

Abstract
Variances, particularly single nucleotide polymorphisms (SNP), in the genomic sequence of individuals are the primary key to understanding gene function as it relates to differences in the susceptibility to disease, environmental influences, and therapy. In this report, the HSP70B′ gene is the target sequence for mutation detection in biopsy samples from human prostate cancer patients undergoing combined hyperthermia and radiation therapy at the Dana-Farber Cancer Institute, using temperature-modulated heteroduplex analysis (TMHA). The underlying principles of TMHA for mutation detection using DHPLC technology are discussed. The procedures involved in amplicon design for mutation analysis by DHPLC are detailed. The melting behavior of the complete coding sequence of the target gene is characterized using WAVEMAKERTM software. Four overlapping amplicons, which span the complete coding region of the HSP70B′ gene, amenable to mutation detection by DHPLC were identified based on the software-predicted melting profile of the target sequence. TMHA was performed on PCR products of individual amplicons of the HSP70B′ gene on the WAVE® Nucleic Acid Fragment Analysis System. The criteria for mutation calling by comparing wild-type and mutant chromatographic patterns are discussed.
INTRODUCTION
Individual human genomes exhibit differences in their DNA sequence. Variations in the genomic sequence between individuals occur at an estimated frequency of at least 1 base per thousand bases. This totals more than 3 million variances per person. Typically, these variances are present in the form of single nucleotide polymorphisms (SNP). The relationship between variability in the sequence of specific genes among individuals to their disease status or susceptibility to disease is a primary key to understanding gene function. Variations, in addition to being critical for identification of disease genes, are also essential in understanding genetic differences in individual responses to the environment, to disease, and to therapies. The challenge of understanding even a fraction of the variations in the genomic sequence, determining which polymorphisms are significant and which are inconsequential, is overwhelming. However, the potential value of such knowledge is great in that it will lead to better diagnostic and prognostic assays and to improved treatment for disease.
A highly sensitive, reproducible, and robust technology for detection of sequence variations is pivotal to exploiting the relationship between sequence variability and function. A recently developed denaturing high-performance liquid chromatography (DHPLC) technology (Oefner and Underhill 1995, 1998; Kuklin et al 1997/98) for the analysis of DNA fragments/PCR products meets the requirements. The technology, which is based on heteroduplex detection, allows for automated identification of SNP and small deletions or insertions. Heteroduplex profiles are easily distinguished from homoduplex peaks (Underhill et al 1997; Kuklin et al 1997/98) and thereby provide a reliable means for mutation scanning and discovery. DHPLC has been shown to clearly resolve mutations in various genes with detection rates ranging from 92.5% to 100% (Liu et al 1998; O'Donovan et al 1998; Choy et al 1999; Jones et al 1999). Furthermore, it is reported in the literature that the sensitivity of detection by DHPLC is higher than for alternative gel-based analysis techniques; that is, mutations not detected by gel-based techniques could be detected by DHPLC (Choy et al 1999; Gross et al 1999; Wagner et al 1999).
In this report, the underlying principles of mutation detection by temperature modulated heteroduplex analysis (TMHA) using DHPLC are discussed. This discussion is followed by a detailed, step-by-step analysis of the procedures involved in mutation detection using the HSP70B’ gene sequence as a specific example. We have chosen to examine HSP70B, as this is the most strongly inducible member of the human HSP70 family. HSP70B structure in control cDNA samples is compared with HSP70B in genomic DNA extracted from the prostate tissue of prostate cancer patients undergoing combined hyperthermia and radiation therapy at the Dana-Farber Cancer Institute. Mutations were detected in HSP70B obtained from 2 out of 3 human tumors.
MATERIALS AND METHODS
Tissue and isolation of genomic DNA
Tissue biopsies of prostate cancer patients undergoing combined hyperthermia and radiation therapy at the Dana-Farber Cancer Institute were immediately frozen in liquid nitrogen. Genomic DNA was extracted using Trizol isolation kit (Life Technologies, Grand Island, NY, USA) according to the manufacturer's instructions.
