to independent discovery of RalBP1 (Ral binding protein 1)/RLIP76 (76-kDa Ral-interacting protein 1)/ RIP (Ral-interacting protein).38-40 In addition to a Ral-GTP binding domain,41 RalBP1 also contains a RhoGAP homology catalytic domain with activity for Rac1 and Cdc42 but not RhoA. GTP-bound RalA and RalB can interact with RalBP1 through a conserved Ral binding domain, regulating RalBP1 subcellular localization but not intrinsic RhoGAP activity.42 Rac1 activation promotes membrane ruffling at the leading edge of migrating cells, whereas Cdc42 promotes filopodia formation.
Figure 4.

Ral effectors. RalA and/or RalB have been determined to interact with a diverse spectrum of downstream effectors. Most bind preferentially to the GTP-bound protein, whereas some are nucleotide independent. Ral effector networks can regulate endocytosis, exocytosis, actin organization, phospholipid metabolism, and generation of second messengers. Ral has been found to activate various transcription factors or promote elements, to regulate gene expression, through pathways that may or may not be distinct from known effectors. PC = phosphatidylcholine; PA = phosphatidic acid.
RalBP1 can also serve as a scaffold and associate with a diversity of other proteins. Two independent yeast 2-hybrid library screening studies identified the closely related Reps1 (RalBP1-associated Eps homology (EH) domain protein 1) and Reps2/POB1 (partner of RalBP1) proteins that interact with RalBP1 C-terminal sequences distinct from the RhoGAP and Ral binding domains.43,44 Reps1 and Reps2 contain EH domains, which are found on proteins involved in endocytosis. Reps1 associates through its EH domain with Rab11-FIP2, a Rab11 binding protein implicated in endocytosis.45 Reps2/POB1, via its EH domain, interacts with Epsin and Eps15,46,47 proteins that regulate receptor-mediated endocytosis. Similarly, a second RalBP1 interaction, through N-terminal sequences, is with the µ2 subunit of the plasma membrane–associated AP-2 tetrameric complex.48 AP-2 promotes clathrin coat formation and specific recognition of membrane receptors for endocytosis. Epsin, Eps15, and Rab11-FIP2 can also interact directly with AP-2. These interactions support a role for RalBP1 in the regulation of receptor-mediated endocytosis.
RalBP1 also interacts with ARIP2 (activin receptor-interacting protein 2), which regulates endocytosis of activin type II receptors,49 HSF1 (heat shock factor 1),50 which regulates expression of heat shock genes in response to stress, cyclin B1 during mitosis,51 and PSD-1, a postsynaptic scaffolding protein implicated in the regulation of excitatory synaptic function.52
RalBP1 was also identified independently as a transporter activity, designated DNP-SG (S-(2, 4-dinitrophenyl)glutathione) ATPase, involved in the active transport of conjugated and unconjugated electrophiles out of cells.53 RalBP1 contains 2 ATP binding sites54 that allow it to function as an ATP-dependent transporter protein and efflux pump for small molecules, including anticancer drugs and endogenous metabolites.55 Inhibition of RalBP1 expression or function has been shown to cause regression of lung, kidney, melanoma, colon, and prostate cancer cell line xenografts, although the significance of these observations for Ral function is not clear.
Perhaps the best characterized of the Ral effectors are 2 components of the octameric exocyst complex (also called Sec6/8 complex), Sec5 and Exo84.56,57 The octameric exocyst complex is involved in the regulation of exocytosis.58,59 The exocyst facilitates the tethering of post-Golgi secretory vesicles to the plasma membrane prior to exocytic fusion, and exocyst function has been implicated in a variety of cellular processes including cell migration and tumor cell invasion. Ral regulates exocyst subcellular localization rather than assembly.60 Studies with Ral effector domain mutants and/or RNA interference have implicated exocyst function in several Ral-mediated functions.12
Other less characterized effectors of RalA include filamin, an actin filament crosslinking protein required for RalA-induced filopodia formation.61 RalA and RalB have also been shown to directly interact with and activate phospholipase C delta 1 (PLCδ1).62 PLCδ1 is not a conventional effector in that interaction with RalB was nucleotide independent and required the N-terminal 11 residues of RalB. Activation of various G protein–coupled receptor signaling pathways stimulates Ral-dependent PLC activation.
Another effector of Ral is phospholipase D (PLD1),63 which leads to the generation of lipid second messengers, including phosphatidic acid, lysophosphatidic acid, and diacylglycerol. However, PLD1 does not function classically as a Ral effector in that PLD1 association is not regulated by GDP/GTP cycling and interaction is through Ral N-terminal sequences distinct from the switch I and II sequences involved in GTP-dependent effector binding. Instead, RalA activation of PLD1 includes the additional association with the Arf6 small GTPase.64
Although RalA and RalB share 100% sequence identity in residues involved in effector interaction (Fig. 2A) and, where studied in vitro, can interact with the same effectors, RalA and RalB can exhibit strikingly different roles in normal and neoplastic cell function. These functional differences are largely because of their distinct subcellular membrane locations. Whereas RalA is found at the plasma membrane and with endosomes, RalB is primarily endosome associated.65 However, Ral subcellular localization is dynamic and can be regulated by its activation state and by phosphorylation.66,67 Ral subcellular localization in turn influences specific effector interactions. Some examples of RalA and RalB differences in oncogenesis are summarized below.
Ral Regulation of Gene Expression
In addition to the specific effector functions described above, RalGEF-Ral signaling can regulate gene expression. RalGEF and/or Ral activation is involved in Ras-mediated activation of various transcription factors (Fig. 4) that include phosphorylation and activation of c-Jun through JNK MAPK activation,68 ATF2,69 STAT3 through Src tyrosine kinase activation,70 NFAT,71 and AFX (FOXO4).72 Ral activation also stimulated transcriptional activation of the c-Fos promoter through the ternary complex factor,73,74 NF-κB binding sites for the human cyclin D1 promoter,75,76 the urokinase plasminogen activator receptor promoter AP-1 binding motifs,69 and the binding site for the Ras-responsive element binding protein 1 (RREB-1).77
The effector signaling that regulates these transcription factors is not well characterized. However, activation of NF-κB has been investigated and found to be regulated downstream of the Ral effector Sec5 and not through PLD1 or RalBP1.75,76 Active RalB signaling causes the association of Sec5 with TBK1 (TANK-binding kinase), an atypical IκB kinase (IKK)–related protein kinase. The recruitment of TBK1 into a complex with Sec5 resulted in increased TBK1 catalytic activity.75 TBK1 can directly phosphorylate and promote the nuclear localization and activation of the c-Rel NF-κB family member.78
Global gene expression profiles have also identified Ral-regulated transcription factors and gene targets.77,79 Microarray analyses of RalA siRNA– and/or RalB siRNA–depleted UM-UC-3 bladder cancer cells, which harbor an activating KRAS mutation, found that a majority of genes identified were dependent on both RalA and RalB (547 genes), but additionally, subsets of RalA (77 genes) isoform– or RalB (85 genes) isoform–specific upregulated or downregulated genes were also identified. Computation analysis identified enrichment of NF-κB as well as RREB-1 binding sites in the Ral gene expression signature. One gene identified with RalA- and RalB-dependent upregulation was CD24, which encodes the metastasis-associated protein CD24,80 a glycosyl phosphatidyl inositol–linked surface protein. Suppression of CD24 expression in UMUC-3 and other tumor cells reduced anchorage-independent proliferation and survival, suggesting that CD24 upregulation may be an important component of Ral-mediated oncogenesis. Taken together, these observations suggest that Ral-regulated genes may be fertile ground for the identification of candidate anti-Ral therapeutic targets.
