

Abstract
Ras genes are frequently activated in cancer. Attempts to develop drugs that target mutant Ras proteins have, so far, been unsuccessful. Tumors bearing these mutations, therefore, remain among the most difficult to treat. Most efforts to block activated Ras have focused on pathways downstream. Drugs that inhibit Raf kinase have shown clinical benefit in the treatment of malignant melanoma. However, these drugs have failed to show clinical benefit in Ras mutant tumors. It remains unclear to what extent Ras depends on Raf kinase for transforming activity, even though Raf proteins bind directly to Ras and are certainly major effectors of Ras action in normal cells and in development. Furthermore, Raf kinase inhibitors can lead to paradoxical activation of the MAPK pathway. MEK inhibitors block the Ras-MAPK pathway, but often activate the PI3’-kinase, and have shown little clinical benefit as single agents. This activation is mediated by EGF-R and other receptor tyrosine kinases through relief of a negative feedback loop from ERK. Drug combinations that target multiple points within the Ras signaling network are likely to be necessary to achieve substantial clinical benefit. Other effectors may also contribute to Ras signaling and provide a source of targets. In addition, unbiased screens for genes necessary for Ras transformation have revealed new potential targets and have added to our understanding of Ras cancer biology.
Keywords: Ras, targeted therapy, signal transduction, Raf, MAPK kinase
Background
Ras genes are frequently mutated in human cancers, and the proteins they encode have been considered drug targets since they were first identified and characterized 30 years ago. Yet, in 2011, no drugs that target Ras proteins directly or act on Ras-driven human cancers have been developed successfully. Indeed, tumors harboring Ras mutations remain the most difficult to treat and are excluded from treatment with specific targeted therapies.
Ras proteins cycle between an inactive GDP-bound “off” state and an active GTP-bound “on” state and function as molecular switches that mediate signal transduction between cell surface growth factor receptors and intracellular signaling pathways.1 The activation of Ras proteins, that is, the exchange of GDP with GTP, is an intrinsically slow process and is catalyzed by guanine nucleotide exchange factors (GEFs). However, this exchange is, in principle, reversible. The inactivation, that is, the hydrolysis of the γ-phosphate of GTP to GDP, is catalyzed by GTPase-activating proteins (GAPs) and is irreversible. Oncogenic mutations occur most frequently in codons 12, 13, and 61 and elsewhere. The resulting oncogenic versions of Ras proteins are resistant to GAP-mediated GTP hydrolysis, which renders them constitutively active. Besides cycling between GDP- and GTP-bound states, Ras proteins undergo posttranslational processing.2-6 These modifications attach the proteins to cellular membranes, which is essential for activity.
The catalytic domain, also referred to as the G domain, is highly homologous between the 3 Ras proteins, K-Ras, H-Ras, and N-Ras, which are activated in human cancer. The first 80 amino acids are identical, and the next 85 amino acids only differ by 5%. Analysis of the approximately 50 crystal structures of H-Ras and the recently solved x-ray structures of K- and N-Ras confirms the remarkable similarity of these proteins.7-9 For this reason, it is obvious that the functional differences between the 3 proteins are not embedded in the G domain but in the C-terminal hypervariable region (HVR), which comprises the last 23 of 24 amino acids. The Ras proteins share only 15% homology in this region. The HVR can be subdivided in 2 parts: the presumed unstructured linker region (AA 166-179 in N- and H-Ras and AA 166-174 in K-Ras) and the membrane-interacting lipid anchor (AA 180-189 in N- and H-Ras and AA 175-188 in K-Ras). Both parts are involved in high affinity interactions with lipid raft and nonraft plasma membrane microdomains.10,11 Determination of x-ray structures of membrane-bound Ras has been impossible. Therefore, biophysical experiments and computer simulations have been carried out in order to study membrane-bound Ras. Computer simulations with full-length H-Ras suggested that the GTP-bound protein undergoes a major conformational change at the membrane, bringing the catalytic domain in contact with the lipid bilayer. Basic residues in α4 have been implicated in these interactions.12
The basic function of nucleotide binding and hydrolysis is carried out by the approximately 20-kDa G domain. The G domain is classified as a α/β protein, typical for nucleotide-binding proteins. The critical regions involve a conserved phosphate-binding loop (P-loop, residues 10-17) and 2 switch regions (switch I, AA 25-40; switch II, AA 57-75) that bind the nucleotide. It is these 3 regions that are affected by oncogenic mutations. The switch regions usually show a high degree of flexibility when analyzed by x-ray diffraction or by nuclear magnetic resonance and electron paramagnetic resonance. The canonical switch mechanism can be considered as a loaded-spring mechanism, where the release of the γ-phosphate after the GTP hydrolysis allows the 2 switch regions to relax into the GDP-specific conformation. Different members of the Ras superfamily show variations of this mechanism.4,13
The release of guanine nucleotides from Ras proteins is a slow process and is accelerated by GEFs, by several orders of magnitude. The catalytic mechanism involves a series of fast reactions, which lead from a binary Ras-nucleotide complex via a trimeric Ras-nucleotide-GEF complex to a binary nucleotide-free complex. These reactions are reversible: GEFs act as catalysts and increase the rate at which equilibrium is reached. The position of the equilibrium is determined by the high affinity of Ras proteins for GDP or GTP, the intracellular concentration of nucleotides, and the affinities and concentrations of effector proteins that shift the equilibrium towards the GTP-bound state. GEFs interact with switch I and II regions and insert residues into the P-loop and the Mg2+-binding area. This perturbation is considered to be the main cause for the decreased affinity between Ras proteins and nucleotides.14,15 Conversely, Ras-GAPs accelerate conversion of active Ras-GTP back to Ras-GDP dramatically. Ras-GAPs insert an arginine side chain into the catalytic site and thereby neutralize developing charges in the transition state. Ras-GAPs stabilize the switch II domain and allow the conserved glutamine 61 to participate in catalysis. Oncogenic mutations of glycine 12 and glutamine 61 cause a perturbation, which renders Ras catalytically insensitive to GAP activity.16
Effector proteins of Ras show an enhanced affinity for the GTP-bound state. Some of the effector-binding domains are preformed and undergo no conformational change upon binding. This means that the recruitment of effector proteins by Ras-GTP at the membrane is considered as the activation process. In those cases where a large conformational change takes place, it has been shown that allosteric regulations of the effector proteins are involved. One of the best-characterized Ras effector proteins is the Raf kinase. Its Ras-binding domain (RBD) is a small domain that contains an ubiquitin fold with an interprotein β-sheet that is Ras-GTP sensitive.17 Other proteins like Ral-GDS have a similar domain and bind to Ras-GTP in a similar fashion. Both Raf and Ral-GDS bind to the switch I domain, as does the γ-subunit of PI3’-kinase. In the latter case, Ras uses also its switch II domain in order to bind the catalytic subunit. These interactions cause a structural change in PI3’-Kγ and affect the binding to phospholipids and the catalytic activity.18 Ras proteins undergo a series of posttranslational processing steps in order to become attached to cellular membranes and become biologically functional,19 as shown in Figure 1.
Figure 1.
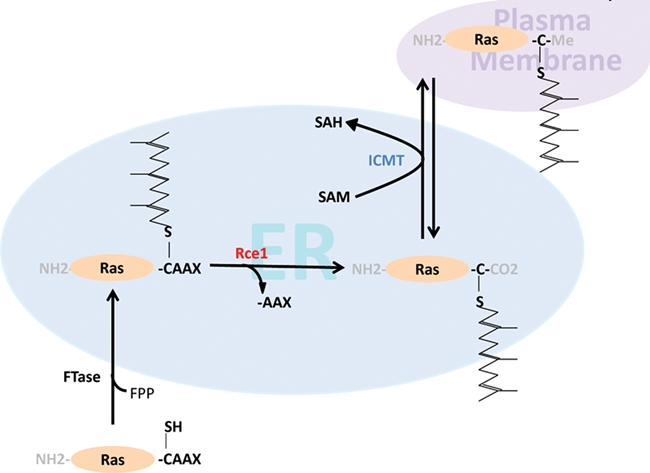
Ras processing.
The C-terminal membrane-interacting lipid anchor region of HVR contains distinct motifs that are subject to these modifications. In all 3 Ras proteins, the cysteine of the CAAX box (Cys185 in K-Ras 4A and 4B and Cys186 in N- and H-Ras) becomes farnesylated under normal conditions (Figure 2). This reaction is catalyzed by the enzyme farnesyl transferase (FTase).20 The farnesyl 15 carbon chain becomes attached to the cysteine via a stable thioether linkage. Ras farnesylation is, therefore, an irreversible reaction. In the presence of farnesyl transferase inhibitors (FTIs) (see below), K-Ras and N-Ras become alternatively prenylated by the attachment of a 20-carbon geranylgeranyl chain through geranylgeranyl transferase I (GGTase I). This has been a major obstacle for the successful application of FTIs in K-Ras– or N-Ras–driven tumors,21 as discussed below. K-Ras 4A and N-Ras have 1 additional cysteine (Cys179 in K-Ras 4A and Cys181 in N-Ras), whereas H-Ras has 2 additional cysteines (Cys181 and Cys184) that become palmitoylated. Ras palmitoylation is reversible and is carried out by palmitoyl transferases (PTase).22 The DHHC family of PTases has been characterized, and it has also been shown that Ras palmitoylation takes place at the endoplasmic reticulum (ER)/Golgi endomembrane system, whereas depalmitoylation, carried out by less-studied acyl protein thioesterases, takes place at the plasma membrane.23
Figure 2.
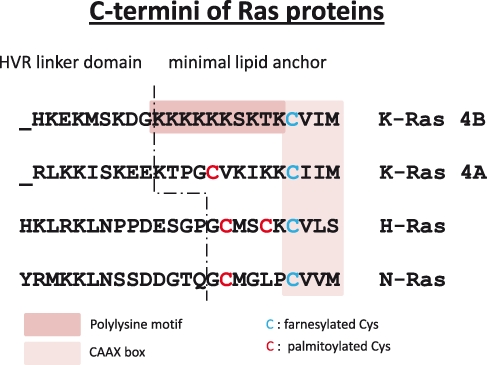
C-termini of Ras proteins.
