

Abstract
Introduction
Four benzamide analogs having a high affinity and selectivity for D3 versus D2 receptors were radiolabeled with 11C or 18F for in vivo evaluation.
Methods
Precursors were synthesized and the four D3 selective benzamide analogs were radiolabeled. The tissue distribution and brain uptake of the four compounds were evaluated in control rats and rats pretreated with cyclosporin A, a modulator of P-glycoprotein and an inhibitor of other ABC efflux transporters that contribute to the blood brain barrier. MicroPET imaging was carried out for [11C]6 in a control and a cyclosporin A pre-treated rat.
Results
All four compounds showed low brain uptake in control rats at 5 and 30 min post-injection; despite recently reported rat behavioral studies conducted on analogs 6 (WC-10) and 7 (WC-44). Following administration of cyclosporin A, increased brain uptake was observed with all four PET radiotracers at both 5 and 30 min post-i.v. injection. An increase in brain uptake following modulation/inhibition of the ABC transporters was also observed in the microPET study.
Conclusions
These data suggest that D3 selective conformationally-flexible benzamide analogs which contain a N-2-methoxyphenylpiperazine moiety are substrates for P-glycoprotein or other ABC transporters expressed at the blood-brain barrier, and that PET radiotracers containing this pharmacophore may display low brain uptake in rodents due to the action of these efflux transporters.
Keywords: D3 receptors, PET, radiotracer, P-glycoprotein
Introduction
The two major classes of dopamine receptors, the D1-like and D2-like receptors, include 5 different subtypes. These G-protein receptors differ in their pharmacology and distribution within the central nervous system. The D1-like receptor subtypes include D1 (rat D1a) and D5 (rat D1b) receptors while the D2-like receptor class consists of D2, D3, and D4 receptors. Stimulation of D1-like receptors results in an activation of adenylate cyclase while agonist stimulation of D2-like receptors results in an inhibition of adenylyl cyclase activity, an increase in the release of arachadonic acid, and an increase in phosphatidylinositol hydrolysis [1–3].
It has been recognized that the D3 subtype receptors play an important role in a number of neurological and neuropsychiatric disorders [4, 5]. The high density of D3 receptors in limbic regions suggests that this receptor may play an important role in pathological abnormalities associated with dysregulation of the dopaminergic system [6–9]. D3-selective antagonists and partial agonists have been proposed as atypical antipsychotics and therapeutics devoid of extrapyramidal effects [10–17] and also as potential therapeutic agents to manage drug dependency [18–21]. Prolonged treatment of 6-hydroxydopamine lesioned rats, a rodent model of Parkinson’s Disease, with L-DOPA results in the development of L-DOPA-induced dyskinesia, which may be caused by an upregulation of D3 receptors [22–24]. D3-selective antagonists and partial agonists have been shown to be effective in blocking L-DOPA-induced dyskinesia in 6-hydroxydopamine lesioned rodents, indicating that the D3 receptor is an important target in the treatment of Parkinson’s Disease [2, 25–28]. Finally, the positive reinforcing effects of psychostimulants such as cocaine and methamphetamine are thought to be primarily mediated by the D3 receptor, and D3-selective partial agonists and antagonists may be useful in the treatment of substance abuse [27, 29–33].
A number of D3 receptor selective ligands described in the literature have served as lead compounds for radiotracer development. Unfortunately, none of the D3-selective PET radiotracers reported to date (Fig. 1) have shown promise in preliminary brain uptake studies in rodents or in in vivo imaging studies [34–38]. Over the past decade, our group has focused on the development of PET radiotracers for imaging the D3 receptor using the benzamide analogue 5 as a lead compound [39]. This research has led to the identification of a series of potential carbon-11 and fluorine-18 labeled PET radiotracers for imaging D3 receptors (Fig. 2) [40, 41]. The advantages of compounds 6 – 9 (Fig. 2 and Table 1) is that, in addition to having a high D3 affinity (0.17 – 2.4 nM) and good selectivity for D3 versus D2 receptors (selectivity ratios ranging from 23 – 163), these compounds have a calculated log P value within the range that is predictive of its ability to cross the blood-brain barrier and bind to dopamine D3 receptors in vivo. [3H]6 has been validated as a useful tool for in vitro studies of the dopamine receptor [42, 43]. Recent behavioral studies also indicate that the antagonist 6 (also known as WC-10) and the agonist 7 (also known as WC-44) are biologically active in rat models of prepulse inhibition and L-DOPA-induced dyskinesia [25, 26, 44].
Fig. 1.

Structures of dopamine D3 receptor PET imaging ligands reported in the literature.
Fig. 2.

Structures of lead compound 5 and the N-phenylpiperazine analogs described in this paper.
Table 1.
In vitro Binding Data
| Compound | D2 | D3 | D4 | D2:D3 | Log P | %IA D2 | %IA D3 |
|---|---|---|---|---|---|---|---|
| 6 (WC-10) | 34.4 ± 3.3 | 0.8 ± 0.1 | 896 ± 193 | 43 | 3.09 | 34 ± 3 | 19 ± 2 |
| 7 (WC-44) | 54.7 ± 4.4 | 2.4 ± 0.4 | 804 ± 33 | 23 | 2.94 | 35 ± 1 | 96 ± 4 |
| 8 | 15.1 ± 1.7 | 0.65 ± 0.2 | 886 ± 101 | 23 | 3.75 | 66.3 ± 1 | 64.5 ± 8 |
| 9 | 27.7 ± 5.4 | 0.17 ± 0.01 | 246 ± 13.3 | 163 | 4.67 | 29.3 ± 7.3 | 34.4 ± 1.7 |
The goal of this study was to prepare the 11C- and 18F-labeled versions of compounds 6 – 9 and to measure their rodent brain uptake as a preliminary step to PET imaging studies in nonhuman primates. Initial studies with [11C]6 showed surprisingly low brain uptake in rats, which suggested that the radiotracer was excluded from the CNS by the blood-brain barrier. However, pretreatment of rats with cyclosporin A (CycA), a nonspecific modulator/inhibitor of the ATP-binding cassette (ABC) transporters including P-glycoprotein or P-gp (ABCB1 transporter), multi-drug resistant protein (MRP) MRP1 (ABCC1 transporter) and MRP2 (ABCC2 transporter) [45], resulted in a dramatic increase in brain uptake of all four D3 radiotracers. These data suggest that the conformationally-flexible benzamide compounds as represented by compounds 6 – 9, are substrates for P-gp or other ABC transporters expressed at the blood-brain barrier, which may explain the negative data obtained with radiolabeled congeners when rats were used for screening these potential D3 receptor radioligands for their ability cross the blood-brain barrier in vivo.