RalGEF-Ral Signaling and Oncogenesis
The RalGEF-Ral pathway was characterized initially to play a relatively minor role in Ras transformation of rodent fibroblasts.81,82 However, subsequent studies by Counter et al. established a very significant role for this effector pathway in Ras transformation of human cells.83 Key support for RalGEF-transforming function came from the use of Ras effector domain binding mutants, in particular the E37G effector domain mutation, which is impaired in Raf and PI3K activation yet exhibits transforming activity in part through retained binding to RalGDS.84-87 Additionally, White et al. identified critical but distinct roles for the related RalA and RalB isoforms in normal and human cancer cell line growth.88 siRNA suppression of RalA impaired tumor cell anchorage-independent but not -dependent growth, whereas suppression of RalB caused tumor but not normal cell apoptosis. Finally, mouse model studies showed that homozygous deletion of RalGDS (a RalGEF) caused resistance to H-Ras–induced skin squamous cell carcinoma formation.89 Ral GTPases have now been implicated in a variety of human cancers. Below, we summarize the data validating Ral GTPases in human cancers associated with frequent RAS mutation (pancreatic, colorectal, and melanoma) as well as human cancers in which RAS mutational activation is infrequent (bladder, prostate).
Ral and Pancreatic Cancer
The most comprehensive and compelling evidence for the importance of RalGEF-Ral signaling in cancer has been seen for pancreatic ductal adenocarcinoma (PDAC), where KRAS is the most frequently mutated gene (>90%) seen.90,91 Although the majority of Ras effector–targeted therapies currently under clinical evaluation are focused on the Raf and PI3K effector pathways,37 it is of interest that the RalGEF-Ral pathway, rather than Raf or PI3K, was found to be more consistently activated in pancreatic patient tumors.92 A panel of 18 matched and unmatched patient pancreatic tumor samples exhibited high levels of both RalA and RalB activation but surprisingly not phosphorylation of ERK1/2 and Akt.92 In addition, the expression of the RalGEF Rgl2 was elevated in matched pancreatic patient tumors, and suppression of Rgl2 expression impaired PDAC growth.93 These studies validate the critical importance of this pathway in patients harboring K-Ras–driven and -dependent pancreatic tumors.
Essential but distinct functions for RalA and RalB have been seen in PDAC growth. Sustained shRNA suppression of RalA but not RalB in 10 of 10 KRAS mutant PDAC cell lines diminished anchorage-independent growth in vitro and primary tumor xenograft growth in immunocompromised mice.92 Conversely, cell lines expressing RalB-specific shRNA exhibited impaired Matrigel (BD Biosciences, Franklin Lakes, NJ) invasion in vitro and lung colony formation in an experimental metastasis model. These studies suggest that RalA is required for the early stages of Ras-driven pancreatic tumorigenesis, whereas RalB is required for later stages of malignant growth.92
Although the basis for these distinct functions, and the effectors involved, remains to be determined, clues are provided by the consequences of Aurora A phosphorylation of RalA. The phosphorylation of RalA at Ser194 by Aurora A kinase disrupted plasma membrane association and resulted in cytoplasmic translocation. In addition, phosphorylation led to preferential association with and activation of RalBP1 and decreased Cdc42 and Rac activity.66 The exocyst, through lipid raft microdomain exocytosis, may also be an important effector of RalA-supported anchorage-independent growth.94
Ral and Colorectal Cancer
Genetic sequencing has verified that KRAS is the most frequently mutated oncogene in colorectal cancer (CRC) (40%-50%).95,96 A critical but distinct role for Ral GTPases has also been described for CRC. As in pancreatic cancer, Ral-GTP levels were found elevated in CRC tissue and cell lines.97 Stable shRNA suppression found that RalA was necessary for the anchorage-independent growth of 8 of 8 CRC cell lines, independent of KRAS mutation status. Surprisingly, stable suppression of RalB was found to greatly enhance the anchorage-independent growth of all 8 CRC tumor cells. This activity was specific because ectopic restoration of RalB expression suppressed soft agar growth. Finally, using Ral effector binding mutants and RNAi, it was determined that both Ral isoforms required the ability to bind RalBP1 but utilized distinct components of the exocyst to mediate their effects on anchorage-independent growth. Specifically, Exo84 binding was required for RalA, while RalB required Sec5 engagement.
That these 2 highly related proteins could serve opposing functions in transformed growth has been described previously in studies with Ras-transformed, immortalized human embryonic kidney epithelial cells.98 Interestingly, these results contrast with observations made with transient RalB suppression in CRC cells that caused apoptosis.88,99 These divergent observations suggest different consequences of short-term versus prolonged loss of Ral function and may reflect compensatory events due to sustained loss. Consistent with this possibility, in CRC tumor cells, loss of one Ral isoform led to an increase in the GTP loading of the other isoform, indicating potential crosstalk between RalA and RalB.
Ral and Melanoma
Oncogenic NRAS mutations occur (15%-30% frequency) in cutaneous malignant melanoma.100,101 It has long been appreciated that oncogenic mutations (V600E) in BRAF are very common (up to 70%) in melanoma and are often mutually exclusive of NRAS mutations, suggesting that the ERK MAPK pathway downstream of Ras is the critical pathway regulating melanoma tumorigenesis.100,102-104 Despite the lack of evidence for mutations in RalGEFs in melanoma,105 recent evidence suggests that the RalGEF-Ral pathway does play an important role in melanoma tumor progression. Genetically engineered, immortalized p19 Arf-deficient primary mouse melanocytes were used to assess the contribution of the 3 major downstream Ras-activated pathways in melanoma tumorigenesis.106 Interestingly, Arf –/– melanocytes expressing constitutively activated Rgl2, but not activated Raf or PI3K, demonstrated the same robust anchorage-independent growth capacity as mutant N-Ras–expressing melanocytes. Furthermore, Ral dominant-negative inhibition of the RalGEF pathway in melanocytes expressing mutant N-Ras impaired growth in soft agar. RalGEF activation alone also phenocopied mutant N-Ras morphological transformation and Matrigel (BD Biosciences) invasion in vitro. Thus, while activated Raf or PI3K also promoted aspects of N-Ras–mediated melanocyte transformation, RalGEF activation was surprisingly the most significant effector pathway.
It was also recently demonstrated that a majority of human melanoma cells with wild-type NRAS and those cells harboring mutations in NRAS and BRAF have high levels of RalA but not RalB activation.107 Expression of RalA-specific shRNA in NRAS and BRAF mutant cell lines, and to a lesser extent NRAS and BRAF wild-type cells, inhibited tumorigenesis in vivo. These observations suggest a role for the RalGEF-Ral pathway in melanoma tumorigenesis, regardless of the mutational status of NRAS and BRAF.
Ral and Bladder Cancer
KRAS and HRAS mutations are found in approximately 13% of bladder cancers.108-110 Microarray gene expression analyses of 65 bladder tumors and 15 normal bladder tissue determined that RalA mRNA expression increased with tumor grade, and expression of RalA and RalB protein levels increased in metastatic patient samples. In contrast, RalB mRNA expression did not significantly associate with higher tumor grade.79 Variable levels of RalA- and RalB-GTP were seen in a panel of bladder carcinoma cell lines.79 Studies with the KRAS mutant UM-UC-3 bladder carcinoma cell line showed that transient siRNA suppression of RalB but not RalA impaired transwell motility.111 However, concurrent suppression of RalA and RalB did not impair motility. Additionally, ectopic expression of constitutively activated RalA impaired motility, while activated RalB stimulated motility. Thus, RalA and RalB play opposing roles in the motility of UM-UC-3 bladder cancer cells. Finally, siRNA suppression of RalB alone or together with RalA, but not RalA alone, disrupted actin stress fibers, and concurrent suppression of both RalA and RalB was required to reduce UM-UC-3 anchorage-dependent growth.
