

Abstract
Heat shock transcription factor 1(HSF1) activation is a multistep process. The conversion of a latent cytoplasmic form to a nuclear, DNA binding state appears to be activated by nonsteroidal anti-inflammatory drugs. In previous studies, we showed that HSF 1 is phosphorylated by the protein kinase RSK2 in vitro and that this effect is inhibited by nonsteroidal anti-inflammatory drugs at the concentration that leads to the activation of HSF1 in vivo (Stevenson et al 1999). In the present study, using cells from a patient with Coffin-Lowry syndrome (deficient in RSK2), we demonstrate that RSK2 slightly represses activation of HSF1 in vivo at 37°C. In Coffin-Lowry syndrome cells, HSF1-HSE DNA binding activity after treatment with sodium salicylate was slightly higher than that in untreated cells, indicating that although RSK2 is involved in HSF1 regulation, it is not the unique protein kinase that suppresses HSF1-HSE binding activity at 37°C. However, heat shock treatment resulted in significantly higher HSF1-HSE binding activity in Coffin-Lowry syndrome cells as compared with normal controls, suggesting that RSK2 represses HSF1-HSE binding activity during heat shock.
INTRODUCTION
Coffin-Lowry syndrome (CLS) is an X-linked disorder that was independently described by Coffin et al (1966) and Lowry et al (1971). These authors documented clinical manifestations comprising moderate to severe mental retardation, progressive skeletal deformity, and facial and digital dysmorphisms in affected patients (Young 1988). Genetic linkage analysis mapped the gene locus to Xp22.2 (Biancalana et al 1994; Bird et al 1995; Hanauer et al 1988). Recently, mutations were found that affect the function of the RSK2 gene, thus providing evidence that abnormalities in the MAPK/RSK signaling pathway cause Coffin-Lowry syndrome (Trivier et al 1996).
RSK2, a 90-kD ribosomal S6 kinase, is a member of a family of growth factor–regulated serine-threonine kinases that has a critical role as an effector of the RAS-MAPK pathway and a regulator of immediate early gene transcription (for review, see Blenis 1993). The RSK kinases are unusual in containing 2 highly conserved kinase domains (Wadzinski et al 1993; Zhao et al 1996). The N-terminal domain, which is apparently active in phosphorylating exogenous substrates of RSK2, is structurally related to protein kinase C and the catalytic domains of protein kinases A and G (Wadzinski et al 1993). The C-terminal domain, which contains sequence similarities with calcium- and calmodulin-activated kinase II and MAPKAP K2, does not directly phosphorylate exogenous substrates but instead plays a role in autophosphorylation and apparently is an essential regulatory subunit of RSK2 (Wadzinski et al 1993). Recently, it was demonstrated that sodium salicylate (NaSal), a nonsteroidal anti-inflammatory drug, inhibits the phosphorylation by the Ser/Thr protein kinase RSK2 of cyclic AMP response element-binding protein (CREB) and I-κBα on residues crucial for their transcriptional activity in vivo and thus repress CREB and NF-κB-dependent transcription (Stevenson et al 1999). Further, human HSF1 was shown to be phosphorylated by RSK2 in vitro, and this effect is inhibited by NSAIDs (Stevenson et al 1999; Housby et al 1999).
The heat shock response is a highly conserved molecular stress response that involves the induction of stress proteins (heat shock proteins or HSPs) that is designed to protect cells against elevated temperatures and a wide variety of stressful stimuli including environmental (UV radiation, heat shock, heavy metals, and amino acids), pathological (viral, bacterial, parasitic infections or fever, inflammation, malignancy, or autoimmunity), or physiological (growth factors, cell differentiation, hormonal stimulation, or tissue development) stimuli (Lindquist and Craig 1988). Indeed, the sensitivity of cancer cells to the chemical stresses encountered in the tumor microenvironment, caused by inadequate tumor perfusion or imposed by chemotherapy, has been shown to be limited by the expression of cellular stress responses (Calderwood 1995). Hyperthermia kills cells largely through the induction of lethal protein aggregation and denaturation cascades and is antagonized by HSP activity, which leads to protein refolding (Schlesinger 1994). The expression of HSPs is regulated through the activity of a sequence specific transcription factor, heat shock factor 1 (HSF1), which senses exposure to heat shock and leads to the transcription of HSP genes (Wu 1995).