2. Materials and Methods
2.1. Chemistry
2.1.1. General
All analytical grade chemicals and reagents were purchased from Sigma-Aldrich (Milwaukee, WI) and were used without further purification unless otherwise stated. Sandimmune cyclosporine (CycA) (Novartis Pharmaceuticals Corporation, New Jersey) was purchased through the institutional veterinary pharmacy. Flash column chromatography was conducted using Scientific Adsorbents, Inc. silica gel, 60 Å, “40 Micron Flash” (32–63 microns). When the reactions involved extraction with dichloromethane (CH2Cl2), ethyl acetate (EtOAc), or ethyl ether (Et2O), the organic solutions were dried with anhydrous Na2SO4 and concentrated with a rotary evaporator under reduced pressure. Yields were not optimized. Melting points were determined on a Haake-Buchler or Mel-Temp melting point apparatus and are uncorrected. 1H NMR spectra were recorded at 300 MHz on a Varian Mercury-VX spectrometer. All chemical shift values are reported in ppm (δ). The following abbreviations were used to describe peak patterns wherever appropriate: b = broad, d = doublet, t = triplet, q = quartet, m=multiplet. Elemental analyses (C, H, N) were determined by Atlantic Microlab, Inc. Log P values were calculated using the program ACD Log P Suite 7.0 (Advanced Chemistry, Toronto, Ontario, Canada).
The synthesis and in vitro characterization of benzamide analogs 4-(dimethylamino)-N-(4-(4-(2-methoxyphenyl)piperazin-1-yl)butyl)benzamide (6), 4-(2-fluoroethyl)-N-(4-(4-(2-methoxyphenyl)piperazin-1-yl)butyl)benzamide (7), 4-(dimethylamino)-N-(4-(4-(2-(2-fluoroethoxy)phenyl)piperazin-1-yl)butyl)benzamide(8) and (N-(4-(4-(2-(2-fluoroethoxy)phenyl)piperazin-1-yl)butyl)-4-(thiophen-3-yl)benzamide (9) have been reported previously [40, 41]. The synthesis of intermediates 2-(4-(4-(2-methoxyphenyl)piperazin-1-yl)butyl)isoindoline-1,3-dione (11a) and 2-(4-(4-(2-hydroxyphenyl)piperazin-1-yl)butyl)isoindoline-1,3-dione (11b), 4-(4-(2-methoxyphenyl)piperazin-1-yl)butan-1-amine (12a), 2-(4-(4-aminobutyl)piperazin-1-yl)phenol (12b) and of the intermediate/precursor 4-(dimethylamino)-N-(4-(4-(2-hydroxyphenyl)piperazin-1-yl)butyl)benzamide (13b) from 1-(2-methoxyphenyl)piperazine (10a) and 1-(2-hydroxyphenyl)piperazine, (10b), were previously reported by our group [41, 42].
4-(2-Hydroxyethyl)-N-(4-(4-(2-methoxyphenyl)piperazin-1-yl)butyl)benzamide (13a)
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) (288 mg, 1.5 mmol) was added to a mixture of 4-(4-(2-methoxyphenyl)piperazin-1-yl)butan-1-amine, 12a (263 mg, 1.0 mmol), 4-(2-hydroxyethyl)benzoic acid (249 mg, 1.5 mmol), and hydroxybenzotriazole (HOBt) (203 mg, 1.5 mmol) in dichloromethane (10 mL) at 0 °C. The reaction mixture was stirred overnight at room temperature until thin-layer chromatography (TLC) indicated the reaction was complete. Ethyl acetate (75 mL) was added into the reaction mixture which was then washed with saturated sodium bicarbonate aqueous solution (30 mL), water (30 mL), and brine (30 mL). The organic solution was dried over anhydrous sodium sulfate. After concentrating in vacuo, the crude product was purified by silica gel column chromatography with ether/methanol (10/1, v/v) as the mobile phase to afford 13a (145 mg, 35%) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 1.66 (m, 4H), 2.45 (t, 2H), 2.64 (m, 4H), 2.89 (t, 2H), 3.05 (m, 4H), 3.47 (m, 2H), 3.84 (t, 2H), 3.85 (s, 3H), 6.75 (br s, 1H), 7.01–6.84 (m, 4H), 7.27 (d, 2H), 7.70 (d, 2H).
4-(4-(4-(2-Methoxyphenyl)piperazin-1-yl)butylcarbamoyl)phenethyl methanesulfonate (14a)
Triethylamine (61 mg, 0.6 mmol) was added to a solution of 13a (125 mg, 0.30 mmol) and methanesulfonyl chloride (52 mg, 0.45 mmol) in dichloromethane (5 mL) at 0 °C. The ice bath was removed and the reaction mixture was stirred at room temperature overnight until TLC indicated the reaction was complete. Ethyl acetate (50 mL) was added into the reaction mixture and the mixture was washed with water (30 mL), brine (30 mL), and dried over anhydrous sodium sulfate. After concentrating in vacuo, the crude product was purified with by silica gel column chromatography using ether/methanol (10/1, v/v) as the mobile phase to afford 14a (82 mg, 55%) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 1.68 (m, 4H), 2.46 (t, 2H), 2.65 (m, 4H), 2.86 (s, 3H), 3.07 (m, 6H), 3.47 (m, 2H), 3.85 (s, 3H), 4.40 (t, 2H), 7.00–6.84 (m, 5H), 7.28 (d, 2H), 7.73 (d, 2H).
2-(2-(4-(4-(4-(Dimethylamino)benzamido)butyl)piperazin-1-yl)phenoxy)ethyl acetate (14b)
A solution of 13b (342 mg, 0.864 mmol) in acetone (60 mL), 2-bromoethyl acetate (937.5 mg, 5.62 mmol) and K2CO3 (775.56 mg, 5.62 mmol) was stirred at reflux overnight. Volatiles were removed in vacuo and ethyl acetate (100 mL) was added to the residue. The organic mixture was washed with water (1 x 50 mL), saturated aqueous sodium carbonate (1 x 50 mL) and brine (1 x 50 mL). The organic solution was dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The crude product was purified by silica gel column chromatography using ether/methanol (100/10, v/v) as the mobile phase to afford the intermediate 14b 2-(2-(4-(4-(4-(dimethylamino)benzamido)butyl)piperazin-1-yl)phenoxy)ethyl acetate (253 mg, 60%) as a white solid which was used without further purification. 1H NMR (300MHz, CDCl3): δ 1.50 – 1.70 (m, 4H), 2.07 (s, 3H), 2.40 – 2.50 (t, 2H), 2.61 (s, 2H), 3.00 (s, 6H), 3.02 – 3.20 (m, 4H), 3.40 – 3.56 (m, 4H), 4.20 – 4.22 (t, 2H), 4.44 – 4.46 (t, 2H), 6.38 (s, 1H), 6.64 – 6.67 (d, 2H), 6.90 – 6.95 (m, 4H), 7.65 – 7.68 (d, 2H).
