

Abstract
The costimulatory receptor CD28 and IL-4Rα-containing cytokine receptors play key roles in controlling the size and quality of pathogen-specific immune responses. Thus, CD28-mediated costimulation is needed for effective primary T-cell expansion and for the generation and activation of regulatory T-cells (Treg cells), which protect from immunopathology. Similarly, IL-4Rα signals are required for alternative activation of macrophages, which counteract inflammation by type 1 responses. Furthermore, immune modulation by CD28 and IL-4 is interconnected through the promotion of IL-4 producing T-helper 2 cells by CD28 signals. Using conditionally IL-4Rα and CD28 deleting mice, as well as monoclonal antibodies, which block or stimulate CD28, or mAb that deplete Treg cells, we have studied the roles of CD28 and IL-4Rα in experimental mouse models of virus (influenza), intracellular bacteria (L. monocytogenes, M. tuberculosis), and parasite infections (T. congolense, L. major). We observed that in some, but not all settings, Treg cells and type 2 immune deviation, including activation of alternative macrophages can be manipulated to protect the host either from infection or from immunopathology with an overall beneficial outcome. Furthermore, we provide direct evidence that secondary CD8 T-cell responses to i.c. bacteria are dependent on CD28-mediated costimulation.
Keywords: CD28, Costimulation, IL-4, IL-4R, Alternatively activated macrophages, Mouse models, Conditional knockout, Monoclonal antibodies, Regulatory T-cells, Influenza, L. monocytogenes, M. tuberculosis, T. congolense, L. major
Introduction
An immune response to an infection can be called successful (a) if the invading pathogen is eliminated or at least effectively controlled and (b) if this aim is reached without excessive damage to the host. To insure success, tight qualitative and quantitative regulation of the immune response is needed. Thus, different types of pathogens call for different arms from the warehouse of immune effector mechanisms. Overshooting responses are avoided by an equally diverse set of counter-regulating measures. The choice of the appropriate weapon against an invader becomes manifested in terms of T-helper subset differentiation. However, this is guided by signals from innate immune cells generated in response to pathogen class recognition via PAMPs. Limitation of immune responses is achieved by antigen and growth factor deprivation (with the exception of chronic infections), the induction and activity of negative regulators such as CTLA-4, BTLA, or PD1 in cis, i.e., on the effector cells themselves, and by “regulatory” T-cells (Treg cells) and “alternatively activated” macrophages (aamMphs) acting as dominant suppressor cells in trans.
Among the cytokines and costimulatory receptors regulating the balance between a highly effective immune response with the smallest possible collateral damage, the Th2 signature cytokine IL-4 and the costimulator CD28 occupy key positions. This is illustrated by the phenotype of the corresponding knockout mice that show imbalanced responses when challenged with pathogens.
IL-4 and IL-13 share a common signaling pathway through the IL-4 receptor α (IL-4Rα) chain. A functional IL-4R (type 1) requires assembly of IL-4Rα with a γc chain, while interaction of IL-4Rα with an IL-13Rα1 subunit leads to formation of a functional IL-13 receptor (type 2) [1]. Therefore, by blocking or deleting IL-4Rα, IL-4 and IL-13 functions are abrogated. While this is the case for most responses, more recent studies showed some evidence that IL-13 bound to IL-13Rα2, which was believed to be a decoy receptor for IL-13 [2] but may also induce signaling in certain cells under particular conditions [3].
More than a decade ago, constitutive IL-4Rα deficient mice were generated by using homologous recombination in embryonic stem cells [4]. Subsequent studies in this mutant mouse strain, including comparative studies in IL-4 and IL-13 deficient mice, uncovered important biological roles for IL-4Rα-responses mediated by IL-4 and IL-13. It turned out that Il-13, believed to be the small brother of IL-13, had overlapping roles, but also strikingly distinct roles, particularly in activating certain effector functions [5].
In the case of CD28, originally defined as the main costimulator of primary T-cell responses, genetic deficiency leads to an impairment of antibody responses (but less of cell-mediated immunity) [6], along with defective Th2 polarization and a marked reduction in regulatory T-cells [7], indicating that CD28-mediated costimulation is of particular importance for Treg and type 2 responses (including alternative macrophage activation via IL-4), while being less critical for cell-mediated immunity.
Importantly, such observations made in constitutively gene-deleted mice are unlikely to reveal the full importance of the relevant molecules in immune regulation due to the enormous plasticity of the immune system during and also after its maturation, which allows adaptational and back-up mechanisms to overcome at least some of the deleterious effects of genetic defects. Furthermore, the role these molecules play when expressed by particular cell types remains unaddressed. Accordingly, interference with the expression or function of immune regulators at the time of infection or in a cell type specific manner has become the most important tool in the analysis of their protective and pathological effects in vivo. Experimentally, this is achieved by “conditional” gene targeting using the Cre/loxP system. This strategy has been successfully employed for the IL-4Rα. Specifically, mice which are selectively IL-4Rα deficient in macrophages and neutrophils [8], smooth muscle cells [9], the CD4+ T-cell subpopulation [10] or T-cells [11] as well as B cells and dendritic cells (unpublished) have been used to probe IL-4Rα function in these cell types after various immune and non-immune challenges. More recently, conditionally deleting CD28 knockout mice have also been generated (F.L. and T.H., unpublished). In addition to this genetic approach, both blocking and stimulatory CD28-specific mAb were developed [12] which allow acute interference with [13] or activation of [14] this receptor during an immune response.