Transcriptional activation of heat shock genes is mediated by the interaction of a sequence specific transcription factor, HSF1, with heat shock elements (HSE) in the promoters of heat shock genes (Hensold et al 1990; Price and Calderwood 1991; Westwood et al 1991; Voellmy 1994; Ruis and Schuller 1995). A potential role of HSF1 gene repression was suggested by the finding that HSF binds to non–heat shock loci on the polytene chromosomes of Drosophila during heat shock (Westwood et al 1991). Binding of HSF to these loci on the chromosomes of heat-shocked cells was correlated with the inhibition of puffing (gene activation) in a large number of developmental loci, suggesting a role for HSF in transcriptional repression (Westwood et al 1991). We have recently demonstrated that HSF1 represses non–heat shock genes in mammalian cells (Cahill et al 1996). The promoter of human prointerleukin-1β (IL-1β) gene in human monocytes was repressed both by elevated temperatures and by heat-independent expression of HSF1 (Cahill et al 1996). Repression was strictly dependent on an HSE-like sequence within the IL-1β promoter, to which HSF1 bound with high affinity (Cahill et al 1996).
In this study, we show that RSK2 slightly represses activation of HSF1 in vivo at 37°C. In sodium salicylate–treated Coffin-Lowry syndrome cells, HSF1-HSE DNA binding activity was slightly higher than that in untreated cells. This indicates that although RSK2 is involved in HSF1 regulation, it is not the unique protein kinase that suppresses HSF1-HSE binding activity at 37°C, and RSK2 is not the only target of sodium salicylate. However, HSF1-HSE binding activity in Coffin-Lowry syndrome cells was significantly higher than that in normal cells after heat shock, suggesting that RSK2 represses HSF1-HSE binding activity during heat shock.
MATERIALS AND METHODS
Cells and growth conditions
Lymphoblast cells that originated from a Coffin-Lowry syndrome patient (RSK−/− cells) and a clinically unaffected person (RSK+/+ cells) (Coriell Institute for Medical Research, Camden, NJ, USA) were grown in RPMI 1640 containing 15% heat-inactivated fetal bovine serum (Coffin-Lowry syndrome patient) or 20% heat-inactivated fetal bovine serum (clinically unaffected person), 100 μg/mL streptomycin, 100 units/mL penicillin, and 2 mMl-glutamine at 37°C in 5% CO2, 95% air atmosphere, and routinely passaged by dilution at a 1:3 ratio. The Coffin-Lowry syndrome patient was an 8-year-old male (−/−). Clinically unaffected lymphoblasts were obtained from an age- and race-matched male (+/+).
Western blot analysis
Cells were harvested in boiling SDS sample buffer after washing twice in PBS, and whole-cell lysates were resolved on 10% SDS-PAGE and electrotransferred onto a PVDF membrane at 10 V in transfer buffer (20% methanol, 150 mM glycine, 25 mM Tris, pH 8.3). After transfer, the nitrocellulose sheet was blocked by 20 mL of 5% nonfat milk in PBS. The nitrocellulose sheet was washed once with PBS and incubated with anti-RSK2 antibody (1:1000) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) in a 0.5% nonfat milk/PBS solution at 4°C overnight. After washing 3 to 5 times with washing buffer (0.05% Tween 20 in PBS), anti-goat IgG peroxidase conjugate secondary antibody (1:2000) (Sigma, St Louis, MO, USA) was added. This incubation was continued at room temperature for 1–2 h. The sheet was then washed 3 to 5 times with washing buffer and incubated with ECL reagent (New England Biolabs, Beverly, MA, USA) at room temperature for 1 minute. X-ray films (Eastman Kodak Company, Rochester, NY, USA) were exposed and developed.