Melting profiles and analysis temperature predictions
WAVEMAKERTM software (Transgenomic) was used for the prediction of melting characteristics and profiles for all sequences analyzed. Analysis temperatures for mutation detection were derived from software-predicted melting profiles of percent helical fraction vs base position. A temperature at which the target fragment, the PCR product being scanned for mutations, is 70% to 85% helical is regarded as the optimal temperature for mutation detection.
Primer selection and PCR conditions
Primers flanking target regions for mutation detection were designed using conventional software packages, such as OLIGO® (Molecular Biology Insights Inc., Cascade, CO, USA) or Primer 3 (http://www-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi) using standard selection criteria. PCR amplification was performed with either AmpliTaq® or AmpliTaq GoldTM (PE—Roche Molecular Systems, Branchburg, NJ, USA) DNA polymerase as recommended by the manufacturer.
HPLC instrumentation and analysis conditions
All HPLC analyses were performed on the WAVE® Nucleic Acid Fragment Analysis System equipped with a DNASep® cartridge (Transgenomic). Chromatography was performed with buffers A, aqueous solution of 0.1 M triethylammonium acetate (TEAA), and B, 0.1 M TEAA in 25% acetonitrile (ACN). ACN (Transgenomic), TEAA (Transgenomic), and water used for buffer preparation were of HPLC grade. Volumetric flasks were used for the preparation of buffers because of the sensitivity of retention times to variations in ACN concentrations. Eluent conditions for analysis were determined using the WAVEMAKERTM software (Transgenomic). The eluent flow rate for all analyses was 0.9 mL/min. Chromatograms were recorded at a wavelength of 260 nm. Analyses under nondenaturing conditions were conducted at 50°C. Mutation detection under partially denaturing conditions was performed at the temperature indicated for each chromatogram.
RESULTS AND DISCUSSION
Screening for mutations by DHPLC involves the analysis of homo- and heteroduplex structures formed between wild-type and mutant sequences (Fig 1) by ion-pair reversed-phase high-performance liquid chromatography (IP RP HPLC) under partially denaturing conditions (Underhill et al 1996; Kuklin et al 1997/98; Liu et al 1998). Hereafter we refer to this technology, which achieves separation of DNA fragments based on fragment size and degree of denaturation, as DNA chromatography.
Fig 1.

Schematic of heteroduplex formation for mutation analysis. The PCR products of wild-type and mutant allele, differing by as little as a single base pair, are denatured by heating and reannealed by slow cooling. The resultant wild-type and mutant homoduplexes melt at higher temperatures than the mismatch containing wild-type/mutant heteroduplexes. The difference in melting temperature between homo- and heteroduplexes is the basis for the identification of mutations by DNA chromatography
A primer pair is designed to flank the DNA sequence of interest. PCR products ranging in size from approximately 150 to 1000 bp may be suitable for analysis. The decisive factors in judging whether a PCR product is suitable for mutation detection are its length, sequence, and melting characteristics, as will be discussed in some detail later. PCR on heterozygous templates generates wild-type and mutant allele products in equimolar amounts. The PCR product generated from homozygous mutant templates requires the addition of wild-type PCR product to permit heteroduplex formation (Kuklin et al 1997/98). Wild-type PCR product is generally added when screening for mutations and for the analysis of unknown samples when the occurrence of homozygous mutants is likely or possible. The latter approach is referred to as mutation detection after post-PCR mixing. As illustrated in Figure 1, a wild-type/mutant mixture is denatured at 95°C. This is followed by slow renaturation, which leads to the formation of wild-type and mutant homoduplexes as well as 2 distinct wild-type/mutant heteroduplexes. The latter are characterized by a mismatch at the site of mutation.