The RalGEF-Ral pathway also appears to be important in anchorage-independent growth and cell survival in HRAS mutant T24 bladder cancer cells. Studies demonstrate that PLD1 activity is dependent on Ras and RalA in bladder cancer cell lines. In addition, expression of RalA-specific shRNA induced apoptosis in response to serum withdrawal, a similar effect to that seen in cells expressing PLD-1–specific shRNA.112
Ral and Prostate Cancer
Overall, RAS mutations are found in 16% of prostate cancers (8% KRAS, 6% HRAS, 2% NRAS; COSMIC). The importance of the RalGEF-Ral pathway in prostate cancer is demonstrated in part by correlation between Ral expression and metastasis. Metastatic patient tumors exhibit increased RalA mRNA expression as compared to primary tumors.79 Higher protein expression of RalA was also seen in patient tumors and was used as part of a gene signature to predict tumor aggressiveness in patients.113
Progression of prostate cancers to androgen independence is an important step in prostate cancer tumorigenesis. RalA appears to have a key role in androgen-independent signaling and gene expression. RalA is activated upon androgen deprivation in a ROS (reactive oxygen species) dependent manner. RalA activation upregulates the transcription of the angiogenic factor VEGF-C.114 Furthermore, loss of NKX3.1 and expression of constitutively activated RalA Q72L synergistically enhanced VEGF-C transcription.115 Androgen-independent tumors rely on growth factor signaling (e.g., epidermal growth factor [EGF]) as opposed to androgen signaling. The RalA effector RalBP1 interacts with REPS2/POB1, which is involved in the endocytosis of the EGF receptor (EGFR). REPS2/POB1 was seen to be downregulated in androgen-independent prostate cancer cell lines and xenografts. Overexpression of REPS2/POB1 in prostate cancer cell lines induced apoptosis and inhibited growth factor signaling.116
Ral GTPases have been implicated in prostate cancer cell migration. In one study, antagonistic roles for RalA and RalB were described for human prostate cancer cell motility. RAS wild-type DU-145 prostate cancer cells expressing RalB but not RalA shRNA had a significant reduction in transwell migration.111 However, concurrent expression of both RalB- and RalA-specific shRNA restored normal motility, indicating RalA dominance over RalB. In a second study of Dunning rat prostate tumor cells, suppression of RalA or RalB altered cell morphology and reduced migration.60 Loss of Ral disrupted subcellular localization of the exocyst complex to paxillin-positive focal complexes, leading to altered migration.
Prostate cancers preferentially metastasize to the bone, but the mechanisms that mediate this specificity are not fully understood. It is apparent that the Ral-GEF-Ral pathway regulates the ability of prostate cancer cells to grow in the bone microenvironment. It was recently demonstrated that the RalGEF-Ral pathway is both necessary and sufficient for prostate cancer bone metastasis.117 Expression of the Ras E37G effector domain mutant that primarily activates the RalGEF-Ral pathway or constitutively activated RalGEF in nonmetastatic DU-145 cells promoted metastasis to the bone. Furthermore, expression of RalA-specific shRNA in the RAS wild-type PC3 metastatic cell lines inhibited bone metastasis but not primary tumor growth. From these studies, it appears that RalA is more important in the growth of the tumor at the secondary site as opposed to the homing and initial colonization.
Ral and Other Cancers
Aberrant Ras activation in malignant peripheral nerve sheath tumors (MPNSTs), which arise from peripheral nerve Schwann cells, is caused by loss-of-function mutations in the neurofibromin RasGAP.118 Farassati et al. recently showed that Ral was overactive in 5 of 5 neurofibromin-deficient mouse MPNST cell lines when compared to nontransformed mouse Schwann cells. Ras-dependent Ral activation was also seen in 2 human MPNST cell lines and 3 tumors. shRNA depletion of RalA was found to inhibit the anchorage-dependent proliferation, Matrigel (BD Biosciences) invasion, and subcutaneous tumor xenograft growth of 35-1-2 mouse MPNST cells.119
Roles for Ral GTPases have been described for ligand-stimulated activities in a number of other cancers. In multiple myeloma (MM) cells, it was found that RalB but not RalA promoted the CXCL12/SDF-1–induced Ral activation and migration of MM cells.120 In MCF-7 breast cancer cells, EGF stimulated Ral activation, and RalA was determined to be important for EGFR promotion of estrogen-independent proliferation.121 More recently, lysophosphatidic acid–stimulated invasion of MDA-MB-231 breast tumor cells was associated with Ral activation, and invasion was found to depend on RalA and RalB protein expression.122
Finally, a tumor suppressor rather than oncogene function for RalA is suggested from studies of squamous cell carcinoma.123 H-Ras–transformed human HaCaT keratinocytes show increased proliferation but not invasion. Instead, further suppression of E-cadherin function is required for invasion. Loss of E-cadherin expression was associated with decreased RalA and RalB expression. Ectopic expression of RalA but not RalB to levels found in parental noninvasive cells reversed the effects of E-cadherin loss, reducing invasion. Using a bioengineered tissue model reflective of the early steps in Ras-induced human squamous cell carcinoma of the skin, RNAi depletion of RalA in H-Ras–transformed HaCaT cells decreased E-cadherin expression and led to enhanced invasion. The potential relevance of these observations for human cancers was suggested by the Oncomine database analyses in which RalA was significantly downregulated (~30%) in head and neck squamous cell carcinoma (HNSCC) when compared to normal oral mucosa. However, a tumor suppressor function for RalA in squamous cell carcinoma is seemingly at odds with the requirement for RalGDS in H-Ras–induced skin carcinomas in mice.89 Further studies of Ral function in human squamous cell carcinoma tumors and cell lines will be needed to clarify its involvement in this cancer type.
Evolutionary Conservation of RalGEF-Ral Signaling in D. melanogaster and C. elegans
Both Drosophila melanogaster and Caenorhabditis elegans express single Ras orthologs (Ras1 and LET-60, respectively) that share extensive identity with human Ras proteins, including 100% identity in the core effector binding regions.124,125 Therefore, Ras1 and LET-60 probably signal through similar suites of effectors as human Ras proteins. Likewise, both flies and worms contain single genes encoding the Ras-dependent RalGEF (RGL and RGL-1) and Ral (Ral and RAL-1), and the predicted proteins are similarly highly conserved. Flies also express an ortholog of Ral-GPS, while worms do not.
In classic eye development experiments, Raf was shown to be the canonical Ras effector in flies.126 However, the relationship of Ras to RalGEF signaling is murky in D. melanogaster. Coexpression experiments with dominant-negative Ral and mutationally activated Ras or Rap (the Ras relative) suggest that Rap, not Ras itself, activates RalGEF-Ral signaling in bristle formation and eye development.28 However, the data are also consistent with multiple Ras and Rap effector pathways signaling in parallel with diverging or competing outcomes. In separate D. melanogaster studies, Ral function was implicated in immune response,75 sensory cell apoptosis,127 and polar cell fate and survival,128 but the role of Ras1 and RGL relative to Ral in these processes is unclear.