4-(Dimethylamino)-N-(4-(4-(2-(2-hydroxyethoxy)phenyl)piperazin-1-yl)butyl)benzamide (15b)
Sodium hydroxide (84 mg, 2.1 mmol) was added into a solution 14b (240 mg, 0.51 mmol) in methanol (6 mL) and water (3 mL) and the mixture was stirred at room temperature for 5 h until TLC indicated the reaction was complete. Water (5 mL) was added and the reaction mixture was extracted with dichloromethane (3 x 20 mL). The organic solution was dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo to give the crude product which was purified by silica gel column chromatography using ether/methanol (85/15, v/v) as the mobile phase to afford 15b (197 mg, 90%) as a white solid. 1H NMR (300 MHz, CDCl3): δ 1.60 – 1.70 (m, 4H), 2.40 – 2.50 (t, 2H), 2.60 – 2.80 (s, 4H), 3.00 (s, 6H), 3.09 (s, 4H), 3.45 – 3.49 (t, 4H), 3.67 (t, 2H), 4.15 (t, 2H), 6.40 (s, 1H), 6.63 – 6.67 (d, 2H), 6.98 – 7.10 (m, 4H), 7.64 – 7.67 (d, 2H), 8.30 (s, 1H).
N-(4-(4-(2-Hydroxyphenyl)piperazin-1-yl)butyl)-4-(thiophen-3-yl)benzamide (13c)
4-(Thiophen-3-yl)benzoic acid (710 mg, 3.48 mmol), N,N'-dicyclohexylcarbodiimide (DCC) (790 mg, 3.83 mmol) and HOBt (520mg, 3.83mmol) were dissolved in 50 ml dichloromethane at room temperature. A solution of 12b (790 mg, 3.16 mmol) in dichloromethane (10 ml) was added into the above solution which was stirred overnight until TLC indicated the reaction was complete. Dichoromethane (150 mL) was added into the above reaction mixture and the solution was washed with saturated sodium carbonate aqueous solution (3 x 60 mL). The organic solution was dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography using dichloromethane/methanol (10/1, v/v) as the mobile phase to afford N-(4-(4-(2-hydroxyphenyl)piperazin-1-yl)butyl)-4-(thiophen-3-yl)benzamide, 13c, (536 mg, 38.7%) as a white solid. Mp: 177–178 °C. 1H NMR (300MHz, CDCl3): δ 1.55 – 1.75 (m, 4H), 2.40 – 2.52 (t, 2H), 2.52 – 2.78 (m, 4H), 2.80 – 2.96 (m, 4H), 3.46 – 3.60 (m, 2H), 6.60 – 6.70 (broad, 1H), 6.77 – 6.86 (t, 1H), 6.90–6.95 (d, 1H), 7.02 – 7.12 (t, 2H), 7.38 – 7.42 (m, 2H), 7.50 – 7.56 (t, 1H), 7.62 – 7.72 (m, 2H), 7.76 – 7.86 (m, 2H).
2-(2-(4-(4-(4-(Thiophen-3-yl)benzamido)butyl)-piperazin-1-yl)phenoxy)ethyl acetate (14c)
2-Bromoethyl acetate (1.33 g, 8 mmol) and K2CO3 (1.10 g, 2.95 mmol) were added to a solution of 13c (0.53 g, 1.22 mmol) in acetone (30 mL) and the mixture was stirred at reflux overnight until TLC indicated the reaction was complete. After concentrating in vacuo, ethyl acetate (100 mL) was added to the residue and the solution was washed with water (50 mL), saturated sodium carbonate aqueous solution (50 mL), and brine (50 mL). The organic solution was dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by silica gel column chromatography with ether/methanol (100/10, v/v) as the mobile phase to afford the intermediate 14c, 2-(2-(4-(4-(4-(thiophen-3-yl)benzamido)butyl)-piperazin-1-yl)phenoxy)ethyl acetate, (0.32 g, 50%) as white solid which was used without further purification. 1H NMR (300 MHz, CDCl3): δ 1.55 – 1.78 (m, 4H), 2.06 (s, 3H), 2.40 – 2.52 (t, 2H), 2.52 – 2.78 (m, 4H), 2.80 – 2.96 (m, 2H), 2.96 – 3.10 (m, 2H), 3.40 – 3.60 (m, 2H), 4.14 – 4.24 (t, 2H), 4.40 – 4.52 (t, 2H), 6.62 – 6.76 (broad, 1H), 6.68 – 7.10 (m, 4H), 7.38 – 7.42 (m, 2H), 7.50 – 7.56 (t, 1H), 7.62 – 7.73 (m, 2H), 7.76 – 7.86 (m, 2H).
N-(4-(4-(2-(2-Hydroxyethoxy)phenyl)piperazin-1-yl)butyl)-4-(thiophen-3-yl)benzamide (15c)
Sodium hydroxide (80 mg, 2 mmol) was added into a solution of 14c (320 mg, 0.57 mmol) in methanol (5 mL) and water (1 mL) and mixture was stirred at room temperature for 5 h until TLC indicated the reaction was complete. Water (5 mL) was added and the mixture was extracted with ether (20 mL x 2). The aqueous layer was acidified with aqueous 6N HCl to pH = 1. The precipitate was filtered to afford 15c (153 mg, 52%) as a white solid which was used without further purification. 1H NMR (300 MHz, CDCl3): δ 1.50 – 1.80 (m, 4H), 2.40 – 2.52 (t, 2H), 2.52 – 2.78 (m, 4H), 2.90 – 3.15 (m, 4H), 3.46 – 3.56 (m, 2H), 3.60 – 3.70 (t, 2H), 4.12 – 4.22 (t, 2H), 5.47 – 5.50 (broad, 1H), 6.66 – 6.76 (broad, 1H), 6.92 – 7.09 (m, 4H), 7.38 – 7.45 (m, 2H), 7.50 – 7.56 (t, 1H), 7.62 – 7.70 (m, 2H), 7.76 – 7.84 (m, 2H).
2-(2-(4-(4-(4-(Dimethylamino)benzamido)butyl)piperazin-1-yl)phenoxy)ethyl methanesulfonate (16b)
Methanesulfonyl chloride (116 mg, 1.01 mmol) was added to a solution of 15b (150 mg, 0.31 mmol) and triethylamine (160 mg, 0.36 mmol) in dichloromethane (20 mL) at 0 °C. The reaction mixture was stirred for 5 h until TLC indicated the reaction was complete. The solution was washed with aqueous sodium bicarbonate solution (50 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography with ether/methanol (10/1, v/v) as the mobile phase to afford 16b (113 mg, 65%) as a colorless solid. Mp: 118–120 °C. 1H NMR (300 MHz, CDCl3): δ 1.50 – 1.80 (m, 2H), 1.80 – 1.95 (m, 2H), 2.60 – 2.70 (t, 2H), 2.72 – 2.90 (m, 4H), 2.99 (s, 3H), 3.00 (s, 3H), 3.08 (s, 3H), 3.32 (br, 4H), 3.48 – 3.51 (t, 2H), 4.26 – 4.28 (t, 2H), 4.58 – 4.62 (t, 2H), 6.65 – 6.68 (d, 2H), 6.80 – 6.90 (d, 2H), 6.96 – 7.10 (m, 4H), 7.77 – 7.80 (d, 2H), 8.27 (s, 1H).