Transient transfection analysis
Cells were seeded in 6-well (2 × 106 cells/well) plates (Becton Dickinson Labware, Franklin Lakes, NJ, USA) just before transfection. Lymphoblast cells were transfected with reporter plasmids, expression plasmids, and control (β-Gal expression plasmids) by using FuGENE6 (Roche Molecular Biochemicals, Indianapolis, IN, USA). A precipitate was formed using 3 μL of FuGENE/μg of transfected DNA, and the transfection mix was made up to 1 mL with serum-free RPMI 1640. After incubation at room temperature for 15 to 45 minutes, the DNA/FuGENE mixture was added to cells, and complete RPMI 1640 (with serum) was added into cells after transfection, 3 to 5 hours. Cells were harvested 24 hours after transfection. Luciferase and β-galactosidase activity assays were performed according to Promega's protocol. Luciferase activity was normalized to β-galactosidase activity, which was used as an internal transfection efficiency control. Results were expressed as fold activity of control.
Electrophoretic mobility shift assay for HSF1-HSE binding
Nuclear extracts were prepared according to Schreiber et al (1989), incubated with a double-stranded 32P-labeled HSE-containing oligonucleotide probe, and analyzed by electrophoretic mobility shift assay (EMSA) as previously described (Cahill et al 1996). Briefly, probe was labeled by end filling with Klenow fragment using [α-32P] dGTP (NEN™ Life Science Products, Boston, MA, USA). We used 2 × 104 cpm/reaction of 32P-labeled HSE-containing oligonucleotide probe to incubate with nuclear extracts (2 ∼ 5 μg) in 20 mM HEPES, 50 mM KCl, 1.5 mM MgCl2, 0.2 mM EDTA, 20% glycerol, 100 μg/mL poly(dI-dC), 300 μg/mL BSA, 2.5 mM DTT, 1 mM PMSF for 30 minutes at room temperature and resolved by electrophoresis on 4% polyacrylamide gels in 15 mM Tris-HCl, 24 mM boric acid, and 1 mM EDTA. Electrophoretic mobility supershift assays were carried out by adding 1:10 dilution of anti-HSF1 antibody 68-3 for 30 minutes on ice prior to addition of the probe. The gels were dried and subjected to autoradiography using X-ray films (Eastman Kodak Company, Rochester, NY, USA).
RESULTS
Determination of RSK2 in normal (RSK2+/+) and CLS (RSK2−/−) cells
Initial experiments were conducted to confirm the absence of functional RSK2 in CLS patients by Western blot analysis. Anti-RSK2 antibody, directed against the C terminus of the protein, failed to resolve a band at 90 kD in whole-cell lysates from the patient with CLS (Fig 1, right lane). However, RSK2 was readily detected as a protein band at 90 kD in the cells originated from a clinically unaffected person (Fig 1, left lane). This demonstrates the absence of functional RSK2 protein in this CLS patient.
Fig 1.
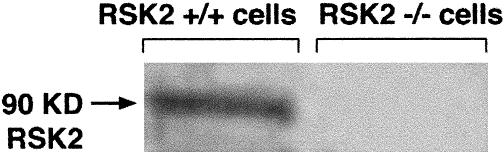
Level of RSK2 in normal (RSK2+/+) and CLS (RSK2−/−) cells. Whole-cell lysates from cells were analyzed by Western blot by using anti-RSK2 antibody (Santa Cruz Biotechnology, Santa Cruz, CA). RSK2 is not detectable in CLS (RSK2−/−) cells (right lane). Anti-RSK2 antibody is directed against the C terminus of the protein
Role of RSK2 in regulating of HSF1 activation in vivo
We next investigated the role of RSK2 in the regulation of HSF1 activity in vivo. Experiments were therefore performed to compare the ability of wild-type HSF1 to activate HSP70B promoter reporter construct (HSP70B-Luc) after cotransfection and expression in normal (RSK2+/+) cells and CLS (RSK2−/−) cells. HSF1 expression activated HSP70B promoter under normal growth conditions and activated HSP70B promoter activity in CLS (RSK2−/−) cells about 25% higher than in normal (RSK2+/+) cells (Fig 2). This result shows that RSK2 slightly repressed activation of HSF1 in vivo at 37°C.
Fig 2.