During C. elegans embryonic morphogenesis, RGL-1-RAL-1 signaling through Sec5 and Exo84 components of the exocyst complex functions redundantly with another Ras family small GTPase, RAP-1, to regulate trafficking of the cadherin adhesion complex to cell junctions,129 but the role of LET-60/Ras in this process is unknown. The canonical worm LET-60/Ras effector pathway is LIN-45/Raf130 that induces the 1° vulval cell fate. While Ras-Raf signal drives 1° vulval fates, Ras-RalGEF-Ral drives the antagonistic 2° vulval fate in support of Notch signaling. The switching of Ras effector utilization from Raf to RalGEF during vulval development is mediated by restriction of RAL-1 expression to presumptive 2° cells29 and concomitant Notch-dependent 2°-specific expression of LIP-1/MAPK phosphatase to quench Ras-MEK-ERK pro-1° signaling.131
Such interplay between Notch and Ras signaling is a common theme in developmental biology,132 and Notch and Ras interplay is also observed in mouse pancreatic cell differentiation and cancer development. Whether this pancreatic Ras-Notch interplay depends on K-Ras activation of the RalGEF-Ral pathway is not known, but it is intriguing that Ral-GEF but not Raf is preferentially activated in pancreatic cancer cells, and Ral activation is necessary for pancreatic cancer growth.
Therapeutic Approaches for Blocking RalGEF-Ral Signaling
Like Ras, Ral GTPases are GTP binding proteins and are therefore not considered tractable targets for inhibitor development. While there has been some success in developing inhibitors of GEFs,37 to date, the most promising directions for inhibitors of RalGEF signaling involve inhibitors of Ral posttranslational processing and effector signaling. Additionally, because protein kinases are tractable targets for drug discovery, the identification of protein kinases that regulate or mediate Ral function also suggests more classic directions for blocking Ral function.
Similar to Ras, Ral GTPases terminate with C-terminal CAAX (C = cysteine, A = aliphatic amino acid, and X = terminal amino acid, which dictates prenyltransferase specificity) tetrapeptide motifs that signal for posttranslational modifications essential for Ral membrane association and subcellular localization133 (Fig. 5). Ral GTPases are substrates for the geranylgeranyltransferase-I (GGTase-1)–catalyzed addition of a C20 geranylgeranyl isoprenoid group to the cysteine residue,134 followed by Rce1 endoprotease cleavage of the AAX residues and carboxylmethylation of the now terminal lipid-modified cysteine residue. Similar to K-Ras4B, sequences upstream of the CAAX motif are rich in basic residues that likely function as a second signal essential for full membrane association. RalA and RalB show greatest divergence in their C-terminal sequences (Fig. 2A), which contribute to their distinct subcellular distributions to the plasma membrane and endomembranes, respectively.65 Inhibitors of GGTase-I (GGTI) were shown to cause growth inhibition of MIA-PaCa2 cells in part by induction of RalB-dependent apotosis and RalA cell cycle perturbation.135 There is currently one GGTI in a phase I clinical trial (GGTI-2418) that is well tolerated with minimal side effects (http://www.tigrispharma.com/), and others are under preclinical evaluation.136 It was originally thought that GGTIs would possess severe off-target normal cell toxicity because of the important role of other GGTase-I substrates in normal cell physiology. However, the recent demonstration of genetic ablation of GGTase-I in mutant KRAS-driven mouse models of cancer argues that GGTI therapy may be feasible and hence a useful approach for anti-Ral therapy.137,138
Figure 5.

Ral posttranslational processing. Ral is synthesized initially as an inactive cytosolic protein. The cytoplasmic (GGTase-I)– or endoplasmic reticulum (Rce1 and ICMT)–associated enzymes recognize the Ral C-terminal CAAX motif, leading to covalent addition of a geranylgeranyl isoprenoid to the cysteine residue of the CAAX motif, followed by proteolytic cleavage of the AAX residues and carboxylmethylation of the now terminal prenylated cysteine. Sequences upstream of the CAAX motif are rich in K or R basic amino acids. These polybasic stretches comprise a second membrane-targeting signal. Reversible phosphorylation of S194 in RalA modulates the targeting activity of the C-terminus, altering subcellular location. CAAL = cysteine–aliphatic amino acid–aliphatic amino acid–leucine; the terminal amino acid of the CAAX tetrapeptide motif dictates prenyltransferase specificity, with leucine recognized by geranylgeranyltransferase-I (GGTase-I); K/R = basic amino acid–rich sequences; Aur-A = Aurora A; GGTI = GGTase-I inhibitor; PKI = protein kinase inhibitor; Rce1 = Ras-converting enzyme 1; Icmt = isoprenylcysteine carboxyl methyltransferase.
An emerging theme in the regulation of small GTPase function involves protein kinase phosphorylation and regulation of subcellular localization and function. This additional level of GTPase regulation beyond the GDP-GTP cycle is demonstrated most dramatically with the finding that protein kinase C alpha (PKCα)–mediated phosphorylation converts K-Ras4B from a growth-promoting protein to an apoptosis-inducing protein.139 PKCα phosphorylation of S181 within the C-terminal polybasic region immediately adjacent to the CAAX motif promotes rapid dissociation of K-Ras4B from the plasma membrane and association with intracellular membranes, including the outer membrane of mitochondria where phosphorylated K-Ras4B interacts with Bcl-XL, causing apoptosis. A similar mode of regulation of RalA has been described in which Aurora A phosphorylation of Ral A140 at a conserved C-terminal S194 residue absent in RalB causes a relocation from the plasma membrane to endomembranes, where RalA associates preferentially with RalBP1/RLIP76.66 The fact that a phospho-deficient S194A mutant of RalA cannot support RalA-dependent PDAC cell anchorage-independent growth and tumorigenicity argues that inhibitors of Aurora A may cause RalA-selective inhibition. The importance of RalA S194 phosphorylation is also supported by the finding that this residue, together with S183, is dephosphorylated by the serine-threonine protein phosphatase 2A tumor suppressor.141
Recently, RalB was shown to be phosphorylated by PKC. PKC-mediated phosphorylation of RalB at the C-terminal S198 residue translocated RalB from the plasma membrane to the perinuclear region of the cell. T24 (mutant HRAS) and UM-UC-3 (mutant KRAS) tumor cells expressing a phosphorylation-deficient (S198A) RalB mutant exhibit impaired anchorage-independent growth, migration, and lung colonization in an experimental metastasis mouse model. Furthermore, enhancement of tumor growth by constitutively activated RalB G23V required S198 phosphorylation.142 As described above, an important component downstream of RalB is Sec5-mediated TBK1 activation.75 TBK1 was subsequently identified in shRNA library screening for synthetic lethal partners of mutant KRAS.99 RalB also showed a synthetic lethal association with mutant KRAS. Thus, inhibitors of TBK1 may be one approach for anti-RalB–selective therapies.
Another protein kinase linked to Ral function is cyclin-dependent kinase 5 (CDK5).143 CDK5 was found to be widely active in pancreatic cancer cells. Inhibition of cyclin-dependent kinase 5 (CDK5) significantly inhibited PDAC cell line tumorigenic growth. CDK5 inhibition correlated with decreased RalA and RalB activation, and expression of constitutively activated Rgl2 (Rlf-CAAX; membrane-targeted Ral-GEF) rescued the reduction in anchorage-independent growth and migration observed with CDK5 inhibition. This suggests that CDK5 mediates PDAC tumorigenesis through Ral-dependent mechanisms. Thus, inhibitors of CDK5 may serve as inhibitors of both RalA and RalB in pancreatic cancer.
Conclusion
The RalGEF-Ral effector signaling network has emerged as an important effector pathway in oncogenic Ras-driven cancer growth. Further studies of RalGEF and Ral function in oncogenesis using mouse models of mutant KRAS-driven pancreatic, lung, colon, or NRAS-driven melanoma will be needed to extend these findings. One striking observation has been the distinct function of RalA and RalB in oncogenesis. The precise basis for their different roles and the effector functions important for Ral-dependent tumor growth remains to be determined. This information will be important for the development of Ral-targeted therapies. It is likely that the combined inhibition of multiple effector pathways will be the most effective avenue for blocking mutant Ras for cancer treatment.