2-(2-(4-(4-(4-(Thiophen-3-yl)benzamido)butyl)piperazin-1-yl)phenoxy)ethyl methanesulfonate (16c)
Methanesulfonyl chloride (72 mg, 0.62 mmol) was added to a solution of 15c (150 mg, 0.31 mmol) and triethylamine (95 mg, 0.94 mmol) in dichloromethane (20 mL) at 0 °C. The reaction mixture was stirred for 8 h at room temperature until TLC indicated the reaction was complete. The solution was washed with aqueous sodium bicarbonate (50 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography using ether/methanol (10/1, v/v) as the mobile phase to afford 16c (115 mg, 66%) as a white solid. Mp: 125–127 °C. 1H NMR (300MHz, CDCl3) is δ 1.60–1.80 (m, 4H), 2.42–2.52 (t, 2H), 2.54–2.74 (m, 4H), 2.94–3.15 (m, 7H), 3.46–3.56 (m, 2H), 4.24–4.32 (t, 2H), 4.56–4.66 (t, 2H), 6.80–7.02 (m, 5H), 7.38–7.45 (m, 2H), 7.50–7.56 (t, 1H), 7.62–7.70 (m, 2H), 7.77–7.84 (m, 2H).
2.2. Radiochemistry
2.2.1. Production of [11C]Methyl Iodide
[11C]CO2 was produced at the Washington University School of Medicine Cyclotron Facility using a JSW BC-16/8 cyclotron by irradiating a gas target of 0.2% O2 in N2 for 15–30 min with a 40 μA beam of 16 MeV protons. [11C]MeI was produced from [11C]CO2 using a GE PETtrace MeI Microlab synthesis module. Approximately 12 min following the end-of-bombardment (E.O.B.), 800–1000 mCi of [11C]MeI was delivered in the gas phase to the hot cell where the radiosynthesis was accomplished.
2.2.2. Radiochemical Synthesis of [11C]6
The synthesis of [11C]6 (Scheme 2) was performed in a gantry system equipped with air-activated valves for remote manipulation of delivery and transfers of solutions. The HPLC system consisted of a Rheodyne injector valve with 2.0 ml sample loop, a Thermo Separations P200 HPLC binary pump, a Spectra Physics Spectra 100UV variable detector (237 nm), a Bioscan Flow-Count radioactivity detector (NaI crystal), a dual-pen chart recorder and a three-way collection valve. [11C]MeI was bubbled for a period of 5–6 min into a solution of 0.7 – 1.0 mg precursor 13b in DMSO (0.18 ml) containing 2 μl 5N NaOH at room temperature. When the transfer of radioactivity was complete, the sealed reaction vessel was heated to 85 °C for 5 min. The reaction vessel was then removed from the oil bath, and 1.8 ml of HPLC mobile phase was added to the reaction vessel. The residue was injected onto a reversed phase Alltech Econosil C-18 semi-preparative HPLC column (250 mm × 10 mm, 10Å) system, UV 237 nm, with a 5 mL injection loop. The HPLC mobile phase was 35% acetonitrile and 65% 0.1 M ammonium formate buffer (0.1 M ammonium formate with 1 ml 90% formic acid dissolved into 1 liter milli-Q water, pH 4.0 – 4.5) at a 4 ml/min flow rate. Under these conditions, the radioactive [11C]6 was collected from 15 to 17 min in a 50 mL round bottom flask. After concentration in vacuo, the residue was diluted with 10% ethanol in saline. The saline solution of [11C]6 was filtered through a 0.22-μm pyrogen-free Millipore filter into a 10 mL dose vial. The total synthesis time was 50–55 min. A 100 μl sample was sent to the quality control (QC) laboratory for determination of purity and specific activity. The QC analytical HPLC system consisted of an Alltech Econosil reversed phase C18 column (250 × 4.6 mm). The mobile phase was 30% acetonitrile and 70% 0.1M ammonium formate buffer (described above) at 1.5 ml/minute flow rate. Under these conditions the radiolabeled product was eluted at 10.5 min and was authenticated by co-eluting with nonradioactive standard solution of 6. The yield was 60% based on the amount of [11C]MeI at the start of the N-alkylation step. The radiochemical purity was > 99%. The specific activity was > 10 Ci/μmol (decay corrected to E.O.B, N = 15).
Scheme 2.

2.2.3. Radiochemical Synthesis of 18F-labeled analogues
Procedure A: General Method for making the Substituted Benzoic Acid Amide Analogues with 18F: 4-(2-[18F]-fluoroethyl)-N-(4-(4-(2-methoxyphenyl)piperazin-1-yl)butyl)benzamide ([18F]7)
[18F]Fluoride was produced from 95% enriched [18O] by the 18O(p, n) 18F nuclear reaction using a JSW BC16/8 cyclotron (The Japan Steel Works Ltd, Tokyo, Japan) or CS-15 cyclotron (The Cyclotron Corp., Berkeley, CA). An anion–exchange column converted into the carbonate ion form was used to separate the enriched [18O]water from the [18F]Fluoride radioactivity.
18F-fluoride (100 – 150 mCi) was added to a 10-mL Pyrex screw cap tube containing 5–6 mg of Kryptofix 222 and 1.5 ~ 2.0 mg of K2CO3. Using HPLC grade acetonitrile (3 x 1.0 mL), the water was azeotropically evaporated from this mixture in an oil bath at 110 °C under a stream of argon. After all water was removed, a solution of the precursor 14a (1.5 – 2.0 mg) in 0.2 mL anhydrous dimethyl sulfoxide (DMSO) was added to a reaction vessel containing the 18F/Kryptofix mixture. A 3 mm glass bead was added to the reaction vessel to insure a more homogeneous heat distribution when the sample was microwave irradiated, and the vessel was firmly capped using a custom designed remotely operated capping station. After vortexing, the reaction mixture was irradiated with microwaves for 30–40 sec at medium power (60 Watts). Radio-TLC (25% methanol and 75% dichloromethane mobile phase) indicated the incorporation yield was 40–60%.