Transcriptional activation of HSP70B promoter by HSF1 in normal (RSK2+/+) and CLS (RSK2−/−) cells at 37°C. Wild-type HSF1 expression plasmid (pcDNA3.1HSF1; 0.2 μg/well) or empty control (pcDNA3.1; 0.2 μg/well) were cotransfected with HSP70B promoter reporter construct (HSP70B-Luc) (0.6 μg/well) along with pSV-β-galactosidase plasmids (0.6 μg/well), respectively, transfected into normal (RSK2+/+) and CLS (RSK2−/−) cells in triplicate and incubated for 20 to 24 h. Wild-type HSF-1 were force-cloned into XhoI/EcoRI sites of the pcDNA3.1 vector as described (Cahill et al 1996). HSP70B-Luc reporter plasmid was constructed as described before (Chen et al 1997). Cells were harvested for luciferase assay. Luciferase activity was normalized to β-galactosidase activity. Luciferase activity is expressed as fold activity over the empty pcDNA3.1 plasmid control
Effect of sodium salicylate on HSF1-HSE binding activity in normal (RSK2+/+) and CLS (RSK2−/−) cells
Normal (RSK2+/+) and CLS (RSK2−/−) cells that were pretreated in the presence or absence of sodium salicylate (NaSal)(20 mM) at 37°C were used to analyze HSF1-HSE binding activity by EMSA. Nuclear HSF was supershifted by anti-HSF1 antibody (Fig 3, lanes 2, 4, 6, 8, and 10). HSF1-HSE binding activity was increased in NaSal-treated normal (RSK2+/+) cells (Fig 3, lanes 5 and 6) as compared with untreated normal (RSK2+/+) cells (Fig 3, lanes 3 and 4). In NaSal treated CLS (RSK2−/−) cells (Fig 3, lanes 9 and 10), HSF1-HSE binding activity was slightly higher than that in untreated CLS (RSK2−/−) cells (Fig 3, lanes 7 and 8), and HSF1-HSE complexes were formed with similar electrophoretic mobility (Fig 3, lanes 5 and 9) to that from heat-shocked Hela cells (Fig 3, lane 1). Since RSK2 phosphorylates HSF1 in vitro, and this effect is inhibited by NaSal (Stevenson et al 1999), we thought that RSK2 might elevate HSF1-HSE binding activity in RSK2−/− cells. However, we found that HSF1-HSE binding activity was not increased in CLS (RSK2−/−) cells (Fig 3, lane 7) as compared with that in normal (RSK2+/+) cells (Fig 3, lane 3). These results indicate that although RSK2 is involved in HSF1 regulation, it is not the unique protein kinase that suppresses HSF1-HSE binding activity at 37°C, and RSK2 is not the only target of sodium salicylate.
Fig 3.
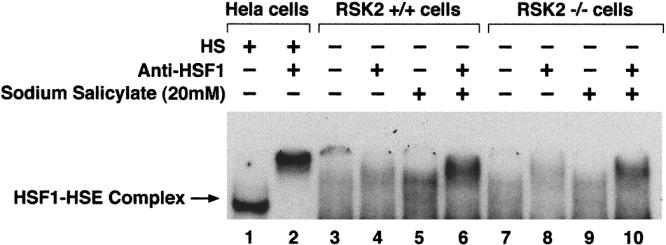
Effect of sodium salicylate on HSF1-HSE binding activity in nuclear extracts from normal (RSK2+/+) and CLS (RSK2−/−) cells at 37°C. Parallel cultures of normal (RSK2+/+) and CLS (RSK2−/−) cells were pretreated for 30 minutes in presence or absence of sodium salicylate (20 mM) (lanes 5, 6, 9, and 10). Nuclear extracts (4 μg) were incubated with 32P-labeled HSE probe and subjected to EMSA. Gel supershift assays were carried out as described before (Cahill et al 1996) (lanes 2, 4, 6, 8, and 10). Heat-shocked Hela cells (lanes 1 and 2) were positive controls. The position of HSF1-HSE complex is indicated by an arrow
Effect of heat shock on HSF1-HSE binding activity in normal (RSK2+/+) and CLS (RSK2−/−) cells
In order to elucidate the role of RSK2 on HSF1-HSE binding activity, nuclear extracts from normal (RSK2+/+) and CLS (RSK2−/−) cells after heat shock treatment (43°C, 1 hour) were analyzed by EMSA (Fig 4). HSF1-HSE complex from normal (RSK2+/+) cells (Fig 4, lane 3) and CLS (RSK2−/−) cells (Fig 4, lane 5) after heat shock led to the formation of a complex of similar electrophoretic mobility to that from heat-shocked Hela cells (Fig 4, lane 1), and the HSF1-HSE complex bands were supershift with anti-HSF1 antibody (Fig 4, lanes 2, 4, and 6). HSF1-HSE binding activity was significantly higher in CLS (RSK2−/−) cells (Fig 4, lane 5) than that in normal control (RSK2+/+) cells (Fig 4, lane 3). Taken together, these results show that RSK2 represses HSF1-HSE binding activity during heat shock.