Acknowledgments
The authors apologize to colleagues whose work was not cited because of space limitations.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This work was supported by the National Cancer Institute (NCI) Specialized Programs of Research Excellence in GI Cancer [grant number CA106991]; the NCI National Cooperative Drug Discovery Groups [grant number CA67771]; the National Institutes of Health (NIH) to C.J.D. [grant number CA042978] and D.J.R. [grant number GM085309]; an American Cancer Society Fellowship to N.F.N.; and a T32 Cancer Cell Biology Training Program Grant Fellowship to J.K.S.
References
- 1. Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cox AD, Der CJ. Ras history: the saga continues. Small GTPases. 2010;1:1-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yeh JJ, Der CJ. Targeting signal transduction in pancreatic cancer treatment. Expert Opin Ther Targets. 2007;11:673-94 [DOI] [PubMed] [Google Scholar]
- 4. Buday L, Downward J. Many faces of Ras activation. Biochim Biophys Acta. 2008;1786:178-87 [DOI] [PubMed] [Google Scholar]
- 5. Repasky GA, Chenette EJ, Der CJ. Renewing the conspiracy theory debate: does Raf function alone to mediate Ras oncogenesis? Trends Cell Biol. 2004;14:639-47 [DOI] [PubMed] [Google Scholar]
- 6. Young A, Lyons J, Miller AL, Phan VT, Alarcon IR, McCormick F. Ras signaling and therapies. Adv Cancer Res. 2009;102:1-17 [DOI] [PubMed] [Google Scholar]
- 7. Schreck R, Rapp UR. Raf kinases: oncogenesis and drug discovery. Int J Cancer. 2006;119:2261-71 [DOI] [PubMed] [Google Scholar]
- 8. Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875-85 [DOI] [PubMed] [Google Scholar]
- 9. Castellano E, Downward J. Role of RAS in the regulation of PI 3-kinase. Curr Top Microbiol Immunol. 2010;346:143-69 [DOI] [PubMed] [Google Scholar]
- 10. Vogt PK, Gymnopoulos M, Hart JR. PI 3-kinase and cancer: changing accents. Curr Opin Genet Dev. 2009;19:12-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20:87-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bodemann BO, White MA. Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nat Rev Cancer. 2008;8:133-40 [DOI] [PubMed] [Google Scholar]
- 13. Chardin P, Tavitian A. The ral gene: a new ras related gene isolated by the use of a synthetic probe. EMBO J. 1986;5:2203-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chardin P, Tavitian A. Coding sequences of human ralA and ralB cDNAs. Nucleic Acids Res. 1989;17:4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Albright CF, Giddings BW, Liu J, Vito M, Weinberg RA. Characterization of a guanine nucleotide dissociation stimulator for a ras-related GTPase. EMBO J. 1993;12:339-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vojtek AB, Hollenberg SM, Cooper JA. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell. 1993;74:205-14 [DOI] [PubMed] [Google Scholar]
- 17. Hofer F, Fields S, Schneider C, Martin GS. Activated Ras interacts with the Ral guanine nucleotide dissociation stimulator. Proc Natl Acad Sci U S A. 1994;91:11089-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Spaargaren M, Bischoff JR. Identification of the guanine nucleotide dissociation stimulator for Ral as a putative effector molecule of R-ras, H-ras, K-ras, and Rap. Proc Natl Acad Sci U S A. 1994;91:12609-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lopez-Barahona M, Bustelo XR, Barbacid M. The TC21 oncoprotein interacts with the Ral guanosine nucleotide dissociation factor. Oncogene. 1996;12:463-70 [PubMed] [Google Scholar]
- 20. Kikuchi A, Demo SD, Ye ZH, Chen YW, Williams LT. ralGDS family members interact with the effector loop of ras p21. Mol Cell Biol. 1994;14:7483-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peterson SN, Trabalzini L, Brtva TR, et al. Identification of a novel RalGDS-related protein as a candidate effector for Ras and Rap1. J Biol Chem. 1996;271:29903-8 [DOI] [PubMed] [Google Scholar]
- 22. Wolthuis RM, Bauer B, van’t Veer LJ, et al. RalGDS-like factor (Rlf) is a novel Ras and Rap 1A-associating protein. Oncogene. 1996;13:353-62 [PubMed] [Google Scholar]
- 23. Shao H, Andres DA. A novel RalGEF-like protein, RGL3, as a candidate effector for rit and Ras. J Biol Chem. 2000;275:26914-24 [DOI] [PubMed] [Google Scholar]
- 24. Xu J, Shi S, Matsumoto N, Noda M, Kitayama H. Identification of Rgl3 as a potential binding partner for Rap-family small G-proteins and profilin II. Cell Signal. 2007;19:1575-82 [DOI] [PubMed] [Google Scholar]
- 25. Rodriguez-Viciana P, Sabatier C, McCormick F. Signaling specificity by Ras family GTPases is determined by the full spectrum of effectors they regulate. Mol Cell Biol. 2004;24:4943-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murphy GA, Graham SM, Morita S, et al. Involvement of phosphatidylinositol 3-kinase, but not RalGDS, in TC21/R-Ras2-mediated transformation. J Biol Chem. 2002;277:9966-75 [DOI] [PubMed] [Google Scholar]
- 27. Ferro E, Trabalzini L. RalGDS family members couple Ras to Ral signalling and that’s not all. Cell Signal. 2010;22:1804-10 [DOI] [PubMed] [Google Scholar]
- 28. Mirey G, Balakireva M, L’Hoste S, Rosse C, Voegeling S, Camonis J. A Ral guanine exchange factor-Ral pathway is conserved in Drosophila melanogaster and sheds new light on the connectivity of the Ral, Ras, and Rap pathways. Mol Cell Biol. 2003;23:1112-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zand TP, Reiner DJ, Der CJ. Ras effector switching promotes divergent cell fates in C. elegans vulva patterning. Dev Cell. 2011;20:84-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ceriani M, Scandiuzzi C, Amigoni L, Tisi R, Berruti G, Martegani E. Functional analysis of RalGPS2, a murine guanine nucleotide exchange factor for RalA GTPase. Exp Cell Res. 2007;313:2293-307 [DOI] [PubMed] [Google Scholar]
- 31. de Bruyn KM, de Rooij J, Wolthuis RM, et al. RalGEF2, a pleckstrin homology domain containing guanine nucleotide exchange factor for Ral. J Biol Chem. 2000;275:29761-6 [DOI] [PubMed] [Google Scholar]
- 32. Martegani E, Ceriani M, Tisi R, Berruti G. Cloning and characterization of a new Ral-GEF expressed in mouse testis. Ann N Y Acad Sci. 2002;973:135-7 [DOI] [PubMed] [Google Scholar]
- 33. Rebhun JF, Chen H, Quilliam LA. Identification and characterization of a new family of guanine nucleotide exchange factors for the ras-related GTPase Ral. J Biol Chem. 2000;275:13406-10 [DOI] [PubMed] [Google Scholar]
- 34. D’Adamo DR, Novick S, Kahn JM, Leonardi P, Pellicer A. rsc: a novel oncogene with structural and functional homology with the gene family of exchange factors for Ral. Oncogene. 1997;14:1295-305 [DOI] [PubMed] [Google Scholar]
- 35. Hernandez-Munoz I, Malumbres M, Leonardi P, Pellicer A. The Rgr oncogene (homologous to RalGDS) induces transformation and gene expression by activating Ras, Ral and Rho mediated pathways. Oncogene. 2000;19:2745-57 [DOI] [PubMed] [Google Scholar]