After cooling the reaction mixture, the radioactivity was loaded onto a normal phase light weight Alumina N Sep-Pak cartridge (Waters). Using 3 ml of the HPLC mobile phase (10% tetrahydrofuran (THF): 12% acetonitrile: 78% 0.1 M ammonium formate buffer (6.32 mg ammonium formate and 1 ml of 90% formic acid dissolved in 1 liter milli-Q water, pH = 4 ~ 4.5), the radioactivity was eluted and the unreacted [18F]Fluoride was retained on the alumina N Sep-Pak. The eluted radioactivity was loaded on a C-18 Alltech Econosil semi-preparative HPLC column (10 μÅ, 250 × 10 mm). The product was eluted with HPLC mobile phase (above) at 4.0 ml/minute flow rate. The retention time of [18F]7 was ~ 21 min and the retention time for the precursor 14a was 27.8 min. 40 ml of Milli-Q water was added into the HPLC collection. The mixture was loaded onto a reverse phase C-18 Sep-Pak Light cartridge (Waters) to remove the HPLC mobile phase. The trapped radioactive product was eluted with 0.6 mL ethanol into a vial and 5.4 mL 0.9% saline was added to get a 6 mL dose of 10% ethanol/saline solution. The solution was filtered through a 0.22-μm sterile filter. A 100 μL aliquot of sample was reserved for quality control. The entire procedure took approximately 100 min.
Quality control analysis was performed on an analytical HPLC system that consisted of an Alltech Econosil reversed phase C-18 column (250 × 4.6 mm), UV 254 nm. The mobile phase consisted of 19% tetrahydrofuran and 81% 0.1 M ammonium formate buffer at pH 4.0 – 4.5. At a flow rate of 1.4 mL/min, [18F]7 eluted at 10.8 min. The identity of [18F]7 was confirmed by co-injection of a nonradioactive standard solution of 7. The radiochemical purity was > 99%. The labeling yield was ~30% (decay corrected), and the specific activity was ~ 2000 Ci/mmol (N =7).
4-(dimethylamino)-N-(4-(4-(2-(2-[18F]fluoroethoxy)phenyl)piperazin-1-yl)butyl)benzamide ([18F]8)
Compound [18F]8 was prepared from precursor 16b as described above for [18F]7 with the following modifications. A capped reaction vessel was heated for 10 min at 110 °C instead of using microwave irradiation. The semi-preparative HPLC column was Phenomenex Luna C-18(2) (10 μÅ, 250 × 10 mm). The mobile phase was 45% methanol and 55% 0.1 M ammonium formate buffer. At a flow rate of 4.0 mL/min, the [18F]8 eluted at ~23 min with a radiochemical purity >99%. The labeling yield was ~20% (decay corrected), and the specific activity was ~ 2000 Ci/mmol (N = 10). The entire procedure took 2 h. The identity of [18F]8 was confirmed by co-injection of a nonradioactive standard solution of 8 on the analytical HPLC system with a mobile phase of 50% methanol and 50% 0.1 M ammonium formate buffer. At a flow rate of 1.0 mL/min, the two compounds co-eluted with a retention time of 6.7 min.
N-(4-(4-(2-(2-[18F]fluoroethoxy)phenyl)piperazin-1-yl)butyl)-4-(thiophen-3-yl)benzamide ([18F]9)
Compound [18F]9 was prepared from precursor 16c as described above for [18F]7 with the following modifications. The capped reaction vessel was heated for 5 min at 85 °C instead of using microwave irradiation. The semi-preparative HPLC column was Phenomenex Luna C-18(2) (10 μÅ, 250 × 10 mm). The mobile phase was 52% methanol and 48% 0.1 M ammonium formate buffer. At a flow rate of 4.0 mL/min, the [18F]9 eluted at 37 min with a radiochemical purity >98%. The labeling yield was approximately 23% (decay corrected), and the specific activity was ~ 2000 Ci/mmol (N = 15). The entire procedure took approximately 2 h. The identity of [18F]9 was confirmed by co-injection of a nonradioactive standard solution of 9 on the analytical HPLC system with a mobile phase of 75% methanol and 25% 0.1 M ammonium formate buffer. At a flow rate of 1.2 mL/min, the two compounds co-eluted with a retention time of ~3.44 min.
2.3. In Vitro Assays
2.3.1. Dopamine receptor binding assays
Receptor binding studies were conducted using a filtration binding assay as previously described [40]. For human D2long, D3, and D4.4 dopamine receptors expressed in HEK 293 cells, 50 μL of membrane homogenates were suspended in 50 mM Tris–HCl/150 mM NaCl/10 mM EDTA buffer, pH = 7.5 and incubated with 50 μL of [125I]-IABN at 37 °C for 60 min, using 20 μM (+)-butaclamol to define the non-specific binding [46]. The radioligand concentration was equal to approximately 0.5 times the Kd value and the concentration of the competitive inhibitor ranged over 5 orders of magnitude for competition experiments. For each competition curve, two concentrations of inhibitor per decade were used and triplicates were performed. Binding was terminated by the addition of the cold wash buffer (10 mM Tris–HCl/150 mM NaCl, pH = 7.5) and filtration over a glass-fiber filter (Schleicher and Schuell No. 32). A Packard Cobra gamma counter was used to measure the radioactivity. The equilibrium dissociation constant and maximum number of binding sites were generated using unweighted non-linear regression analysis of data modeled according to the equation describing mass action binding. The concentration of inhibitor that inhibits 50% of the specific binding of the radioligand (IC50 value) was determined by using nonlinear regression analysis to analyze the data of competitive inhibition experiments. Competition curves were modeled for a single site and the IC50 values were converted to equilibrium dissociation constants (Ki values) using the Cheng and Prusoff correction [47]. Mean Ki values ± S.E.M. are reported for at least three independent experiments.
2.3.2. Whole cell adenylyl cyclase assay
The accumulation of 3H-cyclic AMP in HEK cells was measured by a modification of the method of Shimizu et al. [48] Transfected HEK cells were treated with serum-free medium containing 2,8-3H-adenine (ICN Pharmaceuticals Inc., Costa Rica, CA) and cells were incubated at 37 °C for 75 min. Cells and drugs diluted in serum-free media containing 0.1 mM 3-isobutyl-1-methylxanthine (Sigma-Aldrich, Milwaukee, WI) were mixed to give a final volume of 500 μL and cells were incubated for 20 min at 37 °C. The reaction was stopped by addition of 500 μL of 10% trichloroacetic acid and 1 mM cyclic AMP. After centrifugation, the supernatants were fractionated using Dowex AG1-X8 and neutral alumina to separate the 3H-ATP and the 3H-cyclic AMP. Individual samples were corrected for column recovery by monitoring the recovery of the cyclic AMP using spectrophotometric analysis at OD 259 nm [46, 48].