N-Ras, H-Ras, and likely, K-Ras 4A undergo a palmitoylation/depalmitoylation cycle. This is an efficient way of shuttling these proteins from the ER/Golgi to the plasma membrane and reverse.24 In contrast, K-Ras 4B contains a polybasic lysine stretch in the adjacent upstream region of the CAAX box. This sequence mediates the interaction of K-Ras 4B with acidic phospholipids in the plasma membrane. Furthermore, K-Ras 4B contains a serine residue (Ser181) that is subject to phosphorylation. Phosphorylation by members of the protein kinase C family had been proposed, leading to dissociation of K-Ras 4B from the plasma membrane.25,26
The last 3 amino acids of the CAAX box (–AAX) are subject to proteolytic processing. In humans, the reaction is carried out by a prenyl protein–specific endoprotease known as ras-converting enzyme 1 (RCE1). It is an integral membrane protein of the ER. Rce1-null mice die between embryonic day 15.5 and the first week of life. The reasons for this lethality are not clear.27,28 The final CAAX processing step is carboxyl methylation. Isoprenylcysteine-carboxyl-methyltransferase (ICMT) is also an integral membrane protein of the ER, which is unusual for methyltransferases. After modification by ICMT, the fully processed Ras protein consists of a methyl-esterified farnesyl or geranylgeranyl cysteine. ICMT is also essential for mouse development, as mice lacking the gene die by embryonic day 11.5.29,30
These 2 postprenylation events render the C-termini of Ras proteins even more hydrophobic. Additionally, it has been speculated that carboxyl methylation is also required for the binding with interacting proteins. The carboxyl methylation reaction is reversible, and it is conceivable that this could represent another level of regulating Ras activity or subcellular localization.31 However, a Ras-specific methylesterase has not yet been identified.
Direct Therapeutic Attack on the G Domain
Ras proteins bind GTP with picomolar affinities. Furthermore, the concentration of GTP in cells approaches micromolar levels. Unlike protein kinases, in which phosphoryl transfer from ATP to a substrate is a rapid, catalytic process, the role of GDP or GTP is to stabilize inactive or active states of the Ras protein. For these reasons, targeting mutant Ras with nucleotide analogs does not appear to be a promising approach. The search for small molecules that bind to the surface of Ras proteins has been almost as challenging, as Ras does not have an accessible active site or pocket to which molecules are likely to bind. Nevertheless, Taveras and coworkers at Schering-Plough (Kenilworth, NJ) were able to identify a small molecule that binds to pocket on Ras, without displacing bound nucleotide. The compound, SCH 54292, has not been developed clinically but demonstrates that novel chemical approaches may yet identify ways of targeting Ras directly.32
Another approach to inactivate oncogenic Ras could involve small molecules that restore GTP hydrolysis in mutant Ras. Analysis of the GTPase site suggests this will also be technically extremely challenging, as the GTPase site is occupied by guanine nucleotide, and there appears little room for a small molecule to bind. Ideally, such a compound would mimic the effects of GAP and insert an arginine-like residue that would facilitate GTP hydrolysis in the mutant protein. The capacity of mutant Ras to hydrolyze GTP was demonstrated by Ahmadian and coworkers, who constructed GTP analogs covalently attached to positively charged groups that facilitated GTP hydrolysis. While this approach appears encouraging, identifying a small molecule that can bind and interact with mutant Ras in cells remains a daunting challenge.33
Strategies to Interfere with CAAX Processing
Ras prenylation, proteolytic processing, and carboxyl methylation are mechanisms that appear to represent reasonable targets for therapeutic intervention. Drugs that interfere with Ras prenylation have been developed over the last 2 decades. FTIs can be subdivided in 3 different classes: A) CAAX peptidomimetics that compete with Ras-CAAX for FTase; B) nonpeptidomimetics: farnesylpyrophosphate (FPP) analogs that compete with FPP for binding to FTase; or C) bisubstrate inhibitors, which are combinations of A and B.34 Attempts to covalently modify CAAX cysteines of Ras proteins have also been described with limited results so far.35 The first inhibitor approaches focused on the development of competitors for the protein substrate. However, some of these peptides were not very cell permeable or became rapidly degraded in the cell. CAAX peptidomimetics in which the AA portion was replaced with benzodiazepine (C-BZA-M) or aminomethylbenzoic acid (C-AMBA-M) were good inhibitors of FTase and considerably more stable.36 Later, high-throughput screening led to the identification of small molecule inhibitors. Two of those compounds made it into clinical evaluation: lonafarnib and tipifarnib. Lonafarnib (SCH66336) is a nonpeptidic CAAX-competitive inhibitor that is selective for FTase (IC50 = 1.9 nM). Tipifarnib (R115777) is also selective for FTase with an IC50 of 7.9 nM.37,38
FTIs inhibit cell growth of a large variety of cancer cell lines in vitro and also in vivo as tumor xenografts.39 In particular, FTIs were shown to prevent H-Ras farnesylation and reverse H-Ras–driven cell transformation.40 FTIs induce tumor growth inhibition rather than regression when used as monotherapies. However, K-Ras, the major Ras oncogene in human cancers, and N-Ras are subject to alternative prenylation by GGTase I in FTI-treated cells.21 This resulted in a persistent membrane localization of K-Ras and N-Ras and concomitant upregulation of downstream signaling. The fact that K-Ras and N-Ras became cross-prenylated had been often cited as the main reason why FTI monotherapy showed very poor efficacy in clinical trials. FTIs might be more effective in combination with cytotoxic, STI-571, or hormonal agents.39,41
Another problem hampering development of FTIs is the lack of reliable genetic markers of response. FTI activities do not correlate with K- or N-Ras mutational status. Furthermore, FTIs clearly have targets other than Ras proteins that might be responsible for the effects seen in preclinical models. For example, the inhibition of RhoB farnesylation seems to be important for FTI antitumor activity. FTIs also inhibit farnesylation of Rheb1 and Rheb2 GTPases that play a role in tuberous sclerosis. Furthermore, some FTIs potently inhibit GGTase II (Rab GTPase) as well as farnesylation of other farnesylated proteins like lamins, the centromeric proteins CENPE and CENPF, PxF, and HDJ2.42,43
The alternative prenylation of K-Ras and N-Ras by GGTase I led to the development of GGTase I inhibitors (GGTIs). Because geranylgeranylated proteins are more numerous than farnesylated proteins, this strategy is likely to result in widespread toxicity. Indeed, GGTIs at doses sufficient to prevent K-Ras prenylation in the presence of FTIs were found to be lethal in a mouse model.44 The development of dual prenylation inhibitors led to similar conclusions as with FTIs. Again, the observed antitumor activity did not correlate with the inhibition of K-Ras prenylation, which suggests that these inhibitors have a variety of targets in cells.45
Another strategy to interfere with Ras prenylation is to inhibit the formation of isoprenoids FPP and GGPP in the mevalonate pathway. Statins inhibit the rate-limiting enzyme 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA). Biphos-phonates inhibit 2 other critical enzymes in the pathway: isopentyl diphosphate (IPP) isomerase and FPP synthase that are required for FPP or GPP synthesis. Interference with the mevalonate pathway shows antitumor activity in some cancers, but here also, this effect cannot be solely attributed to the inhibition of Ras prenylation.46,47
The function of the postprenylation processing enzymes RCE1 and ICMT in tumorigenesis has been investigated in cells and mice in which the genes for these enzymes have been disrupted. The lack of RCE1 caused mislocalization of Ras proteins. A conditional deletion of RCE1 in fibroblasts was shown to reduce Ras-induced transformation in cells.48 However, interference with RCE1 function in tumor cells or cancer models has only shown modest effects. In hematopoietic cells of mice, simultaneous inactivation of RCE1 and activation of K-Ras led to acceleration of myeloproliferative disease development.49
The development of RCE1 inhibitors first concentrated on substrate analogs like modified CAAX peptides. A nitrophenyl modification of the second aliphatic amino acid in the CAAX peptide behaves as a competitive inhibitor. The activity of such compounds in cell-based assays has not been reported so far.50 The yeast RCE1 enzyme Rce1p and the nonrelated CAAX-protease Ste24p can independently promote the proteolytic cleavage of –AAX of prenylated CAAX box proteins in yeast. Recently, a class of inhibitors was found that are dual-specificity inhibitors. Such peptidyl (acyloxy)methyl ketones (AOMKs) could be engineered so that they exhibit selectivity for either of these enzymes. The same group screened a small molecule library that yielded 9 compounds being able to inhibit Rce1p in the low micromolar range. Some of these inhibitors were effective in disrupting yeast Ras localization.51,52
The effects of interfering with RCE1 function were modest, but more striking effects were seen through blocking the activity of ICMT. Inactivation of ICMT inhibited cell growth and K-Ras–induced oncogenic transformation both in soft agar assays as well as in a nude mouse model. Cells had a strongly reduced level of RhoA and increased levels of p21. Interference with ICMT function inhibited transformation by B-Raf V600E, an event that is thought to be largely Ras independent.53 Furthermore, disrupting ICMT ameliorated K-Ras–induced myeloproliferative disease.54 The anticancer drug methotrexate induces higher levels of homocysteine that causes hypomethylation in cells: treatment with methotrexate reduced Ras methylation by almost 90% and caused mislocalization and a decreased activity of ERK 1/2 and Akt.55 More recently, synthetic as well as natural small molecule inhibitors were found that block ICMT, induce apoptosis, and reduce tumor growth in a variety of model systems.56
Taken together, strategies that compromise Ras-CAAX box processing have, for the most part, not been rewarding. However, the actual proof of concept that these drugs specifically interfere with Ras membrane interaction and could be clinically useful is lacking. Prenylation and postprenylation inhibitors generally lack the specificity to inhibit signaling of specific Ras proteins, especially K-Ras. However, new approaches to block Ras through provoking mislocalization are being investigated and may still hold promise for the future.