Interference with CD28-mediated costimulation
Conditional CD28 knockout mice were generated by flanking exons 2 and 3 with LoxP sequences, allowing effective deletion when crossed to the appropriate Cre transgenic lines (F.L., unpublished). Importantly, these mice also delete CD28 in an inducible fashion when a tamoxifen-regulated transgene is introduced. For the first time, this will allow to study the importance of CD28 in the recall of memory responses, an area of controversy for many years [15, 16]. Previous attempts to address this issue by monoclonal antibody blockade were hampered by the facts that a) no truly ligand-binding site-specific mAb to CD28 was available (but see below), and b) that blocking of CD80 and CD86, the ligands of CD28, confounded results by also depriving the negative regulator CTLA-4 (CD152) of these same ligands [17]. Similarly, transfer of memory T-cells into CD80/CD86 deficient hosts [17] does not allow to attribute the observed effects upon restimulation with the pathogen to a lack of CD28-mediated costimulation because again, CTLA-4 engagement is also affected.
First results using Listeria monocytogenes infection indicate that indeed, targeted deletion of CD28 between the establishment and recall of memory strongly impairs the secondary CD8 T-cell response to a model antigen expressed by the Listeria (M.F., T.G., F.L., T.H., unpublished). This is most likely due to a direct effect on the CD8 T-cells because the elimination of CD4 T-cells (including Treg cells) was shown to result in an increase rather than a decrease of the secondary CD8 T-cell response in the same system [18]. An example of the results thus obtained is given in Fig. 1a.
Fig. 1.
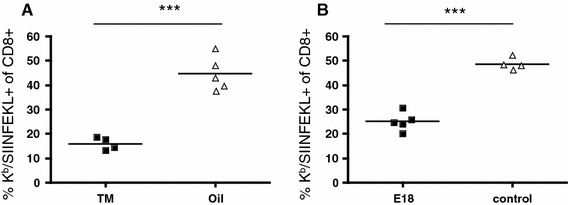
Secondary CD8 T-cell response to Listeria is CD28 dependent. Mice were infected with 5000 CFU Lm-OVA, challenged with 105 CFU on day 30, and analyzed for OVA-specific splenic CD8 cells by staining with Kb/SIINFEKL tetramers 5 days later. In A, CD28−/fl C57BL/6 mice were used, and CD28 deletion was induced by tamoxifen in oil feeding (“TM”, 2.5 mg/dose) on days 24–27; oil only served as control. In B, wt C57BL/6 mice were treated with the blocking CD28-specific mAb E18 (100 μg/dose) on days 29, 31, and 33. ***P < 0.0001
In order to study the importance of CD28-mediated costimulation for secondary T-cell responses, we have not only established inducible gene targeting but also the first mAb to mouse CD28 which fully blocks binding of natural ligands [12, 13]. This mAb was generated in mice, permitting prolonged treatment without the induction of neutralizing antibodies. Indeed, application of this mAb, called E18, in GvHD [13] and in a mouse asthma model (T.G. and T.H, unpublished) has shown high therapeutic efficacy. When again tested for interference with CD8 T-cell memory recall in the Listeria model, a strong reduction of clonal expansion was observed, corroborating our results obtained with conditional gene targeting (Fig. 1b).
Immune deviation and Treg activation with stimulatory CD28-specific antibodies
In the course of generating the first rat CD28-specific mAb, we discovered a novel class of such mAb which is able to trigger T-cell activation without the engagement of the TCR [19]. In contrast to conventional CD28-specific mAb, which bind monovalently close to or at the natural ligand-binding site, such “CD28 superagonists” (CD28SA) bind laterally, allowing lattice formation by crosslinking of individual CD28 homodimers [20, 21]. This correlation between function and topology of binding was observed in rats, mice, and humans [12, 20]. In rodents, CD28 superagonists induce immune deviation to Th2 [22] as well as expansion and functional activation of “natural” Treg cells [23–25]. Accordingly, such mAb have high therapeutic efficacy in a wide range of rodent models of autoimmunity, inflammation, and allograft rejection [25–31]. Moreover, CD28SA application expands the T-cell compartment in lymphopenic hosts, promoting recovery from immunoincompetence [32]. While the disastrous outcome of a first-in-man-study of a human CD28SA [33] has interrupted further development of CD28SA-based therapeuticals, these antibodies continue to be useful tools for the manipulation of the rodent immune system in attempts to study the contribution of immune deviation and/or Treg activation in the control of immunopathology.
With regard to the effects of CD28SA on the course of infections and microbe-induced inflammatory responses in rodent models, the following results have been obtained:
Adjuvant arthritis. In this model, a Th1 response of LEW rats to heat-killed mycobacteria in adjuvant results in swelling and inflammation of joints along with extensive cartilage destruction. Application of the mouse anti-rat CD28SA JJ316 is highly efficacious in preventing and even reversing pathology in both preventive and therapeutic settings ([34] and unpublished).