2.4. In Vivo Studies
All animal experiments were conducted in compliance with the NIH Guidelines for the Care and Use of Research Animals under IACUC approved protocols reviewed by the Washington University Medical School Animal Studies Committee. Mature male Sprague Dawley rats were used for all experiments. Cyclosporin A (CycA) is commonly used as a non-selective competitive inhibitor of P-glycoprotein (P-gp) in rodent studies where it can increase the transport of PET ligands across the blood-brain in a dose-dependent manner [49, 50].
2.4.1 Biodistribution studies
The biodistribution and evaluation of regional brain uptake of the radiolabeled compounds were evaluated in male Sprague Dawley rats (250–450g). A dose of 25 mg/kg CycA (Sandimmune diluted 1:1 with saline) administered i.v. 30 min prior to radiotracer injection was used for modulation of the BBB efflux transporters. Groups (n=3–4) of control or CycA treated rats were injected with radiotracers and euthanized at 5 or 30 min post-injection. Rats were briefly anesthetized with 2–3 % isoflurane/O2 prior to injection with either CycA or radiotracers via the tail vein. [11C]6 (250–300 μCi/150 μL) was administered via intravenous tail vein injection. Due to the short half-life of carbon-11 and the number of samples required to evaluate regional brain uptake, no 5 min CycA group was evaluated for [11C]6. For the studies with the three 18F-labeled radiotracers, rats were injected with 20–30 μCi/150 μL. Rats were sacrificed at 5 or 30 min post injection. Rat brains were rapidly removed, blotted to remove excess blood and the brain stem, cerebellum, cortex, striatum and hippocampus were separated by gross dissection on a chilled glass plate. The remainder of the brain was collected to determine total brain uptake. Samples of clearance and non-target organs including blood, lung, liver, kidney, muscle, fat, and heart were also collected and placed in pre-weighed vials. Bone was taken from all rats for the 18F-labeled radiotracers tracers. All samples were counted in an automated well counter with a standard dilution of the injectate. Tissues were weighed and the percentage of the injected dose per gram of tissue (%ID/g) was calculated. Uptake of radiotracers in both normal and CycA pre-treated rats was evaluated in blood, lung, liver, kidney, muscle, fat, heart, brain stem, striatum, cortex, hippocampus, cerebellum and total brain. Bone uptake was used to estimate metabolic defluorination. The results for all four tracers are presented as % ID/g in Table 2. The effect of efflux transporter modulation is shown in the table as the ratio of uptake at 30 min in CycA treated vs. normal rats. The effect of CycA treatment on regional brain uptake is presented in Fig. 3.
Table 2.
Biodistribution of the four PET tracers and ratio of activity in CycA versus control male SD rats
| [11C]6 %ID/gram | 5 min No Rx | 30 min No Rx | 30 min CycA | CycA:No Rxa | ||
|---|---|---|---|---|---|---|
| Blood | 0.057 ± 0.011 | 0.025 | ± 0.002 | 0.029 | ± 0.004 | 1.16 |
| Lung | 1.964 ± 0.341 | 0.688 | ± 0.076 | 0.647 | ± 0.053 | 0.94 |
| Liver | 1.525 ± 0.331 | 2.098 | ± 0.220 | 2.021 | ± 0.170 | 0.96 |
| Kidney | 2.691 ± 0.330 | 1.959 | ± 0.151 | 1.902 | ± 0.111 | 0.97 |
| Muscle | 0.062 ± 0.012 | 0.071 | ± 0.012 | 0.085 | ± 0.013 | 1.18 |
| Fat | 0.024 ± 0.009 | 0.064 | ± 0.010 | 0.037 | ± 0.003 | 0.58b |
| Total brain | 0.189 ± 0.014 | 0.107 | ± 0.011 | 0.942 | ± 0.141 | 8.82c |
| [18F]7 %ID/gram | 5 min No Rx | 5 min CycA | 30 min No Rx | 30 min CycA | CycA:No Rxa |
|---|---|---|---|---|---|
| Blood | 0.133 ± 0.018 | 0.156 ± 0.033 | 0.167 ± 0.018 | 0.178 ± 0.018 | 1.06 |
| Lung | 1.182 ± 0.086 | 1.423 ± 0.262 | 0.668 ± 0.057 | 0.563 ± 0.016 | 0.84d |
| Liver | 2.035 ± 0.578 | 1.790 ± 0.286 | 2.169 ± 0.113 | 1.870 ± 0.184 | 0.86e |
| Kidney | 1.582 ± 0.293 | 1.782 ± 0.242 | 0.952 ± 0.100 | 0.898 ± 0.076 | 0.94 |
| Muscle | 0.107 ± 0.017 | 0.158 ± 0.055 | 0.143 ± 0.011 | 0.147 ± 0.006 | 1.03 |
| Fat | 0.084 ± 0.036 | 0.062 ± 0.017 | 0.086 ± 0.013 | 0.069 ± 0.011 | 0.81 |
| Bone | 0.243 ± 0.041 | 0.232 ± 0.015 | 0.164 ± 0.018 | 0.168 ± 0.016 | 1.02 |
| Total brain | 0.067 ± 0.011 | 0.421 ± 0.115 | 0.098 ± 0.012 | 0.271 ± 0.068 | 2.77f |
| [18F]8 %ID/gram | 5 min No Rx | 5 min CycA | 30 min No Rx | 30 min CycA | CycA:No Rxa |
|---|---|---|---|---|---|
| Blood | 0.079 ± 0.006 | 0.109 ± 0.009 | 0.046 ± 0.008 | 0.062 ± 0.018 | 1.35 |
| Lung | 2.432 ± 0.211 | 1.893 ± 0.158 | 1.553 ± 0.179 | 1.154 ± 0.102 | 0.74 |
| Liver | 2.124 ± 0.113 | 1.770 ± 0.584 | 2.134 ± 0.313 | 2.431 ± 0.365 | 1.14 |
| Kidney | 2.939 ± 0.544 | 2.235 ± 0.175 | 2.710 ± 0.235 | 2.548 ± 0.490 | 0.94 |
| Muscle | 0.099 ± 0.007 | 0.184 ± 0.014 | 0.111 ± 0.006 | 0.154 ± 0.038 | 1.38 |
| Fat | 0.101 ± 0.046 | 0.116 ± 0.004 | 0.095 ± 0.017 | 0.115 ± 0.030 | 1.20 |
| Bone | 0.490 ± 0.033 | 0.288 ± 0.008 | 0.882 ± 0.153 | 0.599 ± 0.152 | 0.68 |
| Total brain | 0.178 ± 0.030 | 0.895 ± 0.063 | 0.180 ± 0.006 | 0.970 ± 0.077 | 5.38g |
| [18F]9 %ID/gram | 5 min No Rx | 5 min CycA | 30 min No Rx | 30 min CycA | CycA:No Rxa |
|---|---|---|---|---|---|
| Blood | 0.082 ± 0.027 | 0.098 ± 0.009 | 0.104 ± 0.010 | 0.109 ± 0.019 | 1.05 |
| Lung | 4.026 ± 1.177 | 3.356 ± 0.261 | 1.313 ± 0.3574 | 1.063 ± 0.182 | 0.81 |
| Liver | 1.430 ± 0.221 | 1.478 ± 0.336 | 1.162 ± 0.1400 | 1.039 ± 0.124 | 0.89 |
| Kidney | 2.485 ± 0.608 | 1.913 ± 0.037 | 1.363 ± 0.0359 | 1.397 ± 0.260 | 1.03 |
| Muscle | 0.083 ± 0.009 | 0.097 ± 0.0417 | 0.083 ± 0.0139 | 0.108 ± 0.014 | 1.24 |
| Fat | 0.028 ± 0.003 | 0.046 ± 0.028 | 0.091 ± 0.0029 | 0.093 ± 0.017 | 1.02 |
| Bone | 0.253 ± 0.001 | 0.162 ± 0.030 g | 0.220 ± 0.0494 | 0.143 ± 0.016 | 0.65 |
| Total brain | 0.118 ± 0.001 | 0.725 ± 0.022 | 0.095 ± 0.0208 | 0.504 ± 0.023 | 5.33h |
Ratio of tracer uptake in CycA versus control group 30 min post injection
P value = 0.00682
P value = 0.00007
P value = 0.01232
P value = 0.03230
P value = 0.00246
P value < 0.00001
P value < 0.00001
Fig. 3.