- 36. Shirakawa R, Fukai S, Kawato M, et al. Tuberous sclerosis tumor suppressor complex-like complexes act as GTPase-activating proteins for Ral GTPases. J Biol Chem. 2009;284:21580-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010;10:842-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cantor SB, Urano T, Feig LA. Identification and characterization of Ral-binding protein 1, a potential downstream target of Ral GTPases. Mol Cell Biol. 1995;15:4578-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jullien-Flores V, Dorseuil O, Romero F, et al. Bridging Ral GTPase to Rho pathways: RLIP76, a Ral effector with CDC42/Rac GTPase-activating protein activity. J Biol Chem. 1995;270:22473-7 [DOI] [PubMed] [Google Scholar]
- 40. Park SH, Weinberg RA. A putative effector of Ral has homology to Rho/Rac GTPase activating proteins. Oncogene. 1995;11:2349-55 [PubMed] [Google Scholar]
- 41. Fenwick RB, Campbell LJ, Rajasekar K, et al. The RalB-RLIP76 complex reveals a novel mode of ral-effector interaction. Structure. 2010;18:985-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Matsubara K, Hinoi T, Koyama S, Kikuchi A. The post-translational modifications of Ral and Rac1 are important for the action of Ral-binding protein 1, a putative effector protein of Ral. FEBS Lett. 1997;410:169-74 [DOI] [PubMed] [Google Scholar]
- 43. Ikeda M, Ishida O, Hinoi T, Kishida S, Kikuchi A. Identification and characterization of a novel protein interacting with Ral-binding protein 1, a putative effector protein of Ral. J Biol Chem. 1998;273:814-21 [DOI] [PubMed] [Google Scholar]
- 44. Yamaguchi A, Urano T, Goi T, Feig LA. An Eps homology (EH) domain protein that binds to the Ral-GTPase target, RalBP1. J Biol Chem. 1997;272:31230-4 [DOI] [PubMed] [Google Scholar]
- 45. Cullis DN, Philip B, Baleja JD, Feig LA. Rab11-FIP2, an adaptor protein connecting cellular components involved in internalization and recycling of epidermal growth factor receptors. J Biol Chem. 2002;277:49158-66 [DOI] [PubMed] [Google Scholar]
- 46. Morinaka K, Koyama S, Nakashima S, et al. Epsin binds to the EH domain of POB1 and regulates receptor-mediated endocytosis. Oncogene. 1999;18:5915-22 [DOI] [PubMed] [Google Scholar]
- 47. Nakashima S, Morinaka K, Koyama S, et al. Small G protein Ral and its downstream molecules regulate endocytosis of EGF and insulin receptors. EMBO J. 1999;18:3629-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jullien-Flores V, Mahe Y, Mirey G, et al. RLIP76, an effector of the GTPase Ral, interacts with the AP2 complex: involvement of the Ral pathway in receptor endocytosis. J Cell Sci. 2000;113(Pt 16):2837-44 [DOI] [PubMed] [Google Scholar]
- 49. Matsuzaki T, Hanai S, Kishi H, et al. Regulation of endocytosis of activin type II receptors by a novel PDZ protein through Ral/Ral-binding protein 1-dependent pathway. J Biol Chem. 2002;277:19008-18 [DOI] [PubMed] [Google Scholar]
- 50. Hu Y, Mivechi NF. HSF-1 interacts with Ral-binding protein 1 in a stress-responsive, multiprotein complex with HSP90 in vivo. J Biol Chem. 2003;278:17299-306 [DOI] [PubMed] [Google Scholar]
- 51. Rosse C, L’Hoste S, Offner N, Picard A, Camonis J. RLIP, an effector of the Ral GTPases, is a platform for Cdk1 to phosphorylate epsin during the switch off of endocytosis in mitosis. J Biol Chem. 2003;278:30597-604 [DOI] [PubMed] [Google Scholar]
- 52. Han K, Kim MH, Seeburg D, et al. Regulated RalBP1 binding to RalA and PSD-95 controls AMPA receptor endocytosis and LTD. PLoS Biol. 2009;7:e1000187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Awasthi S, Cheng J, Singhal SS, et al. Novel function of human RLIP76: ATP-dependent transport of glutathione conjugates and doxorubicin. Biochemistry. 2000;39:9327-34 [DOI] [PubMed] [Google Scholar]
- 54. Awasthi S, Cheng JZ, Singhal SS, et al. Functional reassembly of ATP-dependent xenobiotic transport by the N- and C-terminal domains of RLIP76 and identification of ATP binding sequences. Biochemistry. 2001;40:4159-68 [DOI] [PubMed] [Google Scholar]
- 55. Vatsyayan R, Lelsani PC, Awasthi S, Singhal SS. RLIP76: a versatile transporter and an emerging target for cancer therapy. Biochem Pharmacol. 2010;79:1699-705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Moskalenko S, Henry DO, Rosse C, Mirey G, Camonis JH, White MA. The exocyst is a Ral effector complex. Nat Cell Biol. 2002;4:66-72 [DOI] [PubMed] [Google Scholar]
- 57. Moskalenko S, Tong C, Rosse C, et al. Ral GTPases regulate exocyst assembly through dual subunit interactions. J Biol Chem. 2003;278:51743-8 [DOI] [PubMed] [Google Scholar]
- 58. Wu H, Rossi G, Brennwald P. The ghost in the machine: small GTPases as spatial regulators of exocytosis. Trends Cell Biol. 2008;18:397-404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. He B, Guo W. The exocyst complex in polarized exocytosis. Curr Opin Cell Biol. 2009;21:537-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Spiczka KS, Yeaman C. Ral-regulated interaction between Sec5 and paxillin targets exocyst to focal complexes during cell migration. J Cell Sci. 2008;121:2880-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ohta Y, Suzuki N, Nakamura S, Hartwig JH, Stossel TP. The small GTPase RalA targets filamin to induce filopodia. Proc Natl Acad Sci U S A. 1999;96:2122-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sidhu RS, Clough RR, Bhullar RP. Regulation of phospholipase C-delta1 through direct interactions with the small GTPase Ral and calmodulin. J Biol Chem. 2005;280:21933-41 [DOI] [PubMed] [Google Scholar]
- 63. Luo JQ, Liu X, Hammond SM, et al. RalA interacts directly with the Arf-responsive, PIP2-dependent phospholipase D1. Biochem Biophys Res Commun. 1997;235:854-9 [DOI] [PubMed] [Google Scholar]
- 64. Xu L, Frankel P, Jackson D, et al. Elevated phospholipase D activity in H-Ras-but not K-Ras-transformed cells by the synergistic action of RalA and ARF6. Mol Cell Biol. 2003;23:645-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shipitsin M, Feig LA. RalA but not RalB enhances polarized delivery of membrane proteins to the basolateral surface of epithelial cells. Mol Cell Biol. 2004;24:5746-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lim KH, Brady DC, Kashatus DF, et al. Aurora-A phosphorylates, activates, and relocalizes the small GTPase RalA. Mol Cell Biol. 2010;30:508-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cascone I, Selimoglu R, Ozdemir C, et al. Distinct roles of RalA and RalB in the progression of cytokinesis are supported by distinct RalGEFs. EMBO J. 2008;27:2375-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. de Ruiter ND, Wolthuis RM, van Dam H, Burgering BM, Bos JL. Ras-dependent regulation of c-Jun phosphorylation is mediated by the Ral guanine nucleotide exchange factor-Ral pathway. Mol Cell Biol. 2000;20:8480-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Okan E, Drewett V, Shaw PE, Jones P. The small-GTPase RalA activates transcription of the urokinase plasminogen activator receptor (uPAR) gene via an AP1-dependent mechanism. Oncogene. 2001;20:1816-24 [DOI] [PubMed] [Google Scholar]