Fig 4.
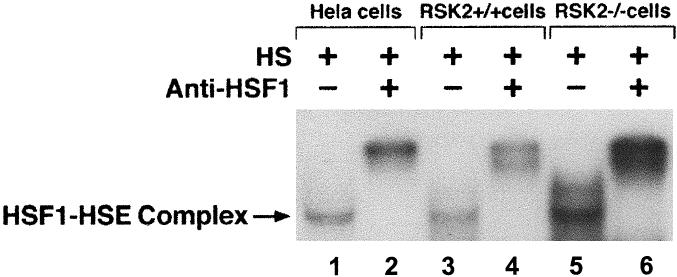
HSF1-HSE binding activity in nuclear extracts from normal (RSK2+/+) cells (lanes 3 and 4), CLS (RSK2−/−) cells (lanes 5 and 6), and Hela (lanes 1 and 2) cells after heat shock (43°C, 1 hour). Nuclear extracts (2 μg) were incubated with 32P-labeled HSE and subjected to EMSA. Gel supershift assays were carried out as described before (Cahill et al 1996). Specific HSF1-HSE complex is indicated by an arrow
DISCUSSION
Our studies show that RSK2 only slightly represses activation of HSF1 in vivo at 37°C. Using CLS (RSK−/−) cells in which RSK2 gene is mutated, thus lacking C terminus catalytic domain, we have been able to show that RSK2 plays a minor role in HSF1 activation regulation at 37°C but significantly represses HSF1-HSE binding activity during heat shock. Previously, we have shown that mitogen-activated protein kinase (MAPK) of the ERK family, glycogen synthase kinase 3 α (GSK3α) and protein kinase C (PKC) phosphorylated HSF1, represses transcriptional function (Chu et al 1996, 1998). This suggested the possibility that constitutive HSF1 repression involves interactions with protein kinase MAPK signaling pathways in the cells. It also indicated that HSF1 is antagonized by RAS-MAPK pathway signaling and that another mechanism for HSF1 repression at 37°C may exist (Engelberg et al 1994; Mivechi et al 1995; Chu et al 1996; Knauf et al 1996; Kline et al 1997).
Sodium salicylate inhibits the phosphorylation of HSF1 on as yet unknown sites and stimulates its ability to bind DNA, although the precise mechanisms involved in this process are unclear (Voellmy 1994; Cahill et al 1996; Chu et al 1998). RSK2 is evidently a repressor of HSF1 activation, and NaSal may function to reverse this repression (Stevenson et al 1999). However, our results from CLS (RSK2−/−) cells indicated that although RSK2 is involved in HSF1 regulation, it is not the unique protein kinase that suppresses HSF1-HSE binding activity at 37°C, and RSK2 is not the only target of NaSal. By inhibiting RSK2, NaSal coordinately influences the activity of at least 2 transcription factors: CREB and NF-κB (Stevenson et al 1999). The effect of NaSal on HSF1 may involve other members of the RSK2 family. We have recently shown that MAPKAP K2 phosphorylates HSF1 in an NaSal sensitive manner (X. Wang and S.K. Calderwood, in preparation). HSF1 activation by NaSal may thus involve RSK2 and other members of this protein kinase family, including MAPKAP K2. Regulation of the transcription factors NF-κB and HSF1 by NaSal thus shows an interesting contrast that NF-κB is activated through the NaSal-dependent phosphorylation of the inhibitory subunit IκBα (Stevenson et al 1999). IκBα phosphorylation involves RSK2 but not MAPKAP K2 (X. Zhao and S.K. Calderwood, in preparation). Although HSF1 is phosphorylated by both RSK2 and MAPKAP K2 in an NaSal-sensitive manner, RSK2 plays a minor role in HSF1 regulation at 37°C (Figs 2 and 3), and our current studies suggest a role for MAPKAP K2 in regulation of HSF1 activity (X. Wang and S.K. Calderwood, in preparation). NaSal may thus exert differential effects on target molecules by inhibiting a range of protein kinases. Whereas our experiments rule out RSK2 as a major regulator of HSF1 at 37°C, they suggest a role for RSK2 in regulation heat shock activation of HSF1.