Mutation analysis by DNA chromatography is based on 2 key features. First, heteroduplexes melt at the mismatch site and generate a single-stranded, partially denatured region at a temperature below the melting temperature of the wild-type and mutant homoduplexes (Oefner and Underhill 1995). Second, single-stranded DNA (ss DNA) is retained less strongly on the DNASep® cartridge, which is used for the analytical separation, than double-stranded DNA (ds DNA) (Huber and Berti 1996). Consequently, at a temperature at which wild-type and mutant homoduplexes are partially denatured to result in up to 2 distinct peaks, the wild-type/mutant heteroduplexes are denatured to a greater extent and are less well retained on the column. Thus, the more denatured heteroduplexes yield up to 2 peaks of equal intensity preceding the homoduplex peaks (Fig 2).
Fig 2.

Mutation detection by DNA chromatography with the WAVE® Nucleic Acid Fragment Analysis System using a DNASep® cartridge. Mutation detection was performed on a 209-bp PCR product in the temperature range from 54° to 61°C in 1°C intervals. The mutated sequence analyzed differs from the wild-type sequence by a single A-to-G transition in position 168 of the 209-bp PCR product. An equimolar mixture of wild-type and mutant sequence was denatured and rehybridized prior to analysis
Mutation detection by DHPLC involves a series of considerations, some of which are of general nature and others of which are specific for mutation detection using DHPLC technology. It is understood that all techniques for mutation detection are dependent on the availability of high-grade genomic template DNA suitable for PCR. Optimized primers for PCR are best selected using software packages such as OLIGO (Molecular Biology Insights) or Primer 3. In addition to primer selection, amplicon design plays a crucial role in determining the success or failure of mutation detection. Amplicon design is defined as the process of selecting regions for PCR amplification and subsequent DHPLC analysis that are most amenable to mutation detection. The process of amplicon design and the criteria involved are outlined in the following on the specific example of the HSP70B′ gene. Mutation detection by any technique requires high-quality starting materials. For DHPLC, this means a PCR product of the correct size, good yield, and high purity.
Mutation detection by DNA chromatography does not require any post-PCR sample preparation other than denaturation and reannealing. Aliquots from PCR are injected directly for analysis. Gradient conditions for DHPLC and the temperature chosen for heteroduplex analysis are crucial factors in determining the success of mutation discovery. The WAVEMAKERTM software determines gradient conditions and analysis temperature(s) when using the WAVE® Nucleic Acid Fragment Analysis System for mutation detection. The final step in mutation detection by DNA chromatography involves data interpretation, namely, the comparison of wild-type peak pattern with sample peak pattern for the purpose of identifying the presence or absence of heteroduplex peaks.
The steps involved in amplicon design are outlined in the following paragraph using the genomic sequence of the HSP70B′ gene, GenBank accession number X51757 (Leung et al 1990). Like the major inducible HSP70 gene, HSP70B′ is devoid of introns. The coding sequence of the gene has a length of 1932 bp.
As a first step in amplicon design, the overall melting behavior of the coding region was assessed using WAVEMAKERTM software (Transgenomic). This software predicts the melting behavior of ds DNA fragments of up to 2000 bp. Predicted melting profiles and temperatures are a function of the sequence analyzed as well as the overall length of the fragment. Fragment length plays a role in melting temperature prediction for DHPLC analysis because longer fragments elute at higher acetonitrile (ACN) concentrations. Since ACN has a denaturing effect on ds DNA, it consequently lowers the predicted melting temperature for analysis of longer ds DNA fragments. Consideration of sequence and the fragment length information increases the accuracy of the predicted melting temperature(s) for TMHA as compared to experimentally determined melting temperature(s). At this point, it is important to emphasize that melting profiles and analysis temperatures for TMHA predicted by WAVEMAKERTM software are specific for mutation detection by DNA chromatography under the conditions outlined in the Materials and Methods section.
The melting behavior of the complete coding region of the HSP70B′ gene is characterized in Figure 3, as determined with the WAVEMAKERTM software. The degree of denaturation, which is expressed as the helical fraction of the fragment as a percentage, is displayed in Figure 3A as a function of temperature. The graph reveals that the HSP70B′ coding region denatures over a range of 61° to 67°C. The coding region contains at least 2 melting domains, indicated by the discontinuous profile of the melting curve.