- 70. Goi T, Shipitsin M, Lu Z, Foster DA, Klinz SG, Feig LA. An EGF receptor/Ral-GTPase signaling cascade regulates c-Src activity and substrate specificity. EMBO J. 2000;19:623-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. de Gorter DJ, Vos JC, Pals ST, Spaargaren M. The B cell antigen receptor controls AP-1 and NFAT activity through Ras-mediated activation of Ral. J Immunol. 2007;178:1405-14 [DOI] [PubMed] [Google Scholar]
- 72. De Ruiter ND, Burgering BM, Bos JL. Regulation of the Forkhead transcription factor AFX by Ral-dependent phosphorylation of threonines 447 and 451. Mol Cell Biol. 2001;21:8225-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Okazaki M, Kishida S, Hinoi T, et al. Synergistic activation of c-fos promoter activity by Raf and Ral GDP dissociation stimulator. Oncogene. 1997;14:515-21 [DOI] [PubMed] [Google Scholar]
- 74. Wolthuis RM, de Ruiter ND, Cool RH, Bos JL. Stimulation of gene induction and cell growth by the Ras effector Rlf. EMBO J. 1997;16:6748-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chien Y, Kim S, Bumeister R, et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell. 2006;127:157-70 [DOI] [PubMed] [Google Scholar]
- 76. Henry DO, Moskalenko SA, Kaur KJ, et al. Ral GTPases contribute to regulation of cyclin D1 through activation of NF-kappaB. Mol Cell Biol. 2000;20:8084-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Oxford G, Smith SC, Hampton G, Theodorescu D. Expression profiling of Ral-depleted bladder cancer cells identifies RREB-1 as a novel transcriptional Ral effector. Oncogene. 2007;26:7143-52 [DOI] [PubMed] [Google Scholar]
- 78. Harris J, Oliere S, Sharma S, et al. Nuclear accumulation of cRel following C-terminal phosphorylation by TBK1/IKK epsilon. J Immunol. 2006;177:2527-35 [DOI] [PubMed] [Google Scholar]
- 79. Smith SC, Oxford G, Baras AS, et al. Expression of ral GTPases, their effectors, and activators in human bladder cancer. Clin Cancer Res. 2007;13:3803-13 [DOI] [PubMed] [Google Scholar]
- 80. Friederichs J, Zeller Y, Hafezi-Moghadam A, Grone HJ, Ley K, Altevogt P. The CD24/P-selectin binding pathway initiates lung arrest of human A125 adenocarcinoma cells. Cancer Res. 2000;60:6714-22 [PubMed] [Google Scholar]
- 81. Urano T, Emkey R, Feig LA. Ral-GTPases mediate a distinct downstream signaling pathway from Ras that facilitates cellular transformation. EMBO J. 1996;15:810-6 [PMC free article] [PubMed] [Google Scholar]
- 82. White MA, Vale T, Camonis JH, Schaefer E, Wigler MH. A role for the Ral guanine nucleotide dissociation stimulator in mediating Ras-induced transformation. J Biol Chem. 1996;271:16439-42 [DOI] [PubMed] [Google Scholar]
- 83. Hamad NM, Elconin JH, Karnoub AE, et al. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev. 2002; 16:2045-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Joneson T, White MA, Wigler MH, Bar-Sagi D. Stimulation of membrane ruffling and MAP kinase activation by distinct effectors of RAS. Science. 1996;271:810-2 [DOI] [PubMed] [Google Scholar]
- 85. Khosravi-Far R, White MA, Westwick JK, et al. Oncogenic Ras activation of Raf/mitogen-activated protein kinase-independent pathways is sufficient to cause tumorigenic transformation. Mol Cell Biol. 1996;16:3923-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Rodriguez-Viciana P, Warne PH, Khwaja A, et al. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457-67 [DOI] [PubMed] [Google Scholar]
- 87. White MA, Nicolette C, Minden A, et al. Multiple Ras functions can contribute to mammalian cell transformation. Cell. 1995;80:533-41 [DOI] [PubMed] [Google Scholar]
- 88. Chien Y, White MA. RAL GTPases are linchpin modulators of human tumour-cell proliferation and survival. EMBO Rep. 2003;4:800-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gonzalez-Garcia A, Pritchard CA, Paterson HF, Mavria G, Stamp G, Marshall CJ. RalGDS is required for tumor formation in a model of skin carcinogenesis. Cancer Cell. 2005;7:219-26 [DOI] [PubMed] [Google Scholar]
- 90. Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549-54 [DOI] [PubMed] [Google Scholar]
- 91. Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lim KH, O’Hayer K, Adam SJ, et al. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr Biol. 2006;16:2385-94 [DOI] [PubMed] [Google Scholar]
- 93. Vigil D, Martin TD, Williams F, Yeh JJ, Campbell SL, Der CJ. Aberrant overexpression of the Rgl2 Ral small GTPase-specific guanine nucleotide exchange factor promotes pancreatic cancer growth through Ral-dependent and Ral-independent mechanisms. J Biol Chem. 2010;285:34729-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Balasubramanian N, Meier JA, Scott DW, Norambuena A, White MA, Schwartz MA. RalA-exocyst complex regulates integrin-dependent membrane raft exocytosis and growth signaling. Curr Biol. 2010;20:75-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268-74 [DOI] [PubMed] [Google Scholar]
- 96. Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108-13 [DOI] [PubMed] [Google Scholar]
- 97. Martin TD, Samuel JC, Routh ED, Der CJ, Yeh JJ. Activation and involvement of Ral GTPases in colorectal cancer. Cancer Res. 2011;71:206-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lim KH, Baines AT, Fiordalisi JJ, et al. Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell. 2005;7:533-45 [DOI] [PubMed] [Google Scholar]
- 99. Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949-54 [DOI] [PubMed] [Google Scholar]
- 101. Tsao H, Goel V, Wu H, Yang G, Haluska FG. Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J Invest Dermatol. 2004;122:337-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Bervoets K, Millan MJ, Colpaert FC. Agonist action at 5-HT1C receptors facilitates 5-HT1A receptor-mediated spontaneous tail-flicks in the rat. Eur J Pharmacol. 1990;191:185-95 [DOI] [PubMed] [Google Scholar]
- 103. Brose MS, Volpe P, Feldman M, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62:6997-7000 [PubMed] [Google Scholar]
- 104. Pollock PM, Harper UL, Hansen KS, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19-20 [DOI] [PubMed] [Google Scholar]
- 105. Omholt K, Hansson J. No evidence of RALGDS mutations in cutaneous melanoma. Melanoma Res. 2007;17:410-2 [DOI] [PubMed] [Google Scholar]
- 106. Mishra PJ, Ha L, Rieker J, et al. Dissection of RAS downstream pathways in melanomagenesis: a role for Ral in transformation. Oncogene. 2010;29:2449-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Zipfel PA, Brady DC, Kashatus DF, Ancrile BD, Tyler DS, Counter CM. Ral activation promotes melanomagenesis. Oncogene. 2010;29:4859-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Platt FM, Hurst CD, Taylor CF, Gregory WM, Harnden P, Knowles MA. Spectrum of phosphatidylinositol 3-kinase pathway gene alterations in bladder cancer. Clin Cancer Res. 2009;15:6008-17 [DOI] [PubMed] [Google Scholar]
- 109. Jebar AH, Hurst CD, Tomlinson DC, Johnston C, Taylor CF, Knowles MA. FGFR3 and Ras gene mutations are mutually exclusive genetic events in urothelial cell carcinoma. Oncogene. 2005;24:5218-25 [DOI] [PubMed] [Google Scholar]