Inhibition of Ras Expression
The idea of preventing Ras expression by antisense or RNA interference is promising, but the successful application of this technology is currently limited by lack of efficient delivery, uptake, and gene silencing. In cell lines, oncogenic mutation–specific, small interfering RNAs were able to silence K-Ras expression and inhibit tumor cell growth.57 A more recent study using anti–K-Ras RNA interference demonstrated that tumor cell lines can be classified as K-Ras dependent or K-Ras independent. This dependency correlated with their state of epithelial-to-mesenchymal transition. Epithelial tumor cells were dependent on K-Ras, whereas cell lines expressing mesenchymal markers were relatively independent on the oncogene. This finding showed that it is important to know the biological phenotype of a tumor in order to apply the correct strategy for Ras interference.58
Targeting Pathways Downstream of Ras
Developing therapeutic agents to directly block oncogenic Ras activity has thus far been a challenging and unsuccessful endeavor, for reasons discussed above. Therefore, a great deal of effort has been applied to developing therapies that target effector pathways downstream of Ras (Fig. 3). Constitutive activation of downstream effector pathways by oncogenic Ras results in the uncontrolled growth, proliferation, and survival of cancer cells. Understanding which effector pathways are required for Ras-driven oncogenesis is critical for determining which pathways should be targeted for therapeutic purposes. Many Ras effector pathways are comprised of kinase cascades, providing multiple nodes for potential therapeutic intervention. While several Ras effectors have been identified and comprehensively described,59-61 below, we discuss 2 of the best-characterized Ras effector pathways: the Raf-MEK-ERK and PI3’K signaling pathways. Importantly, both pathways are integral to Ras-driven transformation, and small-molecule compounds targeting these pathways are currently under clinical investigation.62,63
Figure 3.
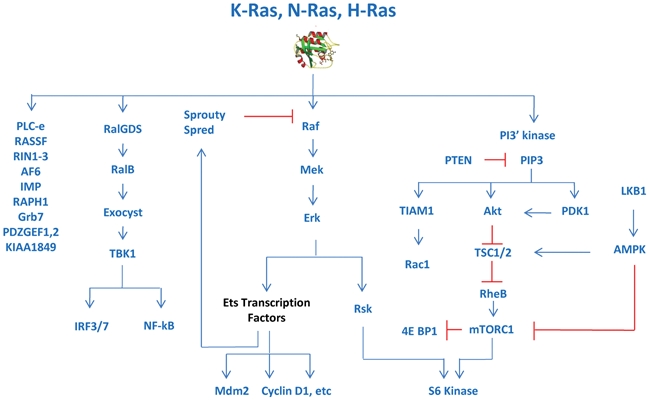
Pathways downstream of Ras.
The Raf-MEK-ERK Pathway
The Raf-MEK-ERK signal transduction pathway, also known as the MAPK cascade, was the first Ras effector signaling pathway identified. Raf serine/threonine kinases (A-Raf, B-Raf, and C-Raf/Raf-1) specifically interact with GTP-bound Ras, resulting in the activation of Raf protein kinase activity.64-67 Upon activation by Ras, Raf phosphorylates and activates the serine/threonine kinase MEK, which in turn phosphorylates and activates the serine/threonine kinase ERK. This series of signaling events results in the activation of transcriptional regulators that promote a wide variety of cellular events, including cell cycle progression and cell proliferation.59,60,68
The requirement for Raf-MEK-ERK signaling in Ras-mediated transformation and tumorigenesis has been well established.60,69-72 Dominant-negative mutants of Raf-1, MEK, and ERK inhibit Ras-driven transformation, highlighting the importance of this signaling cascade downstream of Ras.73-76 In support of these findings, mutations in the effector loop of H-Ras V12 that abrogate its ability to bind Raf-1 eliminate its transforming potential in mammalian cells, demonstrating the requirement for Raf-1 activity downstream of activated Ras.69 In addition, the growth inhibition induced by overexpression of dominant-negative Ras N17 can be overcome by expression of constitutively active Raf-1.77 Finally, cells that lack Ras proteins altogether can be rescued from growth arrest by expression of activated alleles of Raf, MEK, or ERK proteins, again showing that the Raf-MEK-ERK pathway is downstream from Ras in mammalian cells, as it is in Caenorhabditis elegans and Drosophila melanogaster and other model organisms.78
Baccarini and colleagues recently demonstrated that Raf-1 is required for the initiation and maintenance of squamous cell carcinoma in 2 separate models of Ras-driven tumorigenesis.79 In the first model, Ras activation is achieved through a classic chemical carcinogenesis protocol in which tumors are initiated through the topical application of 7,12-dimethylbenz[a]anthracene (DMBA), which causes an activating mutation in codon 61 of H-Ras. Tumor development is then promoted through the topical application of 12-O-tetradecanoylphorbol 13-acetate (TPA). In the second model, activation of the Ras pathway is achieved by expression of a dominant-active form of Son of Sevenless (SOS), specifically in the epidermis. In both models, ablation of Raf-1 leads to the regression of established Ras-driven tumors, suggesting that Raf-1 might serve as an appropriate target of therapeutic intervention downstream of activated Ras. Interestingly, in these models, the ability of Raf-1 to promote and maintain skin tumors is dependent on the inhibition of the RhoGTPase target Rok-α rather than the activation of the canonical MEK/ERK signaling cascade.
More recently, activating mutations in various components of the MAPK signaling cascade have been identified in patients with related genetic developmental disorders.68,80 For example, germline gain-of-function mutations in KRAS, BRAF, MEK1, and MEK2 have been observed in patients with cardiofaciocutaneous (CFC) syndrome.81,82 Activating mutations in Ras-Raf-MEK-ERK pathway components are present in patients with similar neurocardiofaciocutaneous syndromes, including Noonan, LEOPARD, and Costello syndromes.80,83,84 These findings provide genetic evidence that the Raf-MEK-ERK pathway functions downstream of Ras.
The identification of activating BRAF mutations in cancer supports a role for Raf-MEK-ERK signaling in oncogenesis.85,86 Interestingly, in melanoma and colorectal cancer, a pattern of mutual exclusivity between RAS and BRAF mutation has emerged, suggesting that mutation of either gene may be functionally equivalent in the pathogenesis of these malignancies.87 However, in the case of BRAF mutation, activation of additional oncogenic signaling pathways such as the PI3’K pathway may also be required.88
Attempts to target the Raf-MEK-ERK signaling pathway for therapeutic purposes have focused largely on the development of Raf and MEK kinase inhibitors. Sorafenib (Nexavar, Bayer, Leverkusen, Germany) was the first Raf kinase inhibitor to be tested in clinical trials and is now US Food and Drug Administration (FDA) approved for the treatment of renal cell carcinoma and hepatocellular carcinoma.89 Although sorafenib was designed to inhibit Raf-1 kinase activity, it also has activity against additional cancer targets including VEGF-R2, PDGFR, Flt-3, c-kit, and FGFR-1.90 In fact, the success of sorafenib as a cancer therapy has largely been attributed to its inhibitory effects on tumor angiogenesis,89 particularly for renal cell carcinoma, which is largely driven by hyperactive VEGF-R signaling. In support of this, the VEGF-R2 inhibitor Sutent (Pfizer, New York City, NY) is equally effective in treating this disease. In contrast, Sutent (Pfizer) failed to show efficacy in hepatocellular carcinoma, suggesting that sorafenib’s effects in this disease may indeed be mediated through inhibition of Raf kinase. Furthermore, clinical responses to sorafenib correlate well with levels of MAPK signaling in this disease.91 Activation of this pathway in hepatocellular carcinoma is caused by loss of the negative regulatory proteins Spred and Sprouty92,93 rather than by oncogenic Ras.
When B-Raf was identified as a major oncogene in human cancers,85 sorafenib was tested for clinical efficacy in this disease. However, no clinical benefit was observed. This may be because sorafenib interacts with the inactive form of Raf kinase and is less effective against B-Raf V600E than wild-type B-Raf. This prompted the development of second-generation Raf inhibitors, which demonstrate elevated specificity for B-Raf V600E.62,94 While these inhibitors potently suppress Raf-MEK-ERK signaling and cell growth in cancer cells expressing B-Raf V600E, they paradoxically have the opposite effect in cancer cells with wild-type B-Raf, including those with oncogenic Ras mutations.95-98 The promotion of Raf-MEK-ERK signaling in Ras mutant cancer cells by Raf inhibitors has been reviewed extensively62,98,99 and precludes the use of these inhibitors for the treatment of Ras mutant cancers. Furthermore, although the use of the pan-RAF inhibitor PLX-4032 in melanoma patients harboring BRAF V600E mutations produced promising clinical results,100-102 recent studies have identified multiple mechanisms of Raf inhibitor resistance, including enhanced receptor tyrosine kinase signaling as well as mutational activation of NRAS.103-105 The finding that mutational activation of NRAS can bypass the effects of Raf inhibition suggests that targeting Raf in the context of an activating Ras mutation may not be beneficial, despite the evidence in cell culture and animal models that suggest otherwise.
Several small-molecule compounds have been developed to potently and selectively inhibit the activity of MEK. Studies in cancer cell lines and animal models demonstrate that B-Raf mutation predicts sensitivity to these agents, although a subset of Ras mutant cell lines displays sensitivity as well.106,107 Despite these promising preclinical results, the outcome of early clinical trials was underwhelming in part because of the limited bioavailability and dose-limiting toxicity of the compounds.62,108 Several compounds with improved pharmaceutical properties are currently under clinical investigation and hold promise for the treatment of Ras mutant tumors.108 Defining the factors that underlie MEK inhibitor sensitivity and resistance in Ras mutant cancers is of great interest and will aid in determining which patients will benefit most from therapy. Likewise, it will be critical to determine whether toxicities that limited dosing are on target or off target. In this respect, it is of interest that the MEK inhibitor PD0325901, which is considered a relatively specific compound (it acts through an allosteric mechanism rather than as an ATP competitor), was able to block growth of cells in culture that had been engineered to grow in the absence of Ras-Raf-MEK-ERK activity, strongly suggesting that it has off-target effects that could, in principle, have accounted for clinical toxicity.109
The PI3’K Pathway
The phosphoinositide 3-kinase (PI3’K) pathway is another well-studied signaling cascade downstream of Ras. Class IA PI3’Ks are heterodimeric lipid kinases comprised of a p85 regulatory subunit and a p110 catalytic subunit. The p110 catalytic subunit of PI3’K was identified as a Ras effector when it was found to preferentially associate with GTP-bound Ras through its RBD.110,111 Although PI3’K can be activated by upstream receptor tyrosine kinases in a Ras-independent manner, association with and activation by Ras-GTP have proven to be a principal mechanism of PI3’K regulation. PI3’K catalyzes the conversion of phosphatidylinositol (4,5)-bisphosphate (PIP2) to the second-messenger phosphatidylinositol (3,4,5)-trisphosphate (PIP3). A primary downstream effector of PIP3 is the serine/threonine kinase Akt, which activates a host of signaling programs to promote cell growth, survival, and migration.112,113
PI3’K signaling is often upregulated in tumor cells, indicating its importance in the pathology of cancer. Hyperactivation of the pathway can be achieved through a variety of mechanisms, including gain-of-function mutation in PIK3CA, which encodes the p110α catalytic subunit of PI3’K.114-120 PTEN is a lipid phosphatase that negatively regulates PI3’K signaling, and its expression is often lost in cancers, providing yet another method by which PI3’K signaling can be deregulated.121-123 Additionally, increased activity of upstream regulators can also activate the PI3’K signaling pathway, and this can be achieved through amplification or activation of upstream receptor tyrosine kinases or via oncogenic Ras mutation.113,115 Interestingly, although Ras mutation drives PI3’K activity, oncogenic mutations in RAS and PIK3CA often coexist in colorectal cancers.113,124-126 It is unclear whether these coexisting mutations cooperate to amplify common downstream pathways or function independently to activate nonoverlapping pathways.113
Several lines of experimental evidence suggest that Ras mutant tumors depend on the activation of the PI3’K pathway. For example, PI3’K activity is necessary for the transformation of mouse embryonic fibroblasts by Ras.127 In addition, the interaction between Ras and PI3’K is essential in a mouse model of Ras-driven tumor formation.128 Collectively, these studies demonstrate a requirement for PI3’K activity downstream of oncogenic Ras and suggest that targeting PI3’K in Ras mutant cancers may have important antitumor effects.