Mouse Influenza. Intranasal infection of mice with the Influenza A HKx31 (H3N2) leads to transient weight loss, lung pathology, and high levels of pro-inflammatory cytokines in the bronchoalveolar lavage and plasma. Application of the mouse anti-mouse CD28SA D665 alleviates weight loss and inflammation without compromising the control of the infection, an effect mapped to Treg activation by its recapitulation when Treg cells from D665-stimulated cells were transferred (Fig. 2 and unpublished observations). Shown are the marked increases in Treg cells in the bonchoalveolar lavage of flu-infected mice under CD28SA treatment (Fig. 2a), as well as the decrease in TNF and the increase in IL-10 in the lung after D665 stimulation or transfer of Treg cells from D665-stimulated mice (Fig. 2b). In this setting, therefore, it is possible to contain immune-mediated damage to the host by mAb-mediated Treg stimulation.
Mouse trypanosomiasis. A similar protective effect against immunopathology via experimental Treg stimulation was observed in mice infected with Trypanosoma congolense [35]. In this model of African trypanosomiasis, CD28SA treatment down-regulated IFNγ and TNF production by T-cells along with the production of reactive oxygen species by classically activated macrophages. Interestingly, it also triggered the development of alternatively activated macrophages, in line with a CD28SA-induced Th2 shift and IL-4 production [22]. With regard to pathology, CD28SA treatment delayed the onset of liver injury, diminished the anemia burden, and prolonged the survival of infected animals.
Mouse TB. Here, the TH1 response to inhaled Mycobacterium tuberculosis is required for pathogen containment. CD28SA treatment during M. tuberculosis infection increased the percentage of Treg cells systemically as well as in the lung (Fig. 3b), where it also increased IL-10 production (Fig. 3b) but had no effect on bacterial burden in the lung, liver, or spleen (Fig 3c).
Mouse leishmaniasis. As is the case in mouse TB for the intracellular bacterium M. tuberculosis, the control of the parasite burden in Leishmania major depends on a pro-inflammatory Th1 response [36]. We either depleted Treg cells with CD25-specific mAb, or polyclonally stimulated the Treg and Th2 compartment with the CD28SA D665 from week 4 to week 10 post-infection. In both C57BL/6 “healer” and BALB/c “progressor” strains, increased DTH (Th1) responses to Leishmania antigens were observed when Treg cells were depleted after infection, whereas CD28SA treatment decreased the reaction, in line with the role of Treg cells and a type 2 response in this system (Fig. 4a). Furthermore, swelling of the infected footpad (Fig. 4b), parasite load in the infected footpad (Fig. 4c), and antigen-specific IFN-γ production in C57BL/6 mice (Fig. 4d) were negatively influenced by CD28SA treatment and improved by Treg depletion. Progression of L. major infection, DTH responses, and parasite loads in IL-4Rα-/- mice were largely unaffected by CD28SA treatment versus controls (not shown), suggesting that enhancement of type 2 (as compared to Treg) responses may be responsible for the CD28SA effect in C57BL/6 and BALB/c mice (Table 1).
Fig. 2.
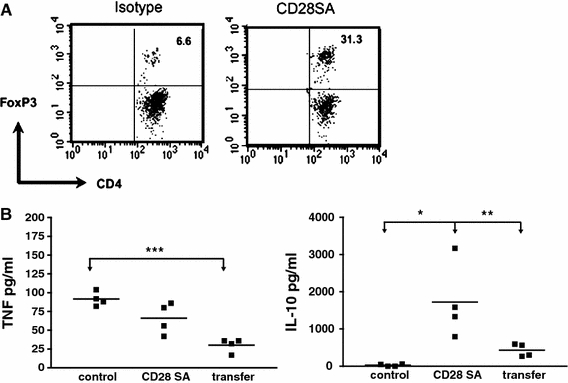
CD28SA-activated Treg cells suppress inflammatory cytokine response in the lung of influenza-infected mice. C57BL/6 mice were i.n. infected with 2 × 105 pfu Influenza A HK × 31. In A, mice were treated with 100 μg of the CD28SA D665 or isotype control antibody on the day of infection, and BAL was obtained after sacrifice on day 6. Treg cells among CD4 cells were quantified by i.c. staining against Foxp3. (b) As in (a), but with an additional group of infected mice receiving 106 purified Treg cells from D665-treated uninfected mice i.v. on day one of infection. BAL was analyzed by ELISA for TNF and IL-10 on day 6
Fig. 3.
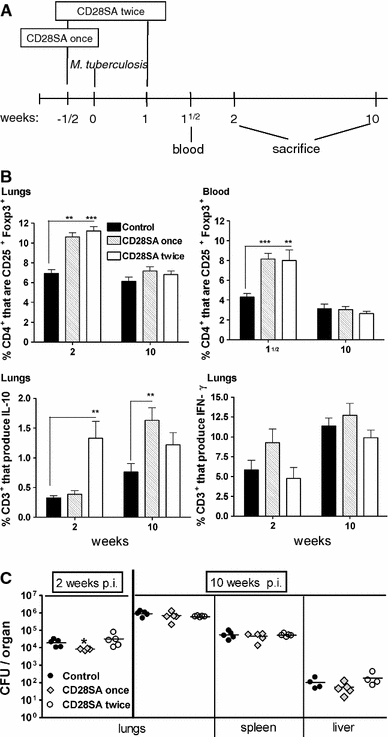
CD28SA treatment during M. tuberculosis infection increases the percentage of CD25+Foxp3+ T-cells but has no major effect on bacterial burden. a BALB/c mice were treated i.p. with CD28 superagonist (CD28SA, D665, 50 μg/mouse) or isotype control (MOCP31) either ½ week prior (CD28SA once) or ½ week prior and 1 week (CD28SA twice) post aerosol with 100 CFU/lung of M. tuberculosis. Mice were bled at 1½ week and killed at 2 and 10 weeks post-infection. b Expression of CD25+ and Foxp3+ on CD4+-gated cells was measured by flow cytometry in collagenase-treated lung and in peripheral blood lymphocytes. The percentage of CD3+ lung cells producing IL-10 and IFN-γ are also represented. Lung cells were harvested following collagenase treatment and stimulated with PMA and ionomycin in the presence of monensin for 4 h and subjected to intracellular staining. c The bacterial load in lungs, spleen, and liver were determined at 2 and 10 weeks post M. tuberculosis infection. Data are presented as log10 CFU per organ. Individual bacterial titers are given together with group medians. Statistical analysis was performed using an unpaired two-tailed Student’s t-test. Data are represented as means ± SEM (n = 5 mice/group; *P < 0.05, **P < 0.01, ***P < 0.001). All data are obtained from one experiment
Fig. 4.