Acknowledgments
This work was supported by National Institutes of Health Grants CA47407, CA31303, CA50642, and CA77465 to S.K.C.
REFERENCES
- Biancalana V, Trivier E, Weber C, et al. Construction of a high-resolution linkage map for Xp22.1-p22.2 and refinement of the genetic localization of the Coffin-Lowry syndrome gene. Genomics. 1994;22:617–625. doi: 10.1006/geno.1994.1435. [DOI] [PubMed] [Google Scholar]
- Bird H, Collins AL, Oley C, Lindsay S. Crossover analysis in a British family suggests that Coffin-Lowry syndrome maps to a 3.4-cM interval in Xp22. Am J Med Genet. 1995;59:512–516. doi: 10.1002/ajmg.1320590420. [DOI] [PubMed] [Google Scholar]
- Blenis J. Signal transduction via the MAP kinases: proceed at your own RSK. Proc Natl Acad Sci USA. 1993;90:5889–5892. doi: 10.1073/pnas.90.13.5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill CM, Waterman WR, Xie Y, Auron PE, Calderwood SK. Transcriptional repression of the prointerleukin 1beta gene by heat shock factor 1. J Biol Chem. 1996;271:24874–24879. [PubMed] [Google Scholar]
- Calderwood SK 1995 Molecular strategies for sensing and responding to stress. Proceedings of the 86th Annual Meeting of the American Association for Cancer Research. 65682. [Google Scholar]
- Chen C, Xie Y, Stevenson MA, Auron PE, Calderwood SK. Heat shock factor 1 represses Ras-induced transcriptional activation of the c-fos gene. J Biol Chem. 1997;272:26803–26806. doi: 10.1074/jbc.272.43.26803. [DOI] [PubMed] [Google Scholar]
- Chu B, Soncin F, Price BD, Stevenson MA, Calderwood SK. Sequential phosphorylation by mitogen-activated protein kinase and glycogen synthase kinase 3 represses transcriptional activation by heat shock factor-1. J Biol Chem. 1996;271:30847–30857. doi: 10.1074/jbc.271.48.30847. [DOI] [PubMed] [Google Scholar]
- Chu B, Zhong R, Soncin F, Stevenson MA, Calderwood SK. Transcriptional activity of heat shock factor 1 at 37 degrees C is repressed through phosphorylation on two distinct serine residues by glycogen synthase kinase 3 and protein kinases Calpha and Czeta. J Biol Chem. 1998;273:18640–18646. doi: 10.1074/jbc.273.29.18640. [DOI] [PubMed] [Google Scholar]
- Coffin GS, Siris E, Wegienka LC. Mental retardation with osteocartilaginous anomalies. Am J Dis Child. 1966;112:205–213. [Google Scholar]
- Engelberg D, Zandi E, Parker CS, Karin M. The yeast and mammalian Ras pathways control transcription of heat shock genes independently of heat shock transcription factor. Mol Cell Biol. 1994;14:4929–4937. doi: 10.1128/mcb.14.7.4929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanauer A, Alembik Y, Gilgenkrantz S, Mujica P, Nivelon-Chevallier A, Pembrey ME, Young ID, Mandel JL. Probable localisation of the Coffin-Lowry locus in Xp22.2-p22.1 by multipoint linkage analysis. Am J Med Genet. 1988;30:523–530. doi: 10.1002/ajmg.1320300154. [DOI] [PubMed] [Google Scholar]
- Hensold JO, Hunt CR, Calderwood SK, Housman DE, Kingston RE. DNA binding of heat shock factor to the heat shock element is insufficient for transcriptional activation in murine erythroleukemia cells. Mol Cell Biol. 1990;10:1600–1608. doi: 10.1128/mcb.10.4.1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Housby JN, Cahill CM, Chu B, Prevelige R, Bickford K, Stevenson MA, Calderwood SK. Non-steroidal anti-inflammatory drugs inhibit the expression of cytokines and induce HSP70 in human monocytes. Cytokine. 1999;11:347–358. doi: 10.1006/cyto.1998.0437. [DOI] [PubMed] [Google Scholar]