Fig 3.
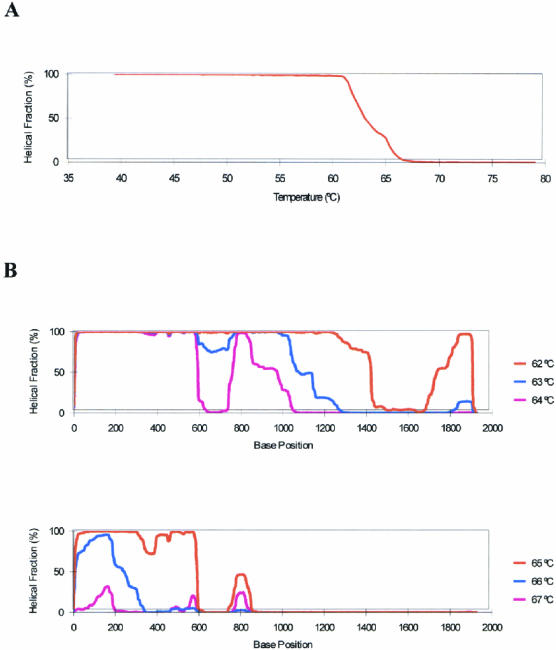
Melting profiles of the complete HSP70B′ coding region. Melting profiles were calculated using the WAVEMAKERTM software. (A) Graph of the degree of naturation (helical fraction of the fragment as a percentage) as a function of temperature. (B) Graph of the degree of naturation as a function of base position at temperatures ranging from 62° to 67°C
A more detailed evaluation of the coding region's melting behavior for the HSP70B′ sequence can be performed if the degree of denaturation at a fixed temperature is plotted as a function of base position. Melting profiles (helical fraction vs base position) for the temperature range from 62° to 67°C are shown in Figure 3B. From these graphs, it is apparent that the sequence contains a 5′-domain, reaching approximately 600 bp into the coding region, which melts at a higher temperature than the remaining part of the gene. A low melting domain is located at the 3′-terminal region of the gene, ranging approximately from base position 1300 to 1800. The central part of the gene melts over a 3°C range with a short, high melting region around position 800.
Amplicon design is guided primarily by the presence of melting domains inherent to the sequence analyzed. A secondary but important consideration is the availability of suitable primer-binding sites for PCR amplification. Melting domains suitable for mutation detection by TMHA are identified first, followed by the design of suitable PCR primers.
Several criteria are applied for the identification of regions suitable for mutation detection by TMHA. Ideally, the target region has a homogeneous melting behavior, which is believed to ensure sensitivity for mutation detection over the whole fragment length at the predicted analysis temperature. Progressive melting of a target region over several degrees is advantageous for mutation detection. A sudden, large drop in the percentage of the fragment's helical fraction is a disadvantage, as it narrows the temperature range in which mutations can be detected. Melting profiles predicted by the WAVEMAKERTM software are those of the homoduplex of the entered sequence. As outlined previously, the mismatch containing heteroduplex will exhibit a lower melting temperature in the region of the mismatch than the corresponding homoduplex, which does not contain a mismatched base pair. The extent of differential melting between the homo- and heteroduplex is determined by the sequence surrounding the mismatch. The latter is directly correlated to resolution of hetero- from homoduplexes by DHPLC because of the differing affinities of the stationary phase of the DNASep® cartridge for ss DNA and ds DNA. SNP and short insertions/deletions are best resolved if mutation detection is performed at the temperature at which the helical fraction for the sequence analyzed ranges from about 70% to 85%. Fragments not exhibiting uniform melting or that contain multiple melting domains need to be analyzed at multiple temperatures corresponding to the melting temperatures of the different domains.