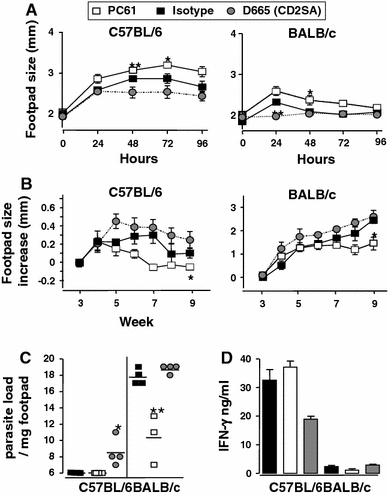
CD28SA dampens while anti-CD25 treatment enhances type-1 immunity during L. major infection. C57BL/6 (n = 4/group) or BALB/c (n = 3 or 4/group) were treated i.p. with CD28 superagonist (D665, 50 μg/mouse), isotype control (MOCP31, 50 μg/mouse) or anti-CD25 (PC61, 100 μg/mouse) from week 4 to week 10 post-infection with 2 × 106L.major LV39 (MRHO/SV/59/P) metacyclic promastigotes into the hind footpad. a DTH responses were assessed every 24 h for 4 d following subcutaneous injection of 10 μg of soluble leishmania antigen (SLA). (B + C) Disease progression was measured by lesion development and parasite load. b Footpad swelling was measured at weekly intervals post-infection and c footpad parasite load determined by limiting dilution of single-cell suspensions from homogenized footpads at week 10 post-infection. d IFN-γ production was measured in popliteal lymphnode cell cultures pulsed with SLA (50 μg/ml) from week 10 L. major–infected mice. Statistical analysis was performed using the Student’s t-test. Data are represented as means ± SD. *P < 0.05, **P < 0.01
Table 1.
Constitutive and conditional IL-4Rα deficient mouse models
| Mutant mouse strains | Cell specificity | Status |
|---|---|---|
| Constitutive mutation | ||
| IL-4Ra−/− | All cells | Mohrs et al. [4] |
| IL-4Ralox/lox | All cells (silent mutation) | Herbert et al. [8] |
| Cell type deficiency | ||
| LysMcreIL-4Ra−/lox | Macrophages and neutrophils | Herbert et al. [8] |
| SMMHCcreIL-4Ra−/lox | Smooth muscle cells | Horsnell et al. [9] |
| LckcreIL-4Ra−/lox | CD4+ T lymphocytes | Leeto et al. [10] |
| iLckcreIL-4Ra−/lox | All T lymphocytes | Dewals et al. [11] |
| CD4creIL-4Ra−/lox | a/b+ TCR T lymphocytes | Unpublished |
| MB1creIL-4Ra−/lox | B lymphocytes | Unpublished |
| CD11ccreIL-4Ra−/lox | Dendritic cells | Unpublished |
| LysMcreLckcreIL-4Ra−/lox | Macrophages/neutrophils/CD4+ T cells | Unpublished |
| Inducible & cell type deficiency | ||
| TetOIL4RaTg | All cells | Unpublished |
| tTA-VAVTgTetOIL4RaTgIL-4Ra−/− | Haematopoetic cells | Unpublished |
Interference with IL-4Rα-mediated cell signaling
The role of alternatively activated macrophages in infectious and inflammatory disease
Macrophage activation can be divided into a classical and an alternative pathway. IFN-γ-induced, classically activated macrophages (caMphs) are indispensable for protective effector responses against intracellular pathogens, particularly due to their highly effective killing effector function. However, excessive inflammatory immune responses mediated by caMphs can also be detrimental to the host. In contrast, IL-4Rα-mediated alternative macrophage activation has been proposed as a possible mechanism to attenuate excessive inflammation. The identification of marker genes [37–42] as well as the generation of macrophage/neutrophil-specific IL-4Rα deficient mice (LysM cre IL-4Rα α-/lox) and subsequent studies of relevant experimental disease models have enabled us to evaluate the role of IL-4Rα-mediated alternative macrophage activation in vivo. Together, these investigations demonstrated the involvement of aaMphs in immuno-modulation and suppression during infectious diseases, like schistosomiasis [8], leishmaniasis [43], cryptococcosis [44–46], as well as during non-infectious inflammatory diseases, like proteoglycan-induced arthritis [46, 47], or experimental autoimmune encephalomyelitis (EAE) [47]. Here, they seem to influence both innate and adaptive immune responses, the latter by suppression of T-cell proliferation [48, 49].