Regional rat brain uptake of the four PET tracers in the absence and presence of CycA modulation of the BBB efflux transporters.
2.4.2. Micro PET imaging of [11C]6 in rats
Brain microPET imaging was performed using two Siemens microPET scanners (Siemens Preclincial Solutions, Knoxville, TN); a microPET-Focus-F220 and a microPET-Inveon MultiModality scanner. Imaging studies were done using 220–225 g male Sprague-Dawley rats under control and CycA treatment conditions (Fig. 4). Animals were anesthetized using 2% isoflurane/oxygen and a tail vein catheter placed in the lateral tail vein. Gas anesthesia was maintained at ~1.5% isoflurane during the imaging session; each rat was positioned on the scanner bed at least 30 minutes prior to tracer injection. Body temperature was maintained using a warming lamp. A dose of 25 mg/kg CycA (Sandimmune diluted 1:1 with saline) administered i.v. 30 min prior to radiotracer injection was used for modulation of the BBB efflux transporters. Both CycA and the radiotracer were administered using an i.v. catheter placed in the lateral tail vein. The rats were anesthetized with isoflurane and injected with ~250 μCi of [11C]6 via the tail vein. The imaging sessions were carried out as 1.5 h dynamic scan using the MicroPET® Focus 220 and Inveon scanners (Siemens Medical Solutions USA, Inc.). Acquired list mode data were histogrammed into a 3D set of sinograms and binned to the following time frames: 12 x 20 sec, 8 x 1 min and 16 x 5 min. Sinogram data was then processed using filter back projection algorithm with attenuation and scatter corrections. Using 0–90 min summarized images (Fig. 4A) as references, regions of interest were manually drawn on the brains of the rats with the software Acquisition Sinogram Image Processing (ASIPro) (Siemens Medical Solutions, Malvern PA) using IDL’s Virtual Machine (ITT Visual Information Solutions, Boulder CO) to obtain the radioactivity uptake (nCi/c.c.) curve over the time course of scan, then the activity was normalized by injected dose as shown in Fig. 4B.
Fig. 4.
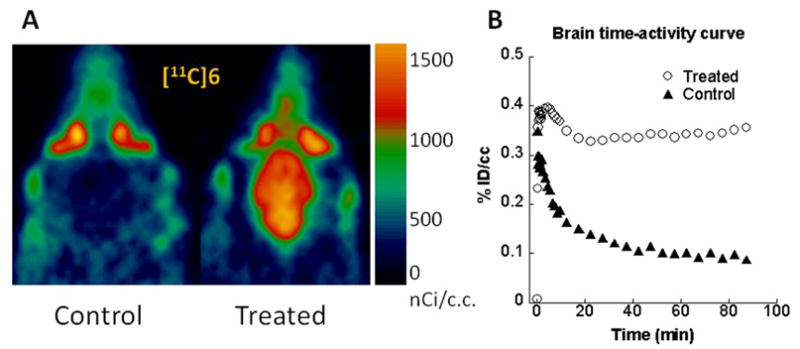
MicroPET imaging studies of [11C]6 in the absence and presence of CycA modulation of the BBB efflux transporters.
3. Results
3.1. Chemistry
The synthesis of the precursors for radiolabeling is shown in Scheme 1. 4-(4-(2-Methoxyphenyl)piperazin-1-yl)butan-1-amine 12a, and its phenol counterpart 12b, were made via N-alkylation of 10a–b with 2-(4-bromobutyl)isoindoline-1,3-dione to give the corresponding 2-(4-(4-(2-hydroxyphenyl)-piperazin-1-yl)butyl)isoindoline-1,3-dione, 11a–b in good yield (70 – 80%). Deprotection of 11a–b with the hydrazine in ethanol gave the corresponding amino analogue 12a–b with high yield (75 – 89%). Condensation of 12a and 4-(2-hydroxyethyl)benzoic acid with DCC as coupling agent gave intermediate 13a. Treatment of 13a with methanesulfonyl chloride in dichloromethane afforded compound 14a, the precursor for making [18F]7. A similar reaction sequence using 12b and either 4-(dimethylamino)benzoic acid or 4-(thiophen-3-yl)benzoic acid gave the phenol precursors 13b–c in about 80% yield [42]. Compound 13b served as the precursor for synthesizing [11C]6. O-alkylation of the phenol group in 13b–c with 2-bromoethyl acetate in the presence of potassium carbonate in acetone gave compounds 14b–c, which were converted to 15b–c by base hydrolysis using sodium hydroxide in aqueous methanol. Treatment of 15b–c with methanesulfonyl chloride in chloromethane with triethylamine as the catalyst afforded 16b–c, which served as precursors for the synthesis of [18F]8–9.
Scheme 1. Reagents.

(a) N-(4-Bromobutyl)phthalimide, triethylamine, CH2Cl2; (b) hydrazine, ethanol; (c) ArCOOH, DCC/EDCI, HOBt; (d) 2-Bromoethyl acetate, K2CO3, acetone; (e) NaOH, CH3OH/H2O; (f) Methanesulfonyl chloride, triethylamine, CH2Cl2.