Based on the melting domains identified in Figure 3, overlapping amplicons spanning the coding region of the HSP70B’ gene were designed for mutation detection by DHPLC. The melting profiles of 4 amplicons covering base positions 165 to 720, 628 to 1177, 1092 to 1623, and 1521 to 2187 are shown in Figure 4. (Numbering of base positions is based on the numbering system of the X51757 sequence.) Amplicons 1 to 4 have lengths of 556, 550, 532, and 667 bp, respectively. The primer-binding site for the upstream primer of amplicon 1 is positioned in the gene's 5′-untranslated region; the downstream primer of amplicon 4 is positioned in its 3′-untranslated region. Thus, the complete coding region of the HSP70B′ gene is covered by the 4 amplicons designed. Based on the melting profiles predicted by the WAVEMAKERTM software for the individual amplicons, the optimal analysis temperatures for mutation detection by TMHA for amplicons 1 to 4 are 67°, 64° and 66°, 64°, and 62°/63°C, respectively.
Fig 4.

Melting profiles of HSP70B′ amplicons 1 to 4. Amplicons are designed to overlap and span the complete coding region of the gene. Graphs of the degree of naturation as a function of base position are shown for each of the amplicons. A 3°C temperature range in which mutations are predicted to be detected is shown for each amplicon. Melting profiles were calculated using the WAVEMAKERTM software
Essential for mutation detection, not only by DHPLC, is the availability of high-quality PCR products devoid of spurious amplification products. The quality, purity, and yield of PCR may be checked on agarose gels. However, HPLC analysis under nondenaturing conditions at 50°C is superior for assessing PCR product quality because of the higher resolving power of HPLC. HPLC has the added advantage of direct product quantification by peak integration. PCR products for mutation detection should appear as single, clean peaks under nondenaturing conditions. The quality of the PCR product can be improved further by the use of a proofreading polymerase or a mixture of a nonproofreading (9 parts) and proofreading (1 part) polymerases (unpublished observations). Polymerases are known to incorporate errors. If errors are incorporated during the initial cycles of PCR, a wild-type sequence may falsely be identified as a mutant sequence. The random incorporation of errors throughout the course of PCR contribute to an elevated baseline preceding the homoduplex peak during mutation detection by DHPLC. Thus, the use of polymerases with proofreading capabilities is beneficial for the amplification of gene fragments to be scanned for mutations by DNA chromatography.
The analysis temperature for TMHA on the WAVE® Nucleic Acid Fragment Analysis System is determined by the WAVEMAKERTM software based on the sequence and length of the fragment scanned for mutations. In addition, the software determines the gradient conditions for the analysis by DHPLC. Under nondenaturing conditions the PCR product for amplicon 1 yields a single peak (data not shown). A temperature titration for the wild-type sequence of amplicon 1 is shown in Figure 5. Temperature titrations are performed under identical gradient conditions at different temperatures flanking the software-predicted analysis temperature. In the absence of a mutation, heteroduplexes are not formed. Increasing temperature during consecutive runs results in a progressive shortening of the retention time. The decrease in retention time is explained by an increase in denaturation of the PCR fragment. The latter increases the portion of the fragment that is present in single-stranded form, which has a decreased affinity for the column matrix as compared to the double-stranded form. The baseline increase preceding the main peak is a result of heteroduplexes formed with PCR products containing randomly incorporated polymerase errors. The extent of this baseline increase is dependent on the fidelity of the polymerase used for amplification.
Fig 5.

Temperature titration of a wild-type PCR product of amplicon 1
The chromatographic profile of the wild-type amplicon at a specific analysis temperature serves as a reference for mutation calling. The presence of a mutant sequence results in the formation of heteroduplexes. As explained previously, heteroduplexes melt at lower temperatures than homoduplexes and exhibit shortened retention times. Chromatographic profiles of samples differing from the wild-type profile are indicative of the presence of a mutation. Differences may be as subtle as peak broadening or the appearance of a shoulder on the leading edge of the homoduplex peak or as pronounced as the appearance of 2 distinct heteroduplex peaks and an additional mutant homoduplex peak. The former scenario is demonstrated in Figure 2 at 56°C and the latter at 58° and 59°C. Resolution between hetero- and homoduplexes is determined by the sequence context in which the mutation occurs and by how the presence of a mismatch in the heteroduplex lowers the melting temperature of the heteroduplex fragment. In general, any difference between wild-type and sample peak pattern is justification for calling a mutation.