The role of IL-4Rα-responsiveness in helminthic diseases
In helminth infection, like Nippostrongylus and Schistosoma, IL-13 turned out to be the essential factor for efficient expulsion and induction of goblet hyperplasia [8, 50], as well as smooth cell hypercontraction [9]. Further studies in experimental schistosomiasis uncovered, however, that IL-4/IL-13-independent hyperplasia is possible [51], and defined the protective role of alternatively activated macrophages by down-regulation of otherwise overshooting T-cell responses [8]. In addition, the use of mice deficient for IL-4Rα on particular T-cell subsets revealed that both CD4+ Th cells and non-CD4+ T-cells are involved in avoiding early mortality and morbidity during schistosomiasis [10, 11].
Beneficial and detrimental IL-4Rα-responsive cell types in leishmaniasis
Infection studies in CD4+ T-cell-specific IL-4Rα deficient BALB/c mice with Leishmania major, which causes cutaneous leishmaniasis, confirmed the detrimental role of IL-4-promoted Th 2 cells, but also suggested that IL-4Rα-responsive cells are involved in protection against cutaneous leishmaniasis [52]. AaMph’s could be excluded as they rather contribute to susceptibility in BALB/c mice by influencing T-helper responses and facilitating evasion by the parasite [43]. Currently, recently established dendritic cell-specific IL-4Rα deficient mice are employed to test the possibility that DC instruction via IL-4 and/or IL-13 may be the missing link.
Concluding remarks
CD28 and the IL-4Rα highlight the potency of costimulatory receptors and cytokines in fine-tuning immune responses to pathogens while avoiding host damage. While “knockout” and blocking experiments defined important roles for CD28 in T-cell memory and for IL-4R signaling in the activation of an anti-inflammatory type of macrophages, the application of stimulatory CD28-specific mAb makes use of two major host-protective pathways by boosting Treg and Th2 activation, which in turn leads to alternative activation of macrophages. Notably, in several of the infectious disease models we have studied, it was possible to use such mAb treatment to protect the host from immunopathology without compromising pathogen control. This is of potential translational interest because we found that high-dose corticosteroid treatment does not interfere with the activation of Treg cells by CD28SA in vivo [14], opening the possibility of controlling a potential cytokine storm while maintaining the beneficial activation of this host-protective T-cell subset.
Acknowledgments
Work of the authors was supported by the Deutsche Forschungsgemeinschaft through Sonderforschungsbereich 479 and International Research Training Group 1522.
Footnotes
This article is published as part of a Special Issue on Pathogen Variation and Host Response in Infectious Disease.
Contributor Information
Thomas Hünig, Email: huenig@vim.uni-wuerzburg.de.
Fred Lühder, Email: fred.luehder@med.uni-goettingen.de.
Karin Elflein, kleinefee@email.de.
Tea Gogishvili, Email: t_gogishvili@vim.uni-wuerzburg.de.
Monika Fröhlich, Email: Monika.Froehlich@stud-mail.uni-wuerzburg.de.
Reto Guler, Email: Reto.Guler@uct.ac.za.
Frank Brombacher, Email: fbrombac@mweb.co.za.
References
- 1.Kelly-Welch AE, Hanson EM, Boothby MR, Keegan AD. Interleukin-4 and interleukin-13 signaling connections maps. Science. 2003;300(5625):1527–1528. doi: 10.1126/science.1085458. [DOI] [PubMed] [Google Scholar]
- 2.Donaldson DD, Whitters MJ, Fitz LJ, Neben TY, Finnerty H, Henderson SL, O’Hara RM, Jr, Beier DR, Turner KJ, Wood CR, Collins M. The murine IL-13 receptor alpha 2: molecular cloning, characterization, and comparison with murine IL-13 receptor alpha 1. J Immunol. 1998;161(5):2317–2324. [PubMed] [Google Scholar]
- 3.Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med. 2006;12(1):99–106. doi: 10.1038/nm1332. [DOI] [PubMed] [Google Scholar]
- 4.Mohrs M, Ledermann B, Kohler G, Dorfmuller A, Gessner A, Brombacher F. Differences between IL-4- and IL-4 receptor alpha-deficient mice in chronic leishmaniasis reveal a protective role for IL-13 receptor signaling. J Immunol. 1999;162(12):7302–7308. [PubMed] [Google Scholar]
- 5.Brombacher F. The role of interleukin-13 in infectious diseases and allergy. Bioessays. 2000;22(7):646–656. doi: 10.1002/1521-1878(200007)22:7<646::AID-BIES7>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 6.Shahinian A, Pfeffer K, Lee KP, Kündig TM, Kishihara K, Wakeham A, Kawai K, Ohashi PS, Thompson CB, Mak TW. Differential T cell costimulatory requirements in CD28-deficient mice. Science. 1993;261:609. doi: 10.1126/science.7688139. [DOI] [PubMed] [Google Scholar]