Although the aforementioned studies emphasize a role for PI3’K signaling in Ras-mediated tumorigenesis, preliminary data suggest that Ras mutant tumors are insensitive to single-agent PI3’K inhibitors. In fact, in vitro experiments have uncovered Ras mutation as a dominant predictor of resistance to PI3’K inhibitors.129-131 In addition, murine lung cancers driven by oncogenic K-Ras do not respond to treatment with a single-agent dual PI3’K-mTOR inhibitor.132 Therefore, while PI3’K activity is an important driver of Ras-mediated transformation and tumorigenesis in cell culture and animal models, inhibition of PI3’K pathway activity alone is likely insufficient for the treatment of established tumors harboring RAS mutations.63,133
Dual Inhibition of the Raf-MEK-ERK and PI3’K Effector Pathways
The limited response of Ras mutant cancer cells to single-agent pathway inhibitors suggests that dual inhibition of Raf-MEK-ERK and PI3’K signaling may be necessary to block the growth of Ras-driven tumors. The efficacy of a single-agent pathway inhibitor is often hindered by the release of negative feedback loops on the reciprocal pathway. For example, treatment of Ras mutant cancer cells with potent and specific MEK inhibitors results in increased phosphorylation of the PI3’K pathway effector Akt.107,134,135 Upregulation of PI3’K signaling in response to MEK inhibition is due to the release of negative feedback from ERK to the EGF receptor.136 In light of the crosstalk between the Raf-MEK-ERK and PI3’K signaling pathways, it has been proposed that dual inhibition of both pathways may be required to evade these feedback loops. In support of this hypothesis, combined inhibition of MEK and PI3’K signaling in Ras mutant cancer cells is superior to single-agent inhibition in vitro and in vivo and results in a synergistic decrease in cell viability and increase in apoptosis.107,134,135 Furthermore, while dual pathway inhibition was also more effective than single-agent pathway inhibition in cancer cells driven by activated receptor tyrosine kinase signaling, the most pronounced synergistic effect was observed in cancer cells harboring oncogenic RAS mutations.107
Studies utilizing transgenic mouse models provide additional support for this therapeutic approach. In a mouse model of lung cancer driven by oncogenic K-Ras, single-agent treatment with a dual PI3’K-mTOR inhibitor had no effect on tumor growth. Additionally, single-agent treatment with a MEK inhibitor caused only modest tumor regression. However, combined treatment with both pathway inhibitors resulted in synergistic tumor regression.132
The necessity for dual pathway inhibition is most evident in cancers that harbor coexisting oncogenic mutations in RAS and PIK3CA. Reports indicate that mutational activation of PIK3CA in KRAS mutant cancer cells confers resistance to MEK inhibition.137,138 Indeed, treatment with single-agent MEK or Akt inhibitors had no significant effect on tumor growth in a xenograft model with coexisting KRAS and PIK3CA mutations. However, combined treatment with both inhibitors was effective at suppressing tumor growth.137
Collectively, these data indicate that dual inhibition of Raf-MEK-ERK and PI3’K signaling might be clinically beneficial in Ras mutant tumors and provide a rationale for the design of future clinical trials to test combinations of pathway inhibitors. Further, these studies emphasize that the efficacy of targeted therapeutics is genotype dependent and underscore the importance of stratifying patients by tumor genotype prior to therapy.
Additional Ras Effectors as Potential Targets in Ras-Driven Tumors
While Raf-MEK-ERK and PI3’K represent the best-characterized effector pathways utilized during tumor development following activation of Ras oncogenes, several additional effectors have been implicated in Ras-driven tumorigenesis including RalGDS, Tiam1, and PLCϵ. Each of these proteins has been shown to interact directly with Ras proteins, thus prompting the question of their role in Ras mutant tumors.60
The discovery that Ral guanine nucleotide dissociation stimulator (RalGDS) was able to interact with activated Ras proteins motivated the initial investigation of this effector arm of Ras signaling during tumorigenesis.139-143 RalGDS is a GEF, stimulating the dissociation of GDP from its target Ral proteins and allowing for binding of GTP and subsequent activation.142 In rodent cells in culture, Ral-GDS or the downstream Ral proteins cooperated with Ras oncogenes to induce transformation but were unable to do so on their own.61,144,145 Subsequent studies in human cells utilizing Ras effector–binding mutants demonstrated that activation of the RalGDS pathway was sufficient to transform human epithelial kidney cells.146 This pathway is thought to play a role in mediating proliferation and cell survival downstream of an activated Ras oncogene, as depletion of RalA impairs anchorage-independent proliferation of Ras mutant pancreatic tumor cells, while RalB was found to be required for survival in a number of tumor cell lines.142,147,148 Recently, Ral proteins have been implicated in the development of melanoma and myeloid malignancies, 2 cancers known to harbor frequent mutations in Ras oncogenes. RalA is activated in several melanoma cell lines with oncogenic N-Ras mutations, and RalA knockdown inhibited the tumorigenicity of these cell lines. Furthermore, studies in immortalized melanocytes showed that RalGEF was able to recapitulate several tumorigenic traits seen after transformation with N-Ras, including anchorage-independent growth, consistent with previous studies of this pathway.149,150 Expression of a Ras effector–binding mutant only able to activate RalGDS in hematopoietic cells demonstrated that activation of this pathway alone was able to inhibit neutrophil differentiation and prolong proliferative potential.151 Finally, in vivo evidence for RalGDS as a relevant Ras effector comes from data generated using a multistage model of skin carcinogenesis, leading to the development of squamous cell tumors harboring H-Ras mutations. Using this protocol in mice with homozygous deletion of RalGDS resulted in reduced tumor incidence, size, and progression to malignancy compared to wild-type mice.61,152 Together, these data support a role for RalGDS both in vitro and in vivo as an important effector pathway utilized by oncogenic Ras to drive tumorigenesis that could potentially be exploited for therapeutic intervention, although the absence of somatic mutations in this effector pathway makes its precise role less clear than the Raf-MEK-ERK and PI3’-kinase pathways.
Activation of Ras signaling has also been linked to breakdown of phosphoinositides through its ability to bind and activate phospholipase Cϵ.153,154 Activated PLCϵ catalyzes cleavage of PI(4,5)P2 into inositol-1,4,5-trisphosphate (IP3) and diacylglycerol (DAG), which subsequently promote the release of Ca2+ and the activation of protein kinase C (PKC), respectively. Although studies have shown conflicting results in vitro,155 the importance of this Ras effector pathway during tumorigenesis is supported in vivo by data generated again using the multistage mouse model of skin carcinogenesis. In these conditions, PLCϵ-null mice showed delayed onset of the characteristic squamous tumors resulting from this protocol as well as markedly reduced tumor incidence.156 Furthermore, tumors that did form in mice lacking PLCϵ also failed to undergo malignant progression to carcinomas. While the understanding of the relationship between Ras oncogenes and the requirement for PLCϵ is incomplete, these data suggest it may be an avenue to pursue to expand the list of potential drug targets in Ras mutant cancers.
Finally, Tiam1, a GEF that stimulates the activation of Rac, has also been implicated in tumorigenesis as an important Ras effector using the DMBA/TPA skin carcinogenesis model.157 Previously, Tiam1 had been shown to bind directly to active Ras, leading to its activation and subsequent stimulation of Rac activity.158-160 Tiam1-deficient mice are resistant to the development of DMBA/TPA-induced, Ras-driven skin tumors. Furthermore, the small number of tumors produced using this protocol grew much slower than the tumors formed in wild-type mice.157 Interestingly, however, the few tumors that grow in Tiam1-null mice show increased invasiveness, suggesting there may be different roles for Tiam1 at different stages of tumor progression. Taken together, each of these effectors represents a potential drug target for Ras-driven tumors, given that deletion of each leads to impaired tumorigenesis in models of Ras-driven cancers. The true promise of each of these effectors, however, will have to be further explored using additional models of Ras-driven tumorigenesis to define the extent to which generalizations can be made about the usage and requirements of these noncanonical effector pathways.
Candidate Synthetic Lethal Targets in Cells with Mutant Ras
While rationally targeting Ras itself or specific Ras effector pathways provides one therapeutic strategy, an alternative is to exploit vulnerabilities created specifically by the presence of a mutant Ras oncogene. Several potential targets have been identified in both cell culture systems and mouse models that are required for the initiation or maintenance of Ras mutant tumor cells. Considering that Tiam1 has been implicated in Ras-driven tumorigenesis, it is perhaps not unexpected that a role for the GTPase for which Tiam1 serves as a GEF, Rac1, has also been identified in Ras mutant tumors. The relevance of Rac1 in Ras transformation was initially described in cell culture systems in which dominant-negative Rac1 was able to inhibit focus formation by Ras oncogenes, while activated Rac1 was able to enhance Ras transformation in addition to growth in soft agar and motility.71,161,162 More recently, mouse models have provided additional support for the role of Rac1 in Ras-driven tumors in vivo. Deletion of Rac1 in keratinocytes and subsequent treatment with the DMBA/TPA skin tumorigenesis protocol demonstrated a role for this GTPase in the development of these H-Ras–driven cancers based on an observed decrease in hyperproliferation of keratinocytes in cells lacking Rac1.163 Furthermore, conditional deletion of Rac1 in combination with activation of K-Ras in a mouse model of lung cancer led to a dramatic reduction in cell proliferation with a reduction in the number of tumors.164 Additionally, loss of Rac1 alone is dispensable for proliferation but is required in the context of activated Ras defining a synthetic lethal interaction in these cells. While Rac1 may be important for the growth of Ras-driven tumors, the exact role and mechanism of activation in these cells remain to be fully elucidated, as several pathways can lead to Rac activity, which in turn can drive a number of downstream cellular behaviors. These data support a role for Rac1 in Ras-driven tumorigenesis, providing another member of a growing list of potential sites for therapeutic intervention and warranting further investigation of this pathway in Ras mutant cells.