Examples of mutation detection in the HSP70B′ gene sequence are shown in Figures 6 and 7. Figure 6A shows a temperature titration of the wild-type amplicon 3 in the range from 61° to 65° C. The analysis temperature predicted by WAVEMAKERTM software at which mutations can be detected for this amplicon is 64°C. Figure 6B shows an identical temperature titration for the PCR product of amplicon 3, which was generated from genomic DNA isolated from a prostate cancer cell line (sample). Comparison of the chromatographic profiles of wild-type and sample PCR products reveal clear differences. Peak broadening of the sample peak is observed at 61°C. The sample chromatogram exhibits increased baseline levels preceding the main peak at 62° and 63°C. The mutation is best resolved in the sample chromatogram at 64° and 65°C, at which wild-type and sample chromatograms differ the most. Specifically, at 64° and 65°C, peaks corresponding to heteroduplexes are observed at shorter retention times than that of the homoduplex peak. The genome of the prostate cancer cell line contains a mutation in the region of amplicon 3.
Fig 6.

Temperature titration of a wild-type PCR product (A) and a wild-type/mutant PCR product (B) of amplicon 3
Fig 7.

Mutation detection on PCR products of amplicon 2 amplified from different template sources. The chromatogram in the center represents the profile of the wild-type homoduplex. Chromatograms at the top and bottom (obtained from prostate cancer patients undergoing combined hyperthermia and radiation therapy at the Dana-Farber Cancer Institute) differ from the wild-type chromatogram and are indicative for the presence of mutations
Additional examples of mutation detection are shown in Figure 7 for amplicon 2 of the HSP70B′ gene sequence. Chromatograms were recorded at a column temperature of 65°C. Samples A and C, which were amplified from genomic DNA isolated from prostate cancer biopsies, show clear differences in their peak pattern compared to the wild-type sample B. Hence, mutations are present in amplicon 2 of samples A and C.
Mutation detection by DHPLC, as it is presented in this context, is a high-throughput, timesaving, and economical tool for mutation screening. DHPLC provides information about whether a mutation is present. In order to determine the specific nature of the mutation, however, PCR products in question need to be sequenced.
In conclusion, detection of unknown mutations by DNA chromatography using DHPLC under partially denaturing conditions is a reliable, reproducible, and sensitive technology. As demonstrated in the case of the HSP70B′ gene, amplicon design for mutation scanning is facilitated by the WAVEMAKERTM software. This software predicts the melting behavior of specific sequences of up to 2000 bp under the analysis conditions of TMHA by DHPLC. Amplicons suitable for mutation detection can be identified based on melting domains within the sequences analyzed. PCR primers for the amplification of identified target regions are selected using conventional primer selection software. After primer selection, the melting profiles of PCR amplified regions are re-evaluated. This allows an optimal temperature for mutation detection to be determined.
Prior to mutation analysis the yield, purity, and quantity of the PCR product are determined by DNA chromatography under nondenaturing conditions. Gradient conditions for mutation analysis are predicted by WAVEMAKERTM software. Mutation analysis is typically performed on a 5-μL aliquot of a PCR directly without any sample preparation or purification. Up to 192 samples can be analyzed in an automated fashion in a single series of runs. About 200 individual samples can be screened for the presence of mutations per day using DHPLC.
Comparing a reference chromatogram of a wild-type standard with that of a sample reliably identifies mutations. Mutation calling is facilitated by the overlay and stacking of chromatograms. The presence of mutations is indicated by deviations of the sample chromatogram from the reference chromatogram.