- 7.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4 + CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12(4):431–440. doi: 10.1016/S1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 8.Herbert DR, Holscher C, Mohrs M, Arendse B, Schwegmann A, Radwanska M, Leeto M, Kirsch R, Hall P, Mossmann H, Claussen B, Forster I, Brombacher F. Alternative macrophage activation is essential for survival during schistosomiasis and downmodulates T helper 1 responses and immunopathology. Immunity. 2004;20(5):623–635. doi: 10.1016/S1074-7613(04)00107-4. [DOI] [PubMed] [Google Scholar]
- 9.Horsnell WG, Cutler AJ, Hoving JC, Mearns H, Myburgh E, Arendse B, Finkelman FD, Owens GK, Erle D, Brombacher F. Delayed goblet cell hyperplasia, acetylcholine receptor expression, and worm expulsion in SMC-specific IL-4Ralpha-deficient mice. PLoS Pathog. 2007;3(1):e1. doi: 10.1371/journal.ppat.0030001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leeto M, Herbert DR, Marillier R, Schwegmann A, Fick L, Brombacher F. TH1-dominant granulomatous pathology does not inhibit fibrosis or cause lethality during murine schistosomiasis. Am J Pathol. 2006;169(5):1701–1712. doi: 10.2353/ajpath.2006.060346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dewals B, Hoving JC, Leeto M, Marillier RG, Govender U, Cutler AJ, Horsnell WG, Brombacher F. IL-4Ralpha responsiveness of non-CD4 T cells contributes to resistance in schistosoma mansoni infection in pan-T cell-specific IL-4Ralpha-deficient mice. Am J Pathol. 2009;175(2):706–716. doi: 10.2353/ajpath.2009.090137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dennehy KM, Elias F, Zeder-Lutz G, Ding X, Altschuh D, Luhder F, Hunig T. Cutting edge: monovalency of CD28 maintains the antigen dependence of T cell costimulatory responses. J Immunol. 2006;176(10):5725–5729. doi: 10.4049/jimmunol.176.10.5725. [DOI] [PubMed] [Google Scholar]
- 13.Beyersdorf N, Ding X, Blank G, Dennehy KM, Kerkau T, Hunig T. Protection from graft-versus-host disease with a novel B7 binding site-specific mouse anti-mouse CD28 monoclonal antibody. Blood. 2008;112(10):4328–4336. doi: 10.1182/blood-2008-03-146662. [DOI] [PubMed] [Google Scholar]
- 14.Gogishvili T, Elias F, Emery JL, McPherson K, Okkenhaug K, Hunig T, Dennehy KM. Proliferative signals mediated by CD28 superagonists require the exchange factor Vav1 but not phosphoinositide 3-kinase in primary peripheral T cells. Eur J Immunol. 2008;38(9):2528–2533. doi: 10.1002/eji.200838223. [DOI] [PubMed] [Google Scholar]
- 15.Dolfi DV, Katsikis PD. CD28 and CD27 costimulation of CD8+ T cells: a story of survival. Adv Exp Med Biol. 2007;590:149–170. doi: 10.1007/978-0-387-34814-8_11. [DOI] [PubMed] [Google Scholar]
- 16.Dooms H, Abbas AK. Control of CD4+ T-cell memory by cytokines and costimulators. Immunol Rev. 2006;211:23–38. doi: 10.1111/j.0105-2896.2006.00387.x. [DOI] [PubMed] [Google Scholar]
- 17.Borowski AB, Boesteanu AC, Mueller YM, Carafides C, Topham DJ, Altman JD, Jennings SR, Katsikis PD. Memory CD8+ T cells require CD28 costimulation. J Immunol. 2007;179(10):6494–6503. doi: 10.4049/jimmunol.179.10.6494. [DOI] [PubMed] [Google Scholar]
- 18.Kursar M, Bonhagen K, Fensterle J, Kohler A, Hurwitz R, Kamradt T, Kaufmann SH, Mittrucker HW. Regulatory CD4 + CD25+ T cells restrict memory CD8+ T cell responses. J Exp Med. 2002;196(12):1585–1592. doi: 10.1084/jem.20011347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tacke M, Hanke G, Hanke T, Hunig T. CD28-mediated induction of proliferation in resting T cells in vitro and in vivo without engagement of the T cell receptor: evidence for functionally distinct forms of CD28. Eur J Immunol. 1997;27(1):239–247. doi: 10.1002/eji.1830270136. [DOI] [PubMed] [Google Scholar]
- 20.Luhder F, Huang Y, Dennehy KM, Guntermann C, Muller I, Winkler E, Kerkau T, Ikemizu S, Davis SJ, Hanke T, Hunig T. Topological requirements and signaling properties of T cell-activating, anti-CD28 antibody superagonists. J Exp Med. 2003;197(8):955–966. doi: 10.1084/jem.20021024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evans EJ, Esnouf RM, Manso-Sancho R, Gilbert RJ, James JR, Yu C, Fennelly JA, Vowles C, Hanke T, Walse B, Hunig T, Sorensen P, Stuart DI, Davis SJ. Crystal structure of a soluble CD28-Fab complex. Nat Immunol. 2005;6(3):271–279. doi: 10.1038/ni1170. [DOI] [PubMed] [Google Scholar]
- 22.Rodriguez-Palmero M, Hara T, Thumbs A, Hünig T. Triggering of T-cell proliferation through CD28 induces GATA-3 and promotes T-helper type 2 differentiation in vitro and in vivo. Eur J Immunol. 1999;29:3914–3924. doi: 10.1002/(SICI)1521-4141(199912)29:12<3914::AID-IMMU3914>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 23.Lin C-H, Hunig T. Efficient expansion of regulatory T-cells in vitro and in vivo with a CD28 superagonist. Eur J Immunol. 2003;33:626–638. doi: 10.1002/eji.200323570. [DOI] [PubMed] [Google Scholar]
- 24.Gogishvili T, Langenhorst D, Luhder F, Elias F, Elflein K, Dennehy KM, Gold R, Hunig T. Rapid regulatory T-cell response prevents cytokine storm in CD28 superagonist treated mice. PLoS ONE. 2009;4(2):e4643. doi: 10.1371/journal.pone.0004643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hunig T. Manipulation of regulatory T-cell number and function with CD28-specific monoclonal antibodies. Adv Immunol. 2007;95:111–148. doi: 10.1016/S0065-2776(07)95004-X. [DOI] [PubMed] [Google Scholar]
- 26.Beaudette-Zlatanova BC, Whalen B, Zipris D, Yagita H, Rozing J, Groen H, Benjamin CD, Hunig T, Drexhage HA, Ansari MJ, Leif J, Mordes JP, Greiner DL, Sayegh MH, Rossini AA. Costimulation and autoimmune diabetes in BB rats. Am J Transplant. 2006;6(5 Pt 1):894–902. doi: 10.1111/j.1600-6143.2005.01227.x. [DOI] [PubMed] [Google Scholar]
- 27.Beyersdorf N, Gaupp S, Balbach K, Schmidt J, Toyka KV, Lin CH, Hanke T, Hunig T, Kerkau T, Gold R. Selective targeting of regulatory T cells with CD28 superagonists allows effective therapy of experimental autoimmune encephalomyelitis. J Exp Med. 2005;202(3):445–455. doi: 10.1084/jem.20051060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Azuma H, Isaka Y, Li X, Hunig T, Sakamoto T, Nohmi H, Takabatake Y, Mizui M, Kitazawa Y, Ichimaru N, Ibuki N, Ubai T, Inamoto T, Katsuoka Y, Takahara S. Superagonistic CD28 antibody induces donor-specific tolerance in rat renal allografts. Am J Transplant. 2008;8(10):2004–2014. doi: 10.1111/j.1600-6143.2008.02358.x. [DOI] [PubMed] [Google Scholar]
- 29.Beyersdorf N, Ding X, Hunig T, Kerkau T. Superagonistic CD28 stimulation of allogeneic T cells protects from acute graft-versus-host disease. Blood. 2009;114(20):4575–4582. doi: 10.1182/blood-2009-04-218248. [DOI] [PubMed] [Google Scholar]
- 30.Kitazawa Y, Fujino M, Li XK, Xie L, Ichimaru N, Okumi M, Nonomura N, Tsujimura A, Isaka Y, Kimura H, Hunig T, Takahara S. Superagonist CD28 antibody preferentially expanded Foxp3-expressing nTreg cells and prevented graft-versus-host diseases. Cell Transplant. 2009;18(5):627–637. doi: 10.1177/096368970901805-619. [DOI] [PubMed] [Google Scholar]
- 31.Kitazawa Y, Fujino M, Sakai T, Azuma H, Kimura H, Isaka Y, Takahara S, Hunig T, Abe R, Li XK. Foxp3-expressing regulatory T cells expanded with CD28 superagonist antibody can prevent rat cardiac allograft rejection. J Heart Lung Transplant. 2008;27(4):362–371. doi: 10.1016/j.healun.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 32.Elflein K, Rodriguez-Palmero M, Kerkau T, Hunig T. Rapid recovery from T lymphopenia by CD28 superagonist therapy. Blood. 2003;102(5):1764–1770. doi: 10.1182/blood-2002-11-3586. [DOI] [PubMed] [Google Scholar]
- 33.Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355(10):1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 34.Rodriguez-Palmero M, Franch A, Castell M, Pelegri C, Perez-Cano FJ, Kleinschnitz C, Stoll G, Hunig T, Castellote C. Effective treatment of adjuvant arthritis with a stimulatory CD28-specific monoclonal antibody. J Rheumatol. 2006;33(1):110–118. [PubMed] [Google Scholar]
- 35.Guilliams M, Bosschaerts T, Herin M, Hunig T, Loi P, Flamand V, Baetselier P, Beschin A. Experimental expansion of the regulatory T cell population increases resistance to African trypanosomiasis. J Infect Dis. 2008;198(5):781–791. doi: 10.1086/590439. [DOI] [PubMed] [Google Scholar]
- 36.Scott P, Natovitz P, Coffman RL, Pearce E, Sher A. Immunoregulation of cutaneous leishmaniasis. T cell lines that transfer protective immunity or exacerbation belong to different T helper subsets and respond to distinct parasite antigens. J Exp Med. 1988;168(5):1675–1684. doi: 10.1084/jem.168.5.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raes G, Baetselier P, Noel W, Beschin A, Brombacher F, Hassanzadeh Gh G. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. J Leukoc Biol. 2002;71(4):597–602. [PubMed] [Google Scholar]
- 38.Mountford AP, Hogg KG, Coulson PS, Brombacher F. Signaling via interleukin-4 receptor alpha chain is required for successful vaccination against schistosomiasis in BALB/c mice. Infect Immun. 2001;69(1):228–236. doi: 10.1128/IAI.69.1.228-236.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raes G, Brys L, Dahal BK, Brandt J, Grooten J, Brombacher F, Vanham G, Noel W, Bogaert P, Boonefaes T, Kindt A, Bergh R, Leenen PJ, Baetselier P, Ghassabeh GH. Macrophage galactose-type C-type lectins as novel markers for alternatively activated macrophages elicited by parasitic infections and allergic airway inflammation. J Leukoc Biol. 2005;77(3):321–327. doi: 10.1189/jlb.0304212. [DOI] [PubMed] [Google Scholar]
- 40.Linehan SA, Coulson PS, Wilson RA, Mountford AP, Brombacher F, Martinez-Pomares L, Gordon S. IL-4 receptor signaling is required for mannose receptor expression by macrophages recruited to granulomata but not resident cells in mice infected with Schistosoma mansoni. Lab Invest. 2003;83(8):1223–1231. doi: 10.1097/01.LAB.0000081392.93701.6F. [DOI] [PubMed] [Google Scholar]
- 41.Noel W, Hassanzadeh G, Raes G, Namangala B, Daems I, Brys L, Brombacher F, Baetselier PD, Beschin A. Infection stage-dependent modulation of macrophage activation in trypanosoma congolense-resistant and -susceptible mice. Infect Immun. 2002;70(11):6180–6187. doi: 10.1128/IAI.70.11.6180-6187.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghassabeh GH, Baetselier P, Brys L, Noel W, Ginderachter JA, Meerschaut S, Beschin A, Brombacher F, Raes G. Identification of a common gene signature for type II cytokine-associated myeloid cells elicited in vivo in different pathologic conditions. Blood. 2006;108(2):575–583. doi: 10.1182/blood-2005-04-1485. [DOI] [PubMed] [Google Scholar]
- 43.Hölscher C, Arendse B, Schwegmann A, Myburgh E, Brombacher F. Impairment of alternative macrophage activation delays cutaneous leishmaniasis in non-healing BALB/c mice. J Immunol. 2005;176:1115–1121. doi: 10.4049/jimmunol.176.2.1115. [DOI] [PubMed] [Google Scholar]
- 44.Muller U, Stenzel W, Kohler G, Werner C, Polte T, Hansen G, Schutze N, Straubinger RK, Blessing M, McKenzie AN, Brombacher F, Alber G. IL-13 induces disease-promoting type 2 cytokines, alternatively activated macrophages and allergic inflammation during pulmonary infection of mice with Cryptococcus neoformans. J Immunol. 2007;179(8):5367–5377. doi: 10.4049/jimmunol.179.8.5367. [DOI] [PubMed] [Google Scholar]
- 45.Stenzel W, Muller U, Kohler G, Heppner FL, Blessing M, McKenzie AN, Brombacher F, Alber G. IL-4/IL-13-dependent alternative activation of macrophages but not microglial cells is associated with uncontrolled cerebral cryptococcosis. Am J Pathol. 2009;174(2):486–496. doi: 10.2353/ajpath.2009.080598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cao Y, Brombacher F, Tunyogi-Csapo M, Glant TT, Finnegan A. Interleukin-4 regulates proteoglycan-induced arthritis by specifically suppressing the innate immune response. Arthritis Rheum. 2007;56(3):861–870. doi: 10.1002/art.22422. [DOI] [PubMed] [Google Scholar]
- 47.Keating P, O’Sullivan D, Tierney JB, Kenwright D, Miromoeini S, Mawasse L, Brombacher F, La Flamme AC. Protection from EAE by IL-4Ralpha(-/-) macrophages depends upon T regulatory cell involvement. Immunol Cell Biol. 2009;87(7):534–545. doi: 10.1038/icb.2009.37. [DOI] [PubMed] [Google Scholar]
- 48.Brys L, Beschin A, Raes G, Ghassabeh GH, Noel W, Brandt J, Brombacher F, Baetselier P. Reactive oxygen species and 12/15-lipoxygenase contribute to the antiproliferative capacity of alternatively activated myeloid cells elicited during helminth infection. J Immunol. 2005;174(10):6095–6104. doi: 10.4049/jimmunol.174.10.6095. [DOI] [PubMed] [Google Scholar]
- 49.Pesce JT, Ramalingam TR, Wilson MS, Mentink-Kane MM, Thompson RW, Cheever AW, Urban JF, Jr, Wynn TA. Retnla (relmalpha/fizz1) suppresses helminth-induced Th2-type immunity. PLoS Pathog. 2009;5(4):e1000393. doi: 10.1371/journal.ppat.1000393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barner M, Mohrs M, Brombacher F, Kopf M. Differences between IL-4R alpha-deficient and IL-4-deficient mice reveal a role for IL-13 in the regulation of Th2 responses. Curr Biol. 1998;8(11):669–672. doi: 10.1016/S0960-9822(98)70256-8. [DOI] [PubMed] [Google Scholar]
- 51.Marillier RG, Michels C, Smith EM, Fick LC, Leeto M, Dewals B, Horsnell WG, Brombacher F. IL-4/IL-13 independent goblet cell hyperplasia in experimental helminth infections. BMC Immunol. 2008;9:11. doi: 10.1186/1471-2172-9-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Radwanska M, Cutler AJ, Hoving JC, Magez S, Holscher C, Bohms A, Arendse B, Kirsch R, Hunig T, Alexander J, Kaye P, Brombacher F. Deletion of IL-4Ralpha on CD4 T cells renders BALB/c mice resistant to Leishmania major infection. PLoS Pathog. 2007;3(5):e68. doi: 10.1371/journal.ppat.0030068. [DOI] [PMC free article] [PubMed] [Google Scholar]