Recently, a synthetic lethal interaction has been defined between the presence of a K-Ras oncogene and genetic deletion of Cdk4. Cdk4 has been implicated previously in models of breast tumorigenesis driven by alternate oncogenes165,166; however, Puyol et al. recently demonstrated that genetic or conditional deletion of Cdk4 led to a senescent response specifically in lung cells expressing an activated K-Ras oncogene.167 Additionally, treatment with a pharmacological inhibitor of Cdk4 showed a reduction in the growth of K-Ras–driven tumors, further supporting a role for Cdk4 as a therapeutic target in this disease. Furthermore, a similar requirement for Cdk4 was identified in a mouse model of H-Ras–driven breast tumorigenesis.168 Conversely, coexpression of Cdk4 and oncogenic Ras in normal epidermal cells leads to the development of squamous cell carcinoma–like invasive neoplasia, and Cdk4 expression was found to circumvent Ras-induced growth arrest in primary human keratinocytes, suggesting a requirement for Cdk4 during Ras-driven tumorigenesis in this system as well.169 Finally, in mouse models, expressing activated H-Ras in melanocytes showed increased incidence of spontaneous cutaneous melanoma when crossed onto a Cdk4(R24C) background, where Cdk4 is insensitive to inhibition by p15INK4B and p16INK4A.170 Furthermore, treatment of these mice with DMBA/TPA led to an increase in the number of nevi and melanomas compared to Cdk4 wild-type mice, suggesting a cooperative interaction between oncogenic Ras and Cdk4 during tumor development. Data from these different systems implicate Cdk4 as a promising therapeutic target in Ras-driven cancers.
Several other candidates have emerged as important mediators of the transforming effects of oncogenic Ras, including NF-κB, cyclin D1, and myc. NF-κB has been recently reported to be required for the development of tumors in mouse models of lung tumorigenesis.171,172 Meylan et al. demonstrated that inhibition of NF-κB signaling led to an apoptotic response in p53-null lung cancer cell lines, while inhibition of the pathway in vivo in the context of K-RasG12D–driven lung tumorigenesis showed reduced tumor development both at the time of tumor initiation or after tumor progression.172 Additionally, Basseres et al. showed that deletion of NF-κB subunit p65/RelA reduced the number of K-Ras–induced lung tumors both in the presence and absence of p53, and tumors that emerged in the absence of p65/RelA showed a higher number of apoptotic cells, reduced spread, and showed lower grade.171 The requirement for cyclin D1 has also been suggested in Ras mutant tumors. Initially, observations that activated Ras led to overexpression of cyclin D1 motivated investigation of the dependence of Ras on cyclin D1.173,174 Subsequently, deficiency in cyclin D1 was shown to decrease tumor development in several different systems of Ras-driven tumorigenesis, including grafting of retroviral Ras-transduced keratinocytes and phorbol ester treatment of Ras transgenic mice, and in a 2-stage model of skin carcinogenesis.174 Finally, a dependence on myc in Ras mutant tumors has been suggested based on data that in a mouse model of Ras-induced lung adenocarcinoma, expression of a dominant-negative myc mutant led to rapid regression of both incipient and established tumors, suggesting a requirement for myc in the development of Ras-driven tumor cells.175 Each of the examples discussed above provides the basis for exploring new avenues of intervention in cells harboring activated Ras alleles that may demonstrate therapeutic efficacy specifically in these tumor cells.
High-Density RNAi Screens for Synthetic Lethality in Ras Mutant Cells
Numerous studies have sought to define the molecular requirements that underlie Ras-driven tumorigenesis in order to inform potential sites of intervention. Recent high-throughput approaches have provided an expanded list of potential therapeutic targets for Ras-driven tumors. Using loss-of-function RNAi screens, several groups have identified proteins that, when lost, elicit a synthetic lethal response with mutant Ras oncogenes while leaving cells with wild-type Ras proteins unaffected. Scholl et al. utilized a subset of the RNAi Consortium Lentiviral shRNA Library targeting 1,011 human genes to identify STK33, a member of the calcium/calmodulin-dependent protein kinase subfamily of serine/threonine protein kinases, as a target that is selectively required for viability and proliferation in the context of mutant KRAS.176 They found that suppression of STK33 across a panel of K-Ras mutant versus wild-type cell lines demonstrated synthetic lethality independently of tissue origin. Furthermore, introduction of exogenous mutant KRAS resulted in newly acquired dependence on STK33. Having never been implicated in cancer previously and being insufficient for tumor initiation and maintenance alone, STK33 represents an example of a component of a signaling pathway that becomes aberrantly required in the presence of KRAS mutations. STK33 may promote viability in KRAS-dependent cells through regulation of S6K1-induced inactivation of the proapoptotic protein BAD.
Using a similar lentiviral shRNA screening strategy in a panel of cancer cell lines, Barbie et al. identified suppression of TBK1, a noncanonical IκB kinase as a second synthetic lethal interaction with K-Ras oncogenes. Viability in cell lines with endogenous mutations in KRAS was selectively reduced following TBK1 knockdown. Furthermore, growth of tumor xenografts was inhibited in KRAS mutant cells expressing TBK1 shRNAs, while TBK1 shRNA showed little to no effect on the tumor-forming ability of cells with wild-type KRAS.177 Introduction of oncogenic KRAS in immortalized human lung epithelial cells led to acquired sensitivity to the knockdown of TBK1. Activation of TBK1 may be linked to NF-κB–driven survival signals downstream of oncogenic K-Ras, consistent with the work discussed previously implicating NF-κB as playing an essential role in K-Ras mutant cells.
Finally, using a retroviral shRNA library, a comparable K-Ras synthetic lethal screen defined several mitotic genes that were selectively required in cells harboring mutations in K-Ras. These included several components of the anaphase-promoting complex, proteasome and polo-like kinase 1 (PLK1), expanding the list of strategies for therapeutic intervention in these cells.178 The authors suggest that based on these findings, Ras oncogenes may lead to increased dependence on key mitotic proteins for survival when compared to non-Ras transformed cells, although the precise mechanism underlying this phenomenon remains to be completely understood. Recently, similar approaches have also identified WT1 and Snail2 as candidate proteins required in Ras mutant cells.179,180 These high-throughput genome-wide screening strategies hold great value in providing rapid exploration of specific requirements of cells for growth and survival and may help to uncover previously unappreciated drug targets in tumors with various genetic lesions.
Future Prospects
Efforts to attack Ras proteins directly are ongoing, based on new ways of developing compounds based on structural considerations and on a better understanding of Ras processing and membrane localization. These efforts are still in early exploratory phases of drug discovery. Targeting downstream pathways, in contrast, is now a major focus of clinical research, as a rich pipeline of drug candidates that target proteins within with MAPK and PI3’-kinase pathways undergoes clinical evaluation. In parallel, new ways of identifying proteins that Ras depends on for malignant transformation are being evaluated, based on recent advances in RNA interference technology. Indeed, siRNA is being developed for systemic therapy and may eventually enter the mainstream of clinical research opportunities. Successful targeting of other major oncogenic drivers, such as EGF-R and BCR-ABL, only serves to underscore the importance of targeting Ras and encourages these exploratory and clinical efforts. Thirty years after Ras genes were identified, the urgency of devising strategies for treating Ras-driven cancers has only increased.
Footnotes
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr. McCormick is a consultant to ONYX, and this review discusses the use of sorafenib. Drs. Gysin, Salt, and Young declare no conflicts of interest.
This work was supported by Daiichi Sankyo Co. Ltd.
References
- 1. Kjeldgaard M, Nyborg J, Clark BF. The GTP binding motif: variations on a theme. FASEB J. 1996;10:1347-68 [PubMed] [Google Scholar]
- 2. Lowy DR, Willumsen BM. Function and regulation of ras. Annu Rev Biochem. 1993;62:851-91 [DOI] [PubMed] [Google Scholar]
- 3. Sprang SR. How Ras works: structure of a Rap-Raf complex. Structure. 1995;3:641-3 [DOI] [PubMed] [Google Scholar]
- 4. Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294:1299-304 [DOI] [PubMed] [Google Scholar]
- 5. Clark R, Wong G, Arnheim N, Nitecki D, McCormick F. Antibodies specific for amino acid 12 of the ras oncogene product inhibit GTP binding. Proc Natl Acad Sci U S A. 1985;82:5280-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Trahey M, McCormick F. A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science. 1987;238:542-5 [DOI] [PubMed] [Google Scholar]
- 7. Gorfe AA, Grant BJ, McCammon JA. Mapping the nucleotide and isoform-dependent structural and dynamical features of Ras proteins. Structure. 2008;16:885-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kouranov A, Xie L, de la Cruz J, et al. The RCSB PDB information portal for structural genomics. Nucleic Acids Res. 2006;34:D302-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deshpande N, Addess KJ, Bluhm WF, et al. The RCSB Protein Data Bank: a redesigned query system and relational database based on the mmCIF schema. Nucleic Acids Res. 2005;33:D233-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hancock JF, Parton RG. Ras plasma membrane signalling platforms. Biochem J. 2005;389:1-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rotblat B, Prior IA, Muncke C, et al. Three separable domains regulate GTP-dependent association of H-ras with the plasma membrane. Mol Cell Biol. 2004;24:6799-810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gorfe AA, Hanzal-Bayer M, Abankwa D, Hancock JF, McCammon JA. Structure and dynamics of the full-length lipid-modified H-Ras protein in a 1,2-dimyristoylglycero-3-phosphocholine bilayer. J Med Chem. 2007;50:674-84 [DOI] [PubMed] [Google Scholar]
- 13. Saraste M, Sibbald PR, Wittinghofer A. The P-loop: a common motif in ATP- and GTP-binding proteins. Trends Biochem Sci. 1990;15:430-4 [DOI] [PubMed] [Google Scholar]
- 14. Lenzen C, Cool RH, Prinz H, Kuhlmann J, Wittinghofer A. Kinetic analysis by fluorescence of the interaction between Ras and the catalytic domain of the guanine nucleotide exchange factor Cdc25Mm. Biochemistry. 1998;37:7420-30 [DOI] [PubMed] [Google Scholar]
- 15. Klebe C, Prinz H, Wittinghofer A, Goody RS. The kinetic mechanism of Ran--nucleotide exchange catalyzed by RCC1. Biochemistry. 1995;34:12543-52 [DOI] [PubMed] [Google Scholar]
- 16. Scheffzek K, Ahmadian MR, Kabsch W, et al. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277:333-8 [DOI] [PubMed] [Google Scholar]
- 17. Nassar N, Horn G, Herrmann C, Scherer A, McCormick F, Wittinghofer A. The 2.2 A crystal structure of the Ras-binding domain of the serine/threonine kinase c-Raf1 in complex with Rap1A and a GTP analogue. Nature. 1995;375:554-60 [DOI] [PubMed] [Google Scholar]
- 18. Walker EH, Perisic O, Ried C, Stephens L, Williams RL. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature. 1999;402:313-20 [DOI] [PubMed] [Google Scholar]
- 19. Willumsen BM, Christensen A, Hubbert NL, Papageorge AG, Lowy DR. The p21 ras C-terminus is required for transformation and membrane association. Nature. 1984;310:583-6 [DOI] [PubMed] [Google Scholar]
- 20. Casey PJ, Solski PA, Der CJ, Buss JE. p21ras is modified by a farnesyl isoprenoid. Proc Natl Acad Sci U S A. 1989;86:8323-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fiordalisi JJ, Johnson RL, 2nd, Weinbaum CA, et al. High affinity for farnesyltransferase and alternative prenylation contribute individually to K-Ras4B resistance to farnesyltransferase inhibitors. J Biol Chem. 2003;278:41718-27 [DOI] [PubMed] [Google Scholar]
- 22. Hancock JF, Magee AI, Childs JE, Marshall CJ. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell. 1989;57:1167-77 [DOI] [PubMed] [Google Scholar]
- 23. Mitchell DA, Vasudevan A, Linder ME, Deschenes RJ. Protein palmitoylation by a family of DHHC protein S-acyltransferases. J Lipid Res. 2006;47:1118-27 [DOI] [PubMed] [Google Scholar]
- 24. Goodwin JS, Drake KR, Rogers C, et al. Depalmitoylated Ras traffics to and from the Golgi complex via a nonvesicular pathway. J Cell Biol. 2005;170:261-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ghomashchi F, Zhang X, Liu L, Gelb MH. Binding of prenylated and polybasic peptides to membranes: affinities and intervesicle exchange. Biochemistry. 1995;34:11910-8 [DOI] [PubMed] [Google Scholar]
- 26. Ashery U, Yizhar O, Rotblat B, Kloog Y. Nonconventional trafficking of Ras associated with Ras signal organization. Traffic. 2006;7:119-26 [DOI] [PubMed] [Google Scholar]
- 27. Boyartchuk VL, Ashby MN, Rine J. Modulation of Ras and a-factor function by carboxyl-terminal proteolysis. Science. 1997;275:1796-800 [DOI] [PubMed] [Google Scholar]
- 28. Bergo MO, Lieu HD, Gavino BJ, et al. On the physiological importance of endoproteolysis of CAAX proteins: heart-specific RCE1 knockout mice develop a lethal cardiomyopathy. J Biol Chem. 2004;279:4729-36 [DOI] [PubMed] [Google Scholar]
- 29. Marr RS, Blair LC, Thorner J. Saccharomyces cerevisiae STE14 gene is required for COOH-terminal methylation of a-factor mating pheromone. J Biol Chem. 1990;265:20057-60 [PubMed] [Google Scholar]
- 30. Bergo MO, Leung GK, Ambroziak P, et al. Isoprenylcysteine carboxyl methyltransferase deficiency in mice. J Biol Chem. 2001;276:5841-5 [DOI] [PubMed] [Google Scholar]
- 31. Philips MR, Pillinger MH, Staud R, et al. Carboxyl methylation of Ras-related proteins during signal transduction in neutrophils. Science. 1993;259:977-80 [DOI] [PubMed] [Google Scholar]
- 32. Taveras AG, Remiszewski SW, Doll RJ, et al. Ras oncoprotein inhibitors: the discovery of potent, ras nucleotide exchange inhibitors and the structural determination of a drug-protein complex. Bioorg Med Chem. 1997;5:125-33 [DOI] [PubMed] [Google Scholar]
- 33. Ahmadian MR, Zor T, Vogt D, et al. Guanosine triphosphatase stimulation of oncogenic Ras mutants. Proc Natl Acad Sci U S A. 1999;96:7065-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Basso AD, Kirschmeier P, Bishop WR. Lipid posttranslational modifications: farnesyl transferase inhibitors. J Lipid Res. 2006;47:15-31 [DOI] [PubMed] [Google Scholar]
- 35. Troutman JM, Subramanian T, Andres DA, Spielmann HP. Selective modification of CaaX peptides with ortho-substituted anilinogeranyl lipids by protein farnesyl transferase: competitive substrates and potent inhibitors from a library of farnesyl diphosphate analogues. Biochemistry. 2007;46:11310-21 [DOI] [PubMed] [Google Scholar]
- 36. James GL, Goldstein JL, Brown MS, et al. Benzodiazepine peptidomimetics: potent inhibitors of Ras farnesylation in animal cells. Science. 1993;260:1937-42 [DOI] [PubMed] [Google Scholar]
- 37. Njoroge FG, Taveras AG, Kelly J, et al. (+)-4-[2-[4-(8-Chloro-3,10-dibromo-6,11-dihydro-5H-benzo[5, 6]cyclohepta[1,2-b]-pyridin-11(R)-yl)-1-piperidinyl]-2-oxo-ethyl]-1-piperidinecarboxamid e (SCH-66336): a very potent farnesyl protein transferase inhibitor as a novel antitumor agent. J Med Chem. 1998;41:4890-902 [DOI] [PubMed] [Google Scholar]
- 38. End DW, Smets G, Todd AV, et al. Characterization of the antitumor effects of the selective farnesyl protein transferase inhibitor R115777 in vivo and in vitro. Cancer Res. 2001;61:131-7 [PubMed] [Google Scholar]
- 39. Liu M, Bryant MS, Chen J, et al. Antitumor activity of SCH 66336, an orally bioavailable tricyclic inhibitor of farnesyl protein transferase, in human tumor xenograft models and wap-ras transgenic mice. Cancer Res. 1998;58:4947-56 [PubMed] [Google Scholar]
- 40. Ashar HR, James L, Gray K, et al. The farnesyl transferase inhibitor SCH 66336 induces a G(2) --> M or G(1) pause in sensitive human tumor cell lines. Exp Cell Res. 2001;262:17-27 [DOI] [PubMed] [Google Scholar]
- 41. Nakajima A, Tauchi T, Sumi M, Bishop WR, Ohyashiki K. Efficacy of SCH66336, a farnesyl transferase inhibitor, in conjunction with imatinib against BCR-ABL-positive cells. Mol Cancer Ther. 2003;2:219-24 [DOI] [PubMed] [Google Scholar]
- 42. Tabancay AP, Jr., Gau CL, Machado IM, et al. Identification of dominant negative mutants of Rheb GTPase and their use to implicate the involvement of human Rheb in the activation of p70S6K. J Biol Chem. 2003;278:39921-30 [DOI] [PubMed] [Google Scholar]
- 43. Lackner MR, Kindt RM, Carroll PM, et al. Chemical genetics identifies Rab geranylgeranyl transferase as an apoptotic target of farnesyl transferase inhibitors. Cancer Cell. 2005;7:325-36 [DOI] [PubMed] [Google Scholar]
- 44. Lobell RB, Omer CA, Abrams MT, et al. Evaluation of farnesyl:protein transferase and geranylgeranyl:protein transferase inhibitor combinations in preclinical models. Cancer Res. 2001;61:8758-68 [PubMed] [Google Scholar]
- 45. Lobell RB, Liu D, Buser CA, et al. Preclinical and clinical pharmacodynamic assessment of L-778,123, a dual inhibitor of farnesyl:protein transferase and geranylgeranyl:protein transferase type-I. Mol Cancer Ther. 2002;1:747-58 [PubMed] [Google Scholar]
- 46. Hindler K, Cleeland CS, Rivera E, Collard CD. The role of statins in cancer therapy. Oncologist. 2006;11:306-15 [DOI] [PubMed] [Google Scholar]
- 47. Caraglia M, Santini D, Marra M, Vincenzi B, Tonini G, Budillon A. Emerging anti-cancer molecular mechanisms of aminobisphosphonates. Endocr Relat Cancer. 2006;13:7-26 [DOI] [PubMed] [Google Scholar]
- 48. Bergo MO, Ambroziak P, Gregory C, et al. Absence of the CAAX endoprotease Rce1: effects on cell growth and transformation. Mol Cell Biol. 2002;22:171-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wahlstrom AM, Cutts BA, Karlsson C, et al. Rce1 deficiency accelerates the development of K-RAS-induced myeloproliferative disease. Blood. 2007;109:763-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hollander IJ, Frommer E, Aulabaugh A, Mallon R. Human Ras converting enzyme endoproteolytic specificity at the P2′ and P3′ positions of K-Ras-derived peptides. Biochim Biophys Acta. 2003;1649:24-9 [DOI] [PubMed] [Google Scholar]
- 51. Porter SB, Hildebrandt ER, Breevoort SR, Mokry DZ, Dore TM, Schmidt WK. Inhibition of the CaaX proteases Rce1p and Ste24p by peptidyl (acyloxy)methyl ketones. Biochim Biophys Acta. 2007;1773:853-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Manandhar SP, Hildebrandt ER, Schmidt WK. Small-molecule inhibitors of the Rce1p CaaX protease. J Biomol Screen. 2007;12:983-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bergo MO, Gavino BJ, Hong C, et al. Inactivation of Icmt inhibits transformation by oncogenic K-Ras and B-Raf. J Clin Invest. 2004;113:539-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wahlstrom AM, Cutts BA, Liu M, et al. Inactivating Icmt ameliorates K-RAS-induced myeloproliferative disease. Blood. 2008;112:1357-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Winter-Vann AM, Kamen BA, Bergo MO, et al. Targeting Ras signaling through inhibition of carboxyl methylation: an unexpected property of methotrexate. Proc Natl Acad Sci U S A. 2003;100:6529-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang M, Hossain MS, Tan W, et al. Inhibition of isoprenylcysteine carboxylmethyltransferase induces autophagic-dependent apoptosis and impairs tumor growth. Oncogene. 2010;29:4959-70 [DOI] [PubMed] [Google Scholar]
- 57. Smakman N, Veenendaal LM, van Diest P, et al. Dual effect of Kras(D12) knockdown on tumorigenesis: increased immune-mediated tumor clearance and abrogation of tumor malignancy. Oncogene. 2005;24:8338-42 [DOI] [PubMed] [Google Scholar]
- 58. Singh A, Greninger P, Rhodes D, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15:489-500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11-22 [DOI] [PubMed] [Google Scholar]
- 60. Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Repasky GA, Chenette EJ, Der CJ. Renewing the conspiracy theory debate: does Raf function alone to mediate Ras oncogenesis? Trends Cell Biol. 2004;14:639-47 [DOI] [PubMed] [Google Scholar]
- 62. Pratilas CA, Solit DB. Targeting the mitogen-activated protein kinase pathway: physiological feedback and drug response. Clin Cancer Res. 2010;16:3329-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28:1075-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Moodie SA, Willumsen BM, Weber MJ, Wolfman A. Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase kinase. Science. 1993;260:1658-61 [DOI] [PubMed] [Google Scholar]
- 65. Warne PH, Viciana PR, Downward J. Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature. 1993;364:352-5 [DOI] [PubMed] [Google Scholar]
- 66. Zhang XF, Settleman J, Kyriakis JM, et al. Normal and oncogenic p21ras proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature. 1993;364:308-13 [DOI] [PubMed] [Google Scholar]
- 67. Vojtek AB, Hollenberg SM, Cooper JA. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell. 1993;74:205-14 [DOI] [PubMed] [Google Scholar]
- 68. Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295-308 [DOI] [PubMed] [Google Scholar]
- 69. White MA, Nicolette C, Minden A, et al. Multiple Ras functions can contribute to mammalian cell transformation. Cell. 1995;80:533-41 [DOI] [PubMed] [Google Scholar]
- 70. Khosravi-Far R, White MA, Westwick JK, et al. Oncogenic Ras activation of Raf/mitogen-activated protein kinase-independent pathways is sufficient to cause tumorigenic transformation. Mol Cell Biol. 1996;16:3923-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Khosravi-Far R, Solski PA, Clark GJ, Kinch MS, Der CJ. Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol Cell Biol. 1995;15:6443-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cuadrado A, Bruder JT, Heidaran MA, App H, Rapp UR, Aaronson SA. H-ras and raf-1 cooperate in transformation of NIH3T3 fibroblasts. Oncogene. 1993;8:2443-8 [PubMed] [Google Scholar]
- 73. Cowley S, Paterson H, Kemp P, Marshall CJ. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841-52 [DOI] [PubMed] [Google Scholar]
- 74. Kolch W, Heidecker G, Lloyd P, Rapp UR. Raf-1 protein kinase is required for growth of induced NIH/3T3 cells. Nature. 1991;349:426-8 [DOI] [PubMed] [Google Scholar]
- 75. Schaap D, van der Wal J, Howe LR, Marshall CJ, van Blitterswijk WJ. A dominant-negative mutant of raf blocks mitogen-activated protein kinase activation by growth factors and oncogenic p21ras. J Biol Chem. 1993;268:20232-6 [PubMed] [Google Scholar]
- 76. Westwick JK, Cox AD, Der CJ, et al. Oncogenic Ras activates c-Jun via a separate pathway from the activation of extracellular signal-regulated kinases. Proc Natl Acad Sci U S A. 1994;91:6030-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Feig LA, Cooper GM. Inhibition of NIH 3T3 cell proliferation by a mutant ras protein with preferential affinity for GDP. Mol Cell Biol. 1988;8:3235-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Drosten M, Dhawahir A, Sum EY, et al. Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J. 2010;29:1091-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ehrenreiter K, Kern F, Velamoor V, et al. Raf-1 addiction in Ras-induced skin carcinogenesis. Cancer Cell. 2009;16:149-60 [DOI] [PubMed] [Google Scholar]
- 80. Chimera JA, Anderson SM, Noell H, Rizk V. Comparison of nucleic acid hybridization and cytologic examination for detection of human papillomavirus infection, with evaluation of two commercially available hybridization kits. Clin Chem. 1991;37:260-2 [PubMed] [Google Scholar]
- 81. Rodriguez-Viciana P, Tetsu O, Tidyman WE, et al. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311:1287-90 [DOI] [PubMed] [Google Scholar]
- 82. Niihori T, Aoki Y, Narumi Y, et al. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet. 2006;38:294-6 [DOI] [PubMed] [Google Scholar]
- 83. Aoki Y, Niihori T, Narumi Y, Kure S, Matsubara Y. The RAS/MAPK syndromes: novel roles of the RAS pathway in human genetic disorders. Hum Mutat. 2008;29:992-1006 [DOI] [PubMed] [Google Scholar]
- 84. Denayer E, de Ravel T, Legius E. Clinical and molecular aspects of RAS related disorders. J Med Genet. 2008;45:695-703 [DOI] [PubMed] [Google Scholar]
- 85. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949-54 [DOI] [PubMed] [Google Scholar]
- 86. Dhomen N, Marais R. New insight into BRAF mutations in cancer. Curr Opin Genet Dev. 2007;17:31-9 [DOI] [PubMed] [Google Scholar]
- 87. Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. [DOI] [PubMed] [Google Scholar]
- 88. Tsao H, Goel V, Wu H, Yang G, Haluska FG. Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J Invest Dermatol. 2004;122:337-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wilhelm S, Carter C, Lynch M, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5:835-44 [DOI] [PubMed] [Google Scholar]
- 90. Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099-109 [DOI] [PubMed] [Google Scholar]
- 91. Abou-Alfa GK, Schwartz L, Ricci S, et al. Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J Clin Oncol. 2006;24:4293-300 [DOI] [PubMed] [Google Scholar]
- 92. Yoshida T, Hisamoto T, Akiba J, et al. Spreds, inhibitors of the Ras/ERK signal transduction, are dysregulated in human hepatocellular carcinoma and linked to the malignant phenotype of tumors. Oncogene. 2006;25:6056-66 [DOI] [PubMed] [Google Scholar]
- 93. Fong CW, Chua MS, McKie AB, et al. Sprouty 2, an inhibitor of mitogen-activated protein kinase signaling, is down-regulated in hepatocellular carcinoma. Cancer Res. 2006;66:2048-58 [DOI] [PubMed] [Google Scholar]
- 94. Li HF, Chen Y, Rao SS, et al. Recent advances in the research and development of B-Raf inhibitors. Curr Med Chem. 2010;17:1618-34 [DOI] [PubMed] [Google Scholar]
- 95. Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431-5 [DOI] [PubMed] [Google Scholar]
- 97. Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Cichowski K, Janne PA. Drug discovery: inhibitors that activate. Nature. 2010;464:358-9 [DOI] [PubMed] [Google Scholar]
- 99. Cox AD, Der CJ. The raf inhibitor paradox: unexpected consequences of targeted drugs. Cancer Cell. 2010;17:221-3 [DOI] [PubMed] [Google Scholar]
- 100. Lee JT, Shan J, Gu W. Targeting the degradation of cyclin D1 will help to eliminate oncogene addiction. Cell Cycle. 2010;9:857-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Villanueva J, Vultur A, Lee JT, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Solit DB, Garraway LA, Pratilas CA, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Sos ML, Fischer S, Ullrich R, et al. Identifying genotype-dependent efficacy of single and combined PI3K- and MAPK-pathway inhibition in cancer. Proc Natl Acad Sci U S A. 2009;106:18351-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Fremin C, Meloche S. From basic research to clinical development of MEK1/2 inhibitors for cancer therapy. J Hematol Oncol. 2010;3:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Urosevic J, Sum EY, Moneo V, et al. Using cells devoid of RAS proteins as tools for drug discovery. Mol Carcinog. 2009;48:1038-47 [DOI] [PubMed] [Google Scholar]
- 110. Rodriguez-Viciana P, Warne PH, Dhand R, et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527-32 [DOI] [PubMed] [Google Scholar]
- 111. Pacold ME, Suire S, Perisic O, et al. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell. 2000;103:931-43 [DOI] [PubMed] [Google Scholar]
- 112. Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20:87-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497-510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. [DOI] [PubMed] [Google Scholar]
- 115. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606-19 [DOI] [PubMed] [Google Scholar]
- 116. Bellacosa A, de Feo D, Godwin AK, et al. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer. 1995;64:280-5 [DOI] [PubMed] [Google Scholar]
- 117. Cheng JQ, Ruggeri B, Klein WM, et al. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc Natl Acad Sci U S A. 1996;93:3636-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Ruggeri BA, Huang L, Wood M, Cheng JQ, Testa JR. Amplification and overexpression of the AKT2 oncogene in a subset of human pancreatic ductal adenocarcinomas. Mol Carcinog. 1998;21:81-6 [PubMed] [Google Scholar]
- 119. Zhao L, Vogt PK. Class I PI3K in oncogenic cellular transformation. Oncogene. 2008;27:5486-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Denley A, Kang S, Karst U, Vogt PK. Oncogenic signaling of class I PI3K isoforms. Oncogene. 2008;27:2561-74 [DOI] [PubMed] [Google Scholar]
- 121. Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943-7 [DOI] [PubMed] [Google Scholar]
- 122. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375-8 [DOI] [PubMed] [Google Scholar]
- 123. Steck PA, Pershouse MA, Jasser SA, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356-62 [DOI] [PubMed] [Google Scholar]
- 124. Ollikainen M, Gylling A, Puputti M, et al. Patterns of PIK3CA alterations in familial colorectal and endometrial carcinoma. Int J Cancer. 2007;121:915-20 [DOI] [PubMed] [Google Scholar]
- 125. Velho S, Oliveira C, Ferreira A, et al. The prevalence of PIK3CA mutations in gastric and colon cancer. Eur J Cancer. 2005;41:1649-54 [DOI] [PubMed] [Google Scholar]
- 126. Barault L, Veyrie N, Jooste V, et al. Mutations in the RAS-MAPK, PI(3)K (phosphatidylinositol-3-OH kinase) signaling network correlate with poor survival in a population-based series of colon cancers. Int J Cancer. 2008;122:2255-9 [DOI] [PubMed] [Google Scholar]
- 127. Rodriguez-Viciana P, Warne PH, Khwaja A, et al. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457-67 [DOI] [PubMed] [Google Scholar]
- 128. Gupta S, Ramjaun AR, Haiko P, et al. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell. 2007;129:957-68 [DOI] [PubMed] [Google Scholar]
- 129. Torbett NE, Luna-Moran A, Knight ZA, et al. A chemical screen in diverse breast cancer cell lines reveals genetic enhancers and suppressors of sensitivity to PI3K isoform-selective inhibition. Biochem J. 2008;415:97-110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Dan S, Okamura M, Seki M, et al. Correlating phosphatidylinositol 3-kinase inhibitor efficacy with signaling pathway status: in silico and biological evaluations. Cancer Res. 2010;70:4982-94 [DOI] [PubMed] [Google Scholar]
- 131. Ihle NT, Lemos R, Jr., Wipf P, et al. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res. 2009;69:143-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550-62 [DOI] [PubMed] [Google Scholar]
- 134. Mirzoeva OK, Das D, Heiser LM, et al. Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition. Cancer Res. 2009;69:565-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Hoeflich KP, O’Brien C, Boyd Z, et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res. 2009;15:4649-64 [DOI] [PubMed] [Google Scholar]
- 136. Yu CF, Liu ZX, Cantley LG. ERK negatively regulates the epidermal growth factor-mediated interaction of Gab1 and the phosphatidylinositol 3-kinase. J Biol Chem. 2002;277:19382-8 [DOI] [PubMed] [Google Scholar]
- 137. Halilovic E, She QB, Ye Q, et al. PIK3CA mutation uncouples tumor growth and cyclin D1 regulation from MEK/ERK and mutant KRAS signaling. Cancer Res. 2010;70:6804-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Wee S, Jagani Z, Xiang KX, et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009;69:4286-93 [DOI] [PubMed] [Google Scholar]
- 139. Hofer F, Fields S, Schneider C, Martin GS. Activated Ras interacts with the Ral guanine nucleotide dissociation stimulator. Proc Natl Acad Sci U S A. 1994;91:11089-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Kikuchi A, Demo SD, Ye ZH, Chen YW, Williams LT. ralGDS family members interact with the effector loop of ras p21. Mol Cell Biol. 1994;14:7483-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Spaargaren M, Bischoff JR. Identification of the guanine nucleotide dissociation stimulator for Ral as a putative effector molecule of R-ras, H-ras, K-ras, and Rap. Proc Natl Acad Sci U S A. 1994;91:12609-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Bodemann BO, White MA. Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nat Rev Cancer. 2008;8:133-40 [DOI] [PubMed] [Google Scholar]
- 143. Campbell PM, Singh A, Williams FJ, et al. Genetic and pharmacologic dissection of Ras effector utilization in oncogenesis. Methods Enzymol. 2006;407:195-217 [DOI] [PubMed] [Google Scholar]
- 144. White MA, Vale T, Camonis JH, Schaefer E, Wigler MH. A role for the Ral guanine nucleotide dissociation stimulator in mediating Ras-induced transformation. J Biol Chem. 1996;271:16439-42 [DOI] [PubMed] [Google Scholar]
- 145. Urano T, Emkey R, Feig LA. Ral-GTPases mediate a distinct downstream signaling pathway from Ras that facilitates cellular transformation. EMBO J. 1996;15:810-6 [PMC free article] [PubMed] [Google Scholar]
- 146. Hamad NM, Elconin JH, Karnoub AE, et al. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev. 2002;16:2045-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Chien Y, White MA. RAL GTPases are linchpin modulators of human tumour-cell proliferation and survival. EMBO Rep. 2003;4:800-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Lim KH, Baines AT, Fiordalisi JJ, et al. Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell. 2005;7:533-45 [DOI] [PubMed] [Google Scholar]
- 149. Zipfel PA, Brady DC, Kashatus DF, Ancrile BD, Tyler DS, Counter CM. Ral activation promotes melanomagenesis. Oncogene. 2010;29:4859-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Mishra PJ, Ha L, Rieker J, et al. Dissection of RAS downstream pathways in melanomagenesis: a role for Ral in transformation. Oncogene. 2010;29:2449-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Omidvar N, Pearn L, Burnett AK, Darley RL. Ral is both necessary and sufficient for the inhibition of myeloid differentiation mediated by Ras. Mol Cell Biol. 2006;26:3966-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Gonzalez-Garcia A, Pritchard CA, Paterson HF, Mavria G, Stamp G, Marshall CJ. RalGDS is required for tumor formation in a model of skin carcinogenesis. Cancer Cell. 2005;7:219-26 [DOI] [PubMed] [Google Scholar]
- 153. Song C, Hu CD, Masago M, et al. Regulation of a novel human phospholipase C, PLCepsilon, through membrane targeting by Ras. J Biol Chem. 2001;276:2752-7 [DOI] [PubMed] [Google Scholar]
- 154. Shibatohge M, Kariya K, Liao Y, et al. Identification of PLC210, a Caenorhabditis elegans phospholipase C, as a putative effector of Ras. J Biol Chem. 1998;273:6218-22 [DOI] [PubMed] [Google Scholar]
- 155. Bunney TD, Katan M. Phospholipase C epsilon: linking second messengers and small GTPases. Trends Cell Biol. 2006;16:640-8 [DOI] [PubMed] [Google Scholar]
- 156. Bai Y, Edamatsu H, Maeda S, et al. Crucial role of phospholipase Cepsilon in chemical carcinogen-induced skin tumor development. Cancer Res. 2004;64:8808-10 [DOI] [PubMed] [Google Scholar]
- 157. Malliri A, van der Kammen RA, Clark K, van der Valk M, Michiels F, Collard JG. Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature. 2002;417:867-71 [DOI] [PubMed] [Google Scholar]
- 158. Mertens AE, Roovers RC, Collard JG. Regulation of Tiam1-Rac signalling. FEBS Lett. 2003;546:11-6 [DOI] [PubMed] [Google Scholar]
- 159. Lambert JM, Lambert QT, Reuther GW, et al. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat Cell Biol. 2002;4:621-5 [DOI] [PubMed] [Google Scholar]
- 160. Strumane K, Rygiel TP, Collard JG. The Rac activator Tiam1 and Ras-induced oncogenesis. Methods Enzymol. 2006;407:269-81 [DOI] [PubMed] [Google Scholar]
- 161. Qiu RG, Chen J, Kirn D, McCormick F, Symons M. An essential role for Rac in Ras transformation. Nature. 1995;374:457-9 [DOI] [PubMed] [Google Scholar]
- 162. Malliri A, Collard JG. Role of Rho-family proteins in cell adhesion and cancer. Curr Opin Cell Biol. 2003;15:583-9 [DOI] [PubMed] [Google Scholar]
- 163. Wang Z, Pedersen E, Basse A, et al. Rac1 is crucial for Ras-dependent skin tumor formation by controlling Pak1-Mek-Erk hyperactivation and hyperproliferation in vivo. Oncogene. 2010;29:3362-73 [DOI] [PubMed] [Google Scholar]
- 164. Kissil JL, Walmsley MJ, Hanlon L, et al. Requirement for Rac1 in a K-ras induced lung cancer in the mouse. Cancer Res. 2007;67:8089-94 [DOI] [PubMed] [Google Scholar]
- 165. Reddy HK, Mettus RV, Rane SG, Grana X, Litvin J, Reddy EP. Cyclin-dependent kinase 4 expression is essential for neu-induced breast tumorigenesis. Cancer Res. 2005;65:10174-8 [DOI] [PubMed] [Google Scholar]
- 166. Yu Q, Sicinska E, Geng Y, et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell. 2006;9:23-32 [DOI] [PubMed] [Google Scholar]
- 167. Puyol M, Martin A, Dubus P, et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010;18:63-73 [DOI] [PubMed] [Google Scholar]
- 168. Reddy HK, Grana X, Dhanasekaran DN, Litvin J, Reddy EP. Requirement of Cdk4 for v-H-ras induced breast tumorigenesis and activation of the v-ras-induced senescence program by the R24C mutation. Genes Cancer. 2010;1:69-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Lazarov M, Kubo Y, Cai T, et al. CDK4 coexpression with Ras generates malignant human epidermal tumorigenesis. Nat Med. 2002;8:1105-14 [DOI] [PubMed] [Google Scholar]
- 170. Chawla R, Procknow JA, Tantravahi RV, Khurana JS, Litvin J, Reddy EP. Cooperativity of Cdk4R24C and Ras in melanoma development. Cell Cycle. 2010;9:3305-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Basseres DS, Ebbs A, Levantini E, Baldwin AS. Requirement of the NF-kappaB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res. 2010;70:3537-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Meylan E, Dooley AL, Feldser DM, et al. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature. 2009;462:104-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Filmus J, Robles AI, Shi W, Wong MJ, Colombo LL, Conti CJ. Induction of cyclin D1 overexpression by activated ras. Oncogene. 1994;9:3627-33 [PubMed] [Google Scholar]
- 174. Robles AI, Rodriguez-Puebla ML, Glick AB, et al. Reduced skin tumor development in cyclin D1-deficient mice highlights the oncogenic ras pathway in vivo. Genes Dev. 1998;12:2469-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Soucek L, Whitfield J, Martins CP, et al. Modelling Myc inhibition as a cancer therapy. Nature. 2008;455:679-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Scholl C, Frohling S, Dunn IF, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137:821-34 [DOI] [PubMed] [Google Scholar]
- 177. Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Luo J, Emanuele MJ, Li D, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Vicent S, Chen R, Sayles LC, et al. Wilms tumor 1 (WT1) regulates KRAS-driven oncogenesis and senescence in mouse and human models. J Clin Invest. 2010;120:3940-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Wang Y, Ngo VN, Marani M, et al. Critical role for transcriptional repressor Snail2 in transformation by oncogenic RAS in colorectal carcinoma cells. Oncogene. 2010;29:4658-70 [DOI] [PMC free article] [PubMed] [Google Scholar]