REFERENCES
- Choy YS, Dabora SL, Hall F, Ramesh V, Niida Y, Franz D, Kasprzyk-Obara J, Reeve MP, Kwiatkowski DJ. Superiority of denaturing high performance liquid chromatography over single-stranded conformation and conformation-sensitive gel electrophoresis for mutation detection in TSC2. Ann Hum Genet. 1999;63:383–391. doi: 10.1046/j.1469-1809.1999.6350383.x. [DOI] [PubMed] [Google Scholar]
- Gross E, Arnold N, Goette J, Schwarz-Boeger U, Kiechle M. A comparison of BRCA1 mutation analysis by direct sequencing, SSCP and DHPLC. Hum Genet. 1999;105:72–78. doi: 10.1007/s004399900092. [DOI] [PubMed] [Google Scholar]
- Huber CG, Berti GN. Detection of partial denaturation in AT-rich DNA fragments by ion-pair reversed-phase chromatography. Anal Chem. 1996;68:2959–2965. doi: 10.1021/ac960037v. [DOI] [PubMed] [Google Scholar]
- Jones AC, Austin J, Hansen N, Hoogendoorn B, Oefner P, Cheadle JP, O'Donovan MC. Optimal temperature selection for mutation detection by denaturing HPLC and comparison to single-stranded conformation polymorphism and heteroduplex analysis. Clin Chem. 1999;45:1133–1140. [PubMed] [Google Scholar]
- Kuklin A, Munson K, Gjerde D, Haefele R, Taylor P. Detection of single-nucleotide polymorphisms with the WAVETM DNA Fragment Analysis System. Genet Test. 1997-98;1:201–206. doi: 10.1089/gte.1997.1.201. [DOI] [PubMed] [Google Scholar]
- Leung TK, Rajendran MY, Monfries C, Hall C, Lim L. The human heat-shock protein family: expression of a novel heat-inducible HSP70 (HSP70B′) and isolation of its cDNA and genomic DNA. Biochem J. 1990;267:125–132. doi: 10.1042/bj2670125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Smith DI, Rechtzigel KJ, Thibodeau SN, James CD. Denaturing high performance liquid chromatography (DHPLC) used in the detection of germline and somatic mutations. Nucleic Acids Res. 1998;26:1396–1400. doi: 10.1093/nar/26.6.1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donovan MC, Oefner PJ, Roberts SC, Austin J, Hoogendoorn B, Guy C, Speight G, Upadhyaya M, Sommer SS, McGuffin P. Blind analysis of denaturing high-performance liquid chromatography as a tool for mutation detection. Genomics. 1998;52:44–49. doi: 10.1006/geno.1998.5411. [DOI] [PubMed] [Google Scholar]
- Oefner PJ, Underhill PA. Comparative DNA sequencing by denaturing high performance liquid chromatography (DHPLC) Am J Hum Genet. 1995;57:A266. doi: 10.1002/0471142905.hg0710s48. [DOI] [PubMed] [Google Scholar]
- Oefner PJ, Underhill PA 1998 DNA mutation detection using denaturing high-performance liquid chromatography (DHPLC). In: Current Protocols in Human Genetics, Supplement 19. Wiley & Sons, New York, 7.10.1–7.10.12. [DOI] [PubMed] [Google Scholar]
- Underhill PA, Jin L, Zemans R, Oefner PJ, Cavalli-Sforza LL. A pre-Columbian Y chromosome–specific transition and its implications for human evolutionary history. Proc Natl Acad Sci USA. 1996;93:196–200. doi: 10.1073/pnas.93.1.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill PA, Jin L, Lin AA, et al. Detection of numerous Y chromosome bialleleic polymorphisms by denaturing high-performance liquid chromatography. Genome Res. 1997;7:996–1005. doi: 10.1101/gr.7.10.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner T, Stoppa-Lyonnet D, Fleischmann E, et al. Denaturing high-performance liquid chromatography detects reliably BRCA1 and BRCA2 mutations. Genomics. 1999;62:369–376. doi: 10.1006/geno.1999.6026. [DOI] [PubMed] [Google Scholar]