- Kline MP, Morimoto RI. Repression of the heat shock factor 1 transcriptional activation domain is modulated by constitutive phosphorylation. Mol Cell Biol. 1997;17:2107–2115. doi: 10.1128/mcb.17.4.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauf U, Newton EM, Kyriakis J, Kingston RE. Repression of human heat shock factor 1 activity at control temperature by phosphorylation. Genes Dev. 1996;10:2782–2793. doi: 10.1101/gad.10.21.2782. [DOI] [PubMed] [Google Scholar]
- Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- Lowry B, Miller JR, Fraser FC. A new dominant gene mental retardation syndrome: association with small stature, tapering fingers, characteristic facies, and possible hydrocephalus. Am J Dis Child. 1971;121:496–500. [PubMed] [Google Scholar]
- Mivechi NF, Giaccia AJ. Mitogen-activated protein kinase acts as a negative regulator of the heat shock response in NIH3T3 cells. Cancer Res. 1995;55:5512–5519. [PubMed] [Google Scholar]
- Price BD, Calderwood SK. Ca2+ is essential for multistep activation of the heat shock factor in permeabilized cells. Mol Cell Biol. 1991;11:3365–3368. doi: 10.1128/mcb.11.6.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruis H, Schuller C. Stress signaling in yeast. Bioessays. 1995;17:959–965. doi: 10.1002/bies.950171109. [DOI] [PubMed] [Google Scholar]
- Schlesinger MJ. How the cell copes with stress and the function of heat shock proteins. Pediatr Res. 1994;36:1–6. doi: 10.1203/00006450-199407001-00001. [DOI] [PubMed] [Google Scholar]
- Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with “mini-extracts”, prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson MA, Zhao M-J, Asea A, Coleman NC, Calderwood SK. Salicylic acid and asprin inhibit the activity of RSK2 kinase and repress RSK2-dependent transcription of CREB and NF-κB responsive genes. J Immunol. 1999;163:5608–5616. [PubMed] [Google Scholar]
- Trivier E, De Cesare D, Jacquot S, et al. Mutations in the kinase Rsk-2 associated with Coffin-Lowry syndrome. Nature. 1996;384:567–570. doi: 10.1038/384567a0. [DOI] [PubMed] [Google Scholar]
- Voellmy R. Transduction of the stress signal and mechanisms of transcriptional regulation of heat shock/stress protein gene expression in higher eukaryotes. Crit Rev Eukaryot Gene Expr. 1994;4:357–401. [PubMed] [Google Scholar]
- Wadzinski BE, Wheat WH, Jaspers S, Peruski LF Jr,, Lickteig RL, Johnson GL, Klemm DJ. Nuclear protein phosphatase 2A dephosphorylates protein kinase A-phosphorylated CREB and regulates CREB transcriptional stimulation. Mol Cell Biol. 1993;13:2822–2834. doi: 10.1128/mcb.13.5.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwood JT, Clos J, Wu C. Stress-induced oligomerization and chromosomal relocalization of heat-shock factor. Nature. 1991;353:822–827. doi: 10.1038/353822a0. [DOI] [PubMed] [Google Scholar]
- Wu C. Heat shock transcription factors: structure and regulation. Annu Rev Cell Dev Biol. 1995;11:441–469. doi: 10.1146/annurev.cb.11.110195.002301. [DOI] [PubMed] [Google Scholar]
- Young ID. The Coffin-Lowry syndrome. J Med Genet. 1988;25:344–348. doi: 10.1136/jmg.25.5.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Bjorbaek C, Moller DE. Regulation and interaction of pp90(rsk) isoforms with mitogen-activated protein kinases. J Biol Chem. 1996;271:29773–29779. doi: 10.1074/jbc.271.47.29773. [DOI] [PubMed] [Google Scholar]