3.2. Radiochemistry
Reaction of the phenol precursor 13b with [11C]MeI in DMSO or DMF containing 5N aqueous NaOH solution (Scheme 2) gave [11C]6 in 40 ~ 60% yield. There was no difference in yield when either DMSO or DMF was used as the reaction solvent. Purification of [11C]6 was accomplished using semi-preparative reversed phase HPLC. The entire synthetic procedure, including production of [11C]MeI, HPLC purification and formulation of the radiotracer for in vivo studies, was complete within 50 – 55 min. [11C]6 was obtained in a specific activity > 10 Ci/mmol (decay corrected to E.O.B. N =15) with radiochemical and chemical purity > 99%, which is sufficient for in vivo biodistribution and imaging studies. For fluorine–18 labeled analogues [18F]7, [18F]8, and [18F]9, the fluoride displacement of the corresponding mesylate precursor was initially achieved using microwave irradiation. The displacement reaction required an irradiation time of only 30 – 40 s for [18F]7. However, when applied to the radiosynthesis of [18F]8 and [18F]9, microwave irradiation was found to give inconsistent radiolabeling yields. This result may be caused by different stabilities of precursors 16b–c versus 14a under the microwave irradiation conditions. We subsequently investigated a thermal displacement reaction and determined that the optimal condition for making [18F]8 was heating at 110 °C for 10 min, and heating at 85 °C for 5 min for making [18F]9. For [18F]7, purification using a three-component solvent system of 10% tetrahydrofuran (THF): 12% acetonitrile: 78% 0.1 M ammonium formate buffer resulted in a clean separation of precursor and UV-positive impurities from [18F]7. A solid phase extraction method was used to separate [18F]7, [18F]8 and [18F]9 from the HPLC solvent, a commonly used procedure which has several advantages over concentration in vacuo. The SPE method is also readily adapted to automated radiosynthesis when needed.
Overall, the procedures used to synthesize [11C]6, [18F]7, [18F]8 and [18F]9 were very straightforward and produced [11C]6, [18F]7, [18F]8 and [18F]9 in good yield, high specific activity and high chemical and radiochemical purity for in vivo studies.
3.3 In vitro receptor binding studies and adenylyl cyclase assays
In vitro binding studies indicate that compounds 6, 7, 8 and 9 each have a subnanomolar affinity for dopamine D3 receptors, reduced affinity for D2 receptors, and a D3: D2 selectivity ratio ranging from 23 – 163. Forskolin-dependent adenylyl cyclase inhibition assays indicate that 6 is a weak partial agonist/antagonist (21% ± 2.6%), 7 is a full agonist (92.1 ± 5.3%), 8 is a partial agonist (64.5 ± 8.3%), and 9 is a weak partial agonist (34.4 ± 1.7%) at D3 dopamine receptors (Table 1) when compared to the full agonist quinpirole.
3.4 Animal Studies 3.4.1 Biodistribution
Biodistribution studies were initially conducted with [11C]6 in male Sprague-Dawley rats. Regional brain uptake and tracer accumulation in clearance and non-target organs were evaluated at 5 and 30 min post-injection. The 5 minute uptake in normal rats (0.189 %ID/g in the whole brain) and 30 min accumulation (0.107 %ID/gram) of [11C]6 in both target and non-target brain regions was relatively low and 43 – 51% washout was observed within 30 min (Table 2 and Fig. 3). The relatively low uptake of [11C]6 in whole brain and D3 receptor-rich regions was unexpected since previous studies have shown 6 (i.e., WC-10) to be pharmacologically active in behavioral studies [26, 44].
P-gp has a high affinity for lipophilic molecules of moderate weight and size that contain cationic centers and planar aromatic domains [51–53]. CycA, a non-specific competitive inhibitor of P-gp, reduces P-glycoprotein-dependent drug efflux from the CNS. If uptake of a radiotracer in the brain is limited by the action of P-gp or other ABC transporters, the rats pretreated with CycA would be expected to show increased uptake of the radiotracer in the CNS [54–60].
To confirm whether the low accumulation of the D3 selective radiotracer [11C]6 in rat brain was due to the action of the ABC transporters, the in vivo studies were repeated in the presence and absence of CycA. The 25 mg/kg dose of CycA used for these studies is approximately the ED50 for blocking P-gp in rat brain [56]. As shown in Table 2 and Fig. 3, pretreatment with CycA caused a large increase in brain uptake of [11C]6. Comparing the 30 min uptakes for control rats versus the CycA pretreated rats, the total brain uptake of [11C]6 showed a CycA effect: the uptake (%ID/g) of [11C]6 increased 8.82 fold from 0.107 ± 0.001 to 0.942 ± 0.141. When compared to the CycA effect on the uptake of [11C]verapamil [49] in rats, our data reveal that CycA pre-treated rats displayed a comparable response in the delivery of [11C]6, although the brain-to-blood ratio at a single time point is not adequate to describe the ligand delivery as reported [55]. In the peripheral organs of lung, liver, kidney, muscle and heart, no significant difference in uptake of [11C]6 between the CycA and the control group was observed 30 min post injection. The results of treatment with CycA on the uptake of [11C]6 in brain, the brain-to-blood ratios (Table 2) and uptake in regional brain areas such as striatum, cerebellum, cortex and hippocampus (Fig. 3) is very comparable with the effect observed for [11C]verapramil [49] and [11C]GR218231 [56], and higher than that observed in studies using [11C]TMSX, [11C]MPDX, [11C]flumazenil, [11C]donepezil, [11C]carazolol [49] [18F]fluorocarazolol [50,57] and [18F]MPPF [50,54].
To further investigate the behavior of this class of compounds, the three 18F-labeled radiotracers [18F]7, [18F]8 and [18F]9 were also evaluated in vivo in the presence and absence of CycA pretreatment and similar results were observed. Because the longer half-life of F-18 allows for more flexibility in study design, the regional brain uptake of the tracers 5 minutes post injection under ABC transporter inhibition/modulation was also investigated. Whole brain activity levels in normal rats for the three 18F-labeled radiotracers [18F]7, [18F]8 and [18F]9 were also <0.2 %ID/gram 5 min post injection, with very little additional washout observed by 30 min post-injection and no evidence of retention in the D3 receptor target region of striatum. As can be seen in Table 2, the biodistribution of the four tracers in normal rats showed rapid clearance from the blood and little accumulation in fat. Initial lung levels of [18F]9 were somewhat higher than the other radiotracers and [18F]8 showed a two-fold increase from 5 min to 30 min in bone activity levels that may represent metabolic defluorination. No accumulation was seen in bone for [18F]7 or [18F]9. The accumulation of all three 18F-labeled radiotracers in the brain increased under ABC transporter inhibition. That is, all four tracers showed increased retention in the brain 30 min post injection under conditions of CycA pretreatment (Table 2 and Fig. 3), though minimal effects were seen in other tissues or organs (Table 2). The reported P values were calculated using a two-sample equal variance, two-tailed Students T-test.
3.4.2. Micro PET scans of [11C]6 in rats
MicroPET imaging studies of rat brain uptake under control conditions and following treatment with CycA were also conducted to further study the effect of ABC transporter-mediated