- 110. Kompier LC, Lurkin I, van der Aa MN, van Rhijn BW, van der Kwast TH, Zwarthoff EC. FGFR3, HRAS, KRAS, NRAS and PIK3CA mutations in bladder cancer and their potential as biomarkers for surveillance and therapy. PLoS One. 2010;5:e13821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Oxford G, Owens CR, Titus BJ, et al. RalA and RalB: antagonistic relatives in cancer cell migration. Cancer Res. 2005;65:7111-20 [DOI] [PubMed] [Google Scholar]
- 112. Shi M, Zheng Y, Garcia A, Xu L, Foster DA. Phospholipase D provides a survival signal in human cancer cells with activated H-Ras or K-Ras. Cancer Lett. 2007;258:268-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Varambally S, Yu J, Laxman B, et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell. 2005;8:393-406 [DOI] [PubMed] [Google Scholar]
- 114. Rinaldo F, Li J, Wang E, Muders M, Datta K. RalA regulates vascular endothelial growth factor-C (VEGF-C) synthesis in prostate cancer cells during androgen ablation. Oncogene. 2007;26:1731-8 [DOI] [PubMed] [Google Scholar]
- 115. Zhang H, Muders MH, Li J, Rinaldo F, Tindall DJ, Datta K. Loss of NKX3.1 favors vascular endothelial growth factor-C expression in prostate cancer. Cancer Res. 2008;68:8770-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Oosterhoff JK, Penninkhof F, Brinkmann AO, Anton Grootegoed J, Blok LJ. REPS2/POB1 is downregulated during human prostate cancer progression and inhibits growth factor signalling in prostate cancer cells. Oncogene. 2003;22:2920-5 [DOI] [PubMed] [Google Scholar]
- 117. Yin J, Pollock C, Tracy K, et al. Activation of the RalGEF/Ral pathway promotes prostate cancer metastasis to bone. Mol Cell Biol. 2007;27:7538-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000;151:33-40 [DOI] [PubMed] [Google Scholar]
- 119. Bodempudi V, Yamoutpoor F, Pan W, et al. Ral overactivation in malignant peripheral nerve sheath tumors. Mol Cell Biol. 2009;29:3964-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. de Gorter DJ, Reijmers RM, Beuling EA, et al. The small GTPase Ral mediates SDF-1-induced migration of B cells and multiple myeloma cells. Blood. 2008;111:3364-72 [DOI] [PubMed] [Google Scholar]
- 121. Yu Y, Feig LA. Involvement of R-Ras and Ral GTPases in estrogen-independent proliferation of breast cancer cells. Oncogene. 2002;21:7557-68 [DOI] [PubMed] [Google Scholar]
- 122. Li TT, Alemayehu M, Aziziyeh AI, et al. Beta-arrestin/Ral signaling regulates lysophosphatidic acid-mediated migration and invasion of human breast tumor cells. Mol Cancer Res. 2009;7:1064-77 [DOI] [PubMed] [Google Scholar]
- 123. Sowalsky AG, Alt-Holland A, Shamis Y, Garlick JA, Feig LA. RalA suppresses early stages of Ras-induced squamous cell carcinoma progression. Oncogene. 2010;29:45-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Fortini ME, Simon MA, Rubin GM. Signalling by the sevenless protein tyrosine kinase is mimicked by Ras1 activation. Nature. 1992;355:559-61 [DOI] [PubMed] [Google Scholar]
- 125. Han M, Sternberg PW. let-60, a gene that specifies cell fates during C. elegans vulval induction, encodes a ras protein. Cell. 1990;63:921-31 [DOI] [PubMed] [Google Scholar]
- 126. Dickson B, Sprenger F, Morrison D, Hafen E. Raf functions downstream of Ras1 in the Sevenless signal transduction pathway. Nature. 1992;360:600-3 [DOI] [PubMed] [Google Scholar]
- 127. Balakireva M, Rosse C, Langevin J, et al. The Ral/ exocyst effector complex counters c-Jun N-terminal kinase-dependent apoptosis in Drosophila melanogaster. Mol Cell Biol. 2006;26:8953-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Ghiglione C, Devergne O, Cerezo D, Noselli S. Drosophila RalA is essential for the maintenance of Jak/Stat signalling in ovarian follicles. EMBO Rep. 2008;9:676-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Frische EW, Pellis-van Berkel W, van Haaften G, et al. RAP-1 and the RAL-1/exocyst pathway coordinate hypodermal cell organization in Caenorhabditis elegans. EMBO J. 2007;26:5083-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Han M, Golden A, Han Y, Sternberg PW. C. elegans lin-45 raf gene participates in let-60 ras-stimulated vulval differentiation. Nature. 1993;363:133-40 [DOI] [PubMed] [Google Scholar]
- 131. Berset T, Hoier EF, Battu G, Canevascini S, Hajnal A. Notch inhibition of RAS signaling through MAP kinase phosphatase LIP-1 during C. elegans vulval development. Science. 2001;291:1055-8 [DOI] [PubMed] [Google Scholar]
- 132. Sundaram MV. The love-hate relationship between Ras and Notch. Genes Dev. 2005;19:1825-39 [DOI] [PubMed] [Google Scholar]
- 133. Reid TS, Terry KL, Casey PJ, Beese LS. Crystallographic analysis of CaaX prenyltransferases complexed with substrates defines rules of protein substrate selectivity. J Mol Biol. 2004;343:417-33 [DOI] [PubMed] [Google Scholar]
- 134. Kinsella BT, Erdman RA, Maltese WA. Carboxyl-terminal isoprenylation of ras-related GTP-binding proteins encoded by rac1, rac2, and ralA. J Biol Chem. 1991;266:9786-94 [PubMed] [Google Scholar]
- 135. Falsetti SC, Wang DA, Peng H, et al. Geranylgeranyltransferase I inhibitors target RalB to inhibit anchorage-dependent growth and induce apoptosis and RalA to inhibit anchorage-independent growth. Mol Cell Biol. 2007;27:8003-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Lu J, Chan L, Fiji HD, Dahl R, Kwon O, Tamanoi F. In vivo antitumor effect of a novel inhibitor of protein geranylgeranyltransferase-I. Mol Cancer Ther. 2009;8:1218-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Liu M, Sjogren AK, Karlsson C, et al. Targeting the protein prenyltransferases efficiently reduces tumor development in mice with K-RAS-induced lung cancer. Proc Natl Acad Sci U S A. 2010;107:6471-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Sjogren AK, Andersson KM, Khan O, Olofsson FJ, Karlsson C, Bergo MO. Inactivating GGTase-I reduces disease phenotypes in a mouse model of K-RAS-induced myeloproliferative disease. Leukemia. 2011;25:186-9 [DOI] [PubMed] [Google Scholar]
- 139. Bivona TG, Quatela SE, Bodemann BO, et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell. 2006;21:481-93 [DOI] [PubMed] [Google Scholar]
- 140. Wu JC, Chen TY, Yu CT, et al. Identification of V23RalA-Ser194 as a critical mediator for Aurora-A-induced cellular motility and transformation by small pool expression screening. J Biol Chem. 2005;280:9013-22 [DOI] [PubMed] [Google Scholar]
- 141. Sablina AA, Chen W, Arroyo JD, et al. The tumor suppressor PP2A Abeta regulates the RalA GTPase. Cell. 2007;129:969-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Wang H, Owens C, Chandra N, Conaway MR, Brautigan DL, Theodorescu D. Phosphorylation of RalB is important for bladder cancer cell growth and metastasis. Cancer Res. 2010;70:8760-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Feldmann G, Mishra A, Hong SM, et al. Inhibiting the cyclin-dependent kinase CDK5 blocks pancreatic cancer formation and progression through the suppression of Ras-Ral signaling. Cancer Res. 2010;70:4460-9 [DOI] [PMC free article] [PubMed] [Google Scholar]