

Abstract
SHP2, encoded by PTPN11, is a non-receptor protein tyrosine phosphatase (PTP) containing two tandem Src homology-2 (SH2) domains. It is expressed ubiquitously and plays critical roles in growth factor mediated processes, primarily by promoting the activation of the RAS/ERK signaling pathway. Genetic and biochemical studies have identified SHP2 as the first bona fide oncoprotein in the PTP superfamily, and a promising target for anti-cancer and anti-leukemia therapy. Here, we report a structure-based approach to identify SHP2 inhibitors with a novel scaffold. Through sequential virtual screenings and in vitro inhibition assays, a reversible competitive SHP2 inhibitor (C21) was identified. C21 is structurally distinct from all known SHP2 inhibitors. Combining molecular dynamics simulation and binding free energy calculation, a most likely binding mode of C21 with SHP2 is proposed, and further validated by site-directed mutagenesis and structure-activity relationship studies. This binding mode is consistent with the observed potency and specificity of C21, and reveals the molecular determinants for further optimization based on the new scaffold.
Keywords: SHP2 inhibitor, Protein tyrosine phosphatase (PTP), Virtual screening, Molecular dynamics simulation, Drug discovery
Protein tyrosine phosphatases (PTPs) play crucial roles in a variety of cellular processes, working in concert with protein tyrosine kinases (PTKs) to control the tyrosine phosphorylation level and regulate key signaling cascades.1, 2 Aberrant tyrosine phosphorylation has been linked to the aetiology of several human diseases (such as cancer, diabetes, obesity, and autoimmune disorders), which makes PTKs and PTPs important therapeutic targets.3–5 Although the PTPs are generally recognized as negative regulators of signaling cascades, increasing evidence suggest that some PTPs, such as SHP2, PTP1B, CDC25 and PRL3, can also potentiate signaling cascades and have oncogenic potentials.5–8 Therefore these PTPs represent promising targets for anti-cancer therapy, and interest in developing specific PTP inhibitors has intensified in recent years.8–11
SHP2, encoded by PTPN11, is a non-receptor PTP containing two tandem Src homology-2 (SH2) domains, one catalytic domain and a C-terminal tail. SHP2 is a positive transducer of growth factor, cytokine, integrin, and hormone signaling pathways regulating a diverse array of processes such as cell proliferation, differentiation, adhesion, migration and apoptosis, due primarily to its ability to promote the activation of the RAS/ERK signaling pathway.12–15 Germline mutations in SHP2 causing hyperactivation of its phosphatase activity (namely gain-of-function mutations) are associated with Noonan syndrome (~50%).16–18 Somatic gain-of-function SHP2 mutations are linked to juvenile myelomonocytic leukemia (JMML, ~34%), myelodysplastic syndrome (~10%), acute lymphoid leukemia (ALL, ~7%), acute myeloid leukemia (AML, ~4%), adult AML (~6%), as well as solid tumors, such as lung adenocarcinoma, colon cancer, neuroblastoma, melanoma and hepatocellular carcinoma.18–24 In addition, SHP2 is implicated in gastric ulcer and gastric carcinoma.25 These genetic and biochemical evidence identify SHP2 as the first bona fide oncoprotein in the PTP superfamily, which makes it a prime target for anti-leukemia and anti-cancer therapy and draws increasing interests in the development of SHP2 inhibitors.26–35 To date, all reported SHP2 inhibitors (Fig. S1, Supplementary Material) display similar micromolar-to-submicromolar in vitro inhibitory activity and most of them lack the selectivity required for mechanistic studies, possibly due to the highly conserved active site pocket shared by all PTPs.36 Consequently, there is still a need to discover new SHP2 inhibitors with novel scaffold, increased potency, selectivity and in vivo activity.
In an effort to discover novel SHP2 inhibitors, we carried out high-throughput virtual screenings on two subsets of ZINC37 database targeting the active pocket of SHP2 catalytic domain (PDBID: 3B7O38). The details of site definitions, structure preparations and screening procedures were described in the Supplementary Material. After the initial screening with DOCK6.239,40 and the secondary screening with AutoDock4.01,41,42 the top-ranked 1,621 molecules with energy score less than −8.5 kcal/mol were selected for further analyses, such as consensus score evaluation, similarity analysis and visual inspection of binding mode. Finally, 35 compounds were purchased and their inhibitory activity against SHP2 was assessed at 50 µM concentration. 9 out of the 35 compounds showed more than 50% inhibition, and another 7 showed inhibition ranging from 30% to 50% (Table S1, Supplementary Material). These 16 compounds were further assayed for IC50 values, and three of them (namely C18, C21 and C30) showed concentration-dependent inhibition, their structures and IC50 are listed in Table 1. Moreover, the IC50 of these three compounds against SHP1 and PTP1B were also determined. C30 and C18 had only moderate inhibitory activity against SHP2 but better inhibition against PTP1B and SHP1 (Table S2, Supplementary Material). Thus they were not pursued further.
Table 1.
Structures and IC50 values of C18, C21, C30, and four analogues of C21 against SHP2.
| Compd. | Structure | IC50 (µM) |
|---|---|---|
| C18 | 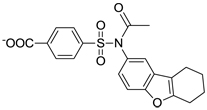 |
181 ± 12 |
| C30 | 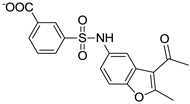 |
176 ± 11 |
| C21 |  |
8.3 ± 0.1 |
| C21-A1 | 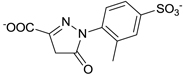 |
41 ± 1 |
| C21-A2 | 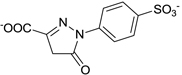 |
155 ± 6 |
| C21-A3 | 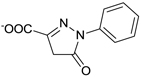 |
NDa |
| C21-A4 | 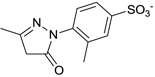 |
NDa |
No detectable inhibition at 1,000 µM
Compound C21, ranked the 20th in the initial screening and the 1st in the secondary screening, inhibited SHP2 with an IC50 of 8.3 µM. C21 is structurally distinct from all known SHP2 inhibitors. Like most known SHP2 inhibitor, C21 did not exhibit significant selectivity for SHP2 over its close homolog SHP1 (IC50 = 14 µM, only 1.7-fold), but it showed considerable selectivity against PTP1B (IC50 = 118 µM, 14.2-fold). To explore the selectivity of C21 more broadly, an additional panel of nine representative PTPs, including cytosolic PTPs (TC-PTP and LYP), the receptor-like PTPs (CD45, LAR and PTPα), the dual specificity phosphatases (VHR, VHX and Cdc14) and the low molecular weight PTP (LMW-PTP) were employed for inhibition assay. C21 showed an IC50 greater than 100 µM against TC-PTP, CD45, LAR, VHX and LMW-PTP, and almost no inhibition at 100 µM against LYP, PTPα, VHR and Cdc14, indicating excellent selectivity of C21 for SHP2 over the panel of PTPs.
Moreover, the mode of inhibition by C21 against SHP2 was also determined through steady-state enzyme kinetic analysis. The results suggest a reversible and competitive pattern for SHP2 inhibition by C21 with an inhibition constant (Ki) of 4.6 µM (Fig. 1). Since the virtual screening was carried out aiming at the catalytic pocket, it was quite reasonable that the identified C21 occupies the active site and acts as a competitive inhibitor. Collectively, the biochemical assays confirmed C21 as a novel SHP2 inhibitor with micromolar potency.
Figure 1.

Kinetic analysis of C21 mediated SHP2 inhibition showed a competitive inhibition mode. The concentrations of C21 were 0 (●), 4 (○), 8 (▼) and 12 µM (▽) respectively.
To define the binding mode between C21 and SHP2, we performed molecular docking with AutoDock4.01. After multiple docking runs and cluster analysis of binding conformations, two possible binding modes with comparable calculated binding free energy (BFE) were found. In mode I (Fig. 2a, BFE = −11.08 kcal/mol), the pyrazoline carboxyl moiety in C21 penetrates into the active site pocket and interacts with residues in the phosphate-binding loop (P-loop); the two sulfonic groups are located at the entrance of the active site and interact with two basic residues K364 and K366. In mode II (Fig. 2b, BFE = −10.22 kcal/mol), C21 binds almost at the same position but with completely opposite orientation: the benzene ring with two sulfonic groups wedge into the active site pocket and strongly interact with the P-loop residues, while the carboxyl on the pyrazoline interacts with K364 and K366 at the entrance of the active site. Considering the inherent standard errors associated with the calculated binding energy in AutoDock4.01, it’s difficult to distinguish which mode is more preferable based purely on the calculated binding energies.
Figure 2.

Binding mode I a) and mode II b) revealed by molecular docking. c) Interaction details of C21 and SHP2 in binding mode II. C21 (green carbon) and residues within 5 Å distance of C21 (cyan carbon) are presented in stick; H-bond interactions are highlighted in yellow dash line.
Molecular dynamics (MD) simulation combined with MM-PBSA/GBSA (Molecular Mechanics - Poisson-Boltzmann Surface Area/Generalized Born Surface Area) has proved to be a reliable method for investigating protein-ligand interaction and discriminating different binding modes.43,44 Therefore 10-ns MD simulations were performed on the complex structures of binding mode I and II respectively. The Root-Mean-Square deviation (RMSD) of backbone atoms of the protein and heavy atoms of C21 were calculated with respect to the initial coordinates (Fig. 3a). The backbone atoms in both binding modes showed almost identical and stable RMSD of ~0.9 Å, indicating that the protein had no obvious conformation changes during the MD simulations. In contrast, the RMSD of C21 in mode II quickly reached the plateau (2.1 ± 0.3 Å), while that in mode I achieved the higher plateau (3.5 ± 0.5 Å) after 0.5 ns. Both RMSDs indicated that shifts, but the lower plateau in mode II suggested that the initial conformation and position of C21 were better kept, supporting the notion that mode II was more stable and preferable than I.
Figure 3.

a) The RMSD of backbone atoms of SHP2 protein and heavy atoms of C21. b) The top sixteen pairwise residue interaction energies in mode II.
Binding free energies from MM-GBSA calculation were used to further discriminate the two binding modes from the energy perspective. Based on 1,800 snapshots extracted from the MD trajectories between 1 and 10 ns at a time interval of 5 ps, the total binding free energy and individual components were calculated (Table 2). The significantly more negative total binding free energy (ΔGMM-GBSA, −63.60 vs. −36.54) in mode II provided further evidence that mode II was more preferable. In detail, the preferred binding came from a remarkable favorable electrostatic interactions (−693.69 vs. −669.36) and a slightly favorable van der Waals interactions (−26.67 vs. −23.26), while the polar and non-polar components of solvation free energy were almost identical in both binding modes.
Table 2.
The calculated binding free energies and individual energy components (kcal/mol) for binding mode I and II.
| Mode | ΔEVDWa | ΔEELEb | ΔEPc | ΔGNPd | ΔGMM-GBSAe |
|---|---|---|---|---|---|
| I | −23.26±0.08 | −669.36±1.01 | 660.59±0.93 | −4.51±0.01 | −36.54±0.13 |
| II | −26.67±0.09 | −693.69±0.91 | 660.87±0.84 | −4.12±0.01 | −63.60±0.15 |
Van der Waals interaction energy in gas phase.
Electrostatic interaction energy in gas phase.
Polar component in solvation free energy.
Non-polar component in solvation free energy.
Total binding free energy.
To get a better understanding of the energy profile, the total binding free energy was decomposed into pairwise residue interaction. Sixteen residues with interaction energy less than - C21 underwent remarkable conformation changes and/or position 0.50 kcal/mol in mode II were shown in Fig. 3b. It was clear that residues in the P-loop provided the most contribution to the binding, with R465 contributing ~20% and other P-loop residues (H458 to G464) contributing ~57%. Two residues K366 and K364, located at the entrance of active site pocket, contributed another ~20%. As shown in Fig. 2c, the two SO3− groups interacted with the P-loop residues through seven H-bonds, three of them from the first SO3− with ε-N and η-N of R465 and the backbone N of S460, and the other four from the second SO3− with the backbone N of A461, G464, R465 and γ-O of S460. At the same time, all P-loop residues as well as Q506, Q510 and Y279 were within 5 Å distance of C21, resulting in abundant van der Waals interactions. These H-bond and van der Waals interactions tied the benzene moiety at the active site and contribute to ~80% total binding free energy. On the other side, the carboxyl group made H-bonds with the ζ-N of K366 and K364, as well as the long range electrostatic interactions with R362; the pyrazoline moiety also made van der Waals interactions with these three residues. Thus the interactions from pyrazoline moiety further enhanced the binding affinity of C21 with SHP2.
Mapping the residues interacting with C21 onto the amino acid sequence of SHP2 and making alignment with SHP1 and PTP1B (Fig. 4), we found that almost all contact residues were conserved among these three enzymes except for R362 and K364, suggesting that the region around these two residues might contribute to the selectivity of C21 over PTP1B and SHP1. Since K364 is unique to SHP2 (Fig. 4) and contributes more binding free energy than R362 (Fig. 3b), we studied two SHP2 mutants bearing substitutions at residue K364, which corresponds to an Arg in SHP1 and a Ser in PTP1B. C21 was 3-fold less active against SHP2/K364R (IC50 = 26 µM) and 7-fold less active against SHP2/K364S (IC50 = 56 µM) as compared to the wild type SHP2. In fact, the IC50 values for SHP2/K364R and SHP2/K364S approach those observed for SHP1 and PTP1B. These results are consistent with the predictions from molecular modeling indicating that K364 is indeed involved in binding C21 and contributes to the selectivity of SHP2 for compound C21. Although the rest of the contact residues are the same for SHP2, SHP1, and PTP1B (Fig. 4), their immediate neighboring residues and residues that form the second shell of the C21 binding pocket are different (the sequence identities among the three PTPs are 40–50%). These structural differences will result in non-identical C21 binding pocket and likely also contribute to the specificity of C21 for SHP2. Further structural study of the complexes of C21 with SHP2 and/or PTP1B/SHP1 will be required to fully understand the determinants for C21 recognition.
Figure 4.

Sequence alignment of four segments of SHP2 interacting with C21. Residue numbers in SHP2 and PTP1B are labeled at the top and bottom, respectively. Residues within 5 Å of C21 in binding mode II are marked by *.
Other than identifying the interaction residues in SHP2 from docking and mutagenesis studies, we also sought to identify the key interaction points in C21 through classical structure-activity relationship (SAR) investigation so as to guide future optimizations. Four analogues of C21 were purchased and evaluated for inhibitory activity against SHP2 (Table 1). Compound C21-A1, lacking the SO3− group at position 1 (namely 1-SO3−), showed ~5-fold decreased inhibition (IC50 = 41 µM). By removing the 4-CH3 from C21-A1, the affinity of compound C21-A2 for SHP2 further decreased ~4-fold (IC50 = 155 µM). When the 2-SO3− in C21-A2 was removed or the 4’-COO− in C21-A1 was replaced with methyl, the resulting C21-A3 or C21-A4 failed to inhibit SHP2 at 1 mM compound concentration. These SAR results indicated that: (1) The simultaneous presence of the 2-SO3− and 4’-COO− at both sides is essential for SHP2 inhibition, and the lack of either negatively-charged center will abolish the binding affinity. (2) Both the 1-SO3− and 4-CH3 groups contribute to SHP2 binding, and together they enhance the inhibitory activity about 20-fold (C21 vs. C21-A2). These findings also agree well with the proposed binding mode II (Fig. 2b and 2c): the two negatively-charged centers on the two rings in C21 simultaneously interact with the two positively-charged sites in SHP2 (the active site and a peripheral site defined by residues K364 and K366) through six H-bonds (four from 2-SO3− and two from 4’-COO−), which precisely position C21 at the active pocket. Then the 1-SO3− (additional 3 H-bonds with the P-loop) and 4-CH3 (hydrophobic interaction with Y279) further enhance the binding affinity and increase the inhibition potency.
In summary, we identified a novel SHP2 inhibitor (C21) with micromolar inhibition potency (Ki = 4.6 µM) and good selectivity against a panel of mammalian PTPs. Through molecular docking, MD simulation and MM-GBSA binding free energy calculation, a most likely binding mode was proposed and subsequently validated from both the receptor (mutagenesis study) and ligand (SAR study) perspectives. Our study provided a novel scaffold upon which more potent and selective SHP2 inhibitors could be developed through structural modifications, such as extending the 4-CH3 to hydrophobic chains for more interactions with the pTyr-loop; substituting the 2’-carbonyl with bulky and hydrophobic groups to complement the free space around the WPD-loop; replacing the sulfonic groups with trifluoromethyl or trifluoromethyl sulfonyl to improve the cell permeability without total loss of electronegativity at this site. The current structure-based drug discovery approach, involving multiple computational techniques, classical inhibition analysis, site-directed mutagenesis and SAR study, should also be applicable to the identification of small molecule inhibitors for other PTPs.
Supplementary Material
Acknowledgments
The virtual screenings, MD simulations and MM-GBSA calculations were carried out on the BigRed supercomputer in Indiana University. This work was supported in part by National Institutes of Health Grants CA126937 and CA152194.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Supplementary materials associated with this article can be found in the online version.
References and notes
- 1.Hunter T. Philos. Trans. R. Soc. Lond. Ser. B-Biol. Sci. 1998;353:583. doi: 10.1098/rstb.1998.0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tonks NK, Neel BG. Curr. Opin. Cell Biol. 2001;13:182. doi: 10.1016/s0955-0674(00)00196-4. [DOI] [PubMed] [Google Scholar]
- 3.Zhang ZY. Curr. Opin. Chem. Biol. 2001;5:416. doi: 10.1016/s1367-5931(00)00223-4. [DOI] [PubMed] [Google Scholar]
- 4.Tonks NK. Nat. Rev. Mol. Cell Biol. 2006;7:833. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 5.Ventura JJ, Nebreda AR. Clin Transl Oncol. 2006;8:153. doi: 10.1007/s12094-006-0005-0. [DOI] [PubMed] [Google Scholar]
- 6.Ostman A, Hellberg C, Bohmer FD. Nat. Rev. Cancer. 2006;6:307. doi: 10.1038/nrc1837. [DOI] [PubMed] [Google Scholar]
- 7.Tautz L, Pellecchia M, Mustelin T. Expert Opin. Ther. Targets. 2006;10:157. doi: 10.1517/14728222.10.1.157. [DOI] [PubMed] [Google Scholar]
- 8.Scott LM, Lawrence HR, Sebti SM, Lawrence NJ, Wu J. Curr. Pharm. Design. 2010;16:1843. doi: 10.2174/138161210791209027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang ZX, Zhang ZY. Cancer Metastasis Rev. 2008;27:263. doi: 10.1007/s10555-008-9113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blaskovich MAT. Curr. Med. Chem. 2009;16:2095. doi: 10.2174/092986709788612693. [DOI] [PubMed] [Google Scholar]
- 11.Vintonyak VV, Antonchick AP, Rauh D, Waldmann H. Curr. Opin. Chem. Biol. 2009;13:272. doi: 10.1016/j.cbpa.2009.03.021. [DOI] [PubMed] [Google Scholar]
- 12.Deb TB, Wong L, Salomon DS, Zhou GC, Dixon JE, Gutkind JS, Thompson SA, Johnson GR. J. Biol. Chem. 1998;273:16643. doi: 10.1074/jbc.273.27.16643. [DOI] [PubMed] [Google Scholar]
- 13.Qu CK. Biochim. Biophys. Acta-Mol. Cell Res. 2002;1592:297. doi: 10.1016/s0167-4889(02)00322-1. [DOI] [PubMed] [Google Scholar]
- 14.Neel BG, Gu HH, Pao L. Trends Biochem.Sci. 2003;28:284. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 15.Dance M, Montagner A, Salles JP, Yart A, Raynal P. Cell. Signal. 2008;20:453. doi: 10.1016/j.cellsig.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, Ion A, Jeffery S, Kalidas K, Patton MA, Kucherlapati RS, Gelb BD. Nature Genet. 2001;29:465. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 17.Zenker M, Buheitel G, Rauch R, Koenig R, Bosse K, Kress W, Tietze HU, Doerr HG, Hofbeck M, Singer H, Reis A, Rauch A. J. Pediatr. 2004;144:368. doi: 10.1016/j.jpeds.2003.11.032. [DOI] [PubMed] [Google Scholar]
- 18.Tartaglia M, Gelb BD. Annu. Rev. Genomics Hum. Genet. 2005;6:45. doi: 10.1146/annurev.genom.6.080604.162305. [DOI] [PubMed] [Google Scholar]
- 19.Tartaglia M, Niemeyer CM, Fragale A, Song XL, Buechner J, Jung A, Hahlen K, Hasle H, Licht JD, Gelb BD. Nature Genet. 2003;34:148. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- 20.Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K, Maris JM, Richardson A, Bardelli A, Sugarbaker DJ, Richards WG, Du JY, Girard L, Minna JD, Loh ML, Fisher DE, Velculescu VE, Vogelstein B, Meyerson M, Sellers WR, Neel BG. Cancer Res. 2004;64:8816. doi: 10.1158/0008-5472.CAN-04-1923. [DOI] [PubMed] [Google Scholar]
- 21.Loh ML, Reynolds MG, Vattikuti S, Gerbing RB, Alonzo TA, Carlson E, Cheng JW, Lee CM, Lange BJ, Meshinchi S. Leukemia. 2004;18:1831. doi: 10.1038/sj.leu.2403492. [DOI] [PubMed] [Google Scholar]
- 22.Loh ML, Vattikuti S, Schubbert S, Reynolds MG, Carlson E, Lieuw KH, Cheng JW, Lee CM, Stokoe D, Bonifas JM, Curtiss NP, Gotlib J, Meshinchi S, Le Beau MM, Emanuel PD, Shannon KM. Blood. 2004;103:2325. doi: 10.1182/blood-2003-09-3287. [DOI] [PubMed] [Google Scholar]
- 23.Chan G, Kalaitzidis D, Neel BG. Cancer Metastasis Rev. 2008;27:179. doi: 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]
- 24.Miyamoto D, Miyamoto M, Takahashi A, Yomogita Y, Higashi H, Kondo S, Hatakeyama M. Oncogene. 2008;27:3508. doi: 10.1038/sj.onc.1211019. [DOI] [PubMed] [Google Scholar]
- 25.Hatakeyama M. Nat. Rev. Cancer. 2004;4:688. doi: 10.1038/nrc1433. [DOI] [PubMed] [Google Scholar]
- 26.Chen LW, Sung SS, Yip MLR, Lawrence HR, Ren Y, Guida WC, Sebti SM, Lawrence NJ, Wu J. Mol. Pharmacol. 2006;70:562. doi: 10.1124/mol.106.025536. [DOI] [PubMed] [Google Scholar]
- 27.Noren-Muller A, Reis-Correa I, Prinz H, Rosenbaum C, Saxena K, Schwalbe HJ, Vestweber D, Cagna G, Schunk S, Schwarz O, Schiewe H, Waldmann H. Proc. Natl. Acad. Sci. U. S. A. 2006;103:10606. doi: 10.1073/pnas.0601490103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geronikaki A, Eleftheriou P, Vicini P, Alam I, Dixit A, Saxena AK. J. Med. Chem. 2008;51:5221. doi: 10.1021/jm8004306. [DOI] [PubMed] [Google Scholar]
- 29.Hellmuth K, Grosskopf S, Lum CT, Wurtele M, Roder N, von Kries JP, Rosario M, Rademann J, Birchmeier W. Proc. Natl. Acad. Sci. U. S. A. 2008;105:7275. doi: 10.1073/pnas.0710468105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lawrence HR, Pireddu R, Chen LW, Luo YT, Sung SS, Szymanski AM, Yip MLR, Guida WC, Sebti SM, Wu J, Lawrence NJ. J. Med. Chem. 2008;51:4948. doi: 10.1021/jm8002526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu WM, Guvench O, MacKerell AD, Qu CK. J. Med. Chem. 2008;51:7396. doi: 10.1021/jm800229d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu D, Pang Y, Ke Y, Yu J, He Z, Tautz L, Mustelin T, Ding S, Huang Z, Feng GS. PLoS One. 2009;4:e4914. doi: 10.1371/journal.pone.0004914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen LW, Pernazza D, Scott LM, Lawrence HR, Ren YA, Luo YT, Wu X, Sung SS, Guida WC, Sebti SM, Lawrence NJ, Wu J. Biochem. Pharmacol. 2010;80:801. doi: 10.1016/j.bcp.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scott LM, Chen L, Daniel KG, Brooks WH, Guida WC, Lawrence HR, Sebti SM, Lawrence NJ, Wu J. Bioorg Med Chem Lett. 2010;21:730. doi: 10.1016/j.bmcl.2010.11.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang X, He YT, Liu SJ, Yu ZH, Jiang ZX, Yang ZY, Dong YS, Nabinger SC, Wu L, Gunawan AM, Wang LN, Chan RJ, Zhang ZY. J. Med. Chem. 2010;53:2482. doi: 10.1021/jm901645u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang S, Zhang ZY. Drug Discov. Today. 2007;12:373. doi: 10.1016/j.drudis.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 37.Irwin JJ, Shoichet BK. J. Chem Inf. Model. 2005;45:177. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barr AJ, Ugochukwu E, Lee WH, King ONF, Filippakopoulos P, Alfano I, Savitsky P, Burgess-Brown NA, Muller S, Knapp S. Cell. 2009;136:352. doi: 10.1016/j.cell.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moustakas DT, Lang PT, Pegg S, Pettersen E, Kuntz ID, Brooijmans N, Rizzo RC. J. Comput.-Aided Mol. Des. 2006;20:601. doi: 10.1007/s10822-006-9060-4. [DOI] [PubMed] [Google Scholar]
- 40.Lang PT, Brozell SR, Mukherjee S, Pettersen EF, Meng EC, Thomas V, Rizzo RC, Case DA, James TL, Kuntz ID. RNA-Publ. RNA Soc. 2009;15:1219. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. J. Comput. Chem. 1998;19:1639. [Google Scholar]
- 42.Huey R, Morris GM, Olson AJ, Goodsell DS. J. Comput. Chem. 2007;28:1145. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 43.Kollman PA, Massova I, Reyes C, Kuhn B, Huo SH, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE. Accounts Chem. Res. 2000;33:889. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 44.Wang JM, Morin P, Wang W, Kollman PA. J. Am. Chem. Soc. 2001;123:5221. doi: 10.1021/ja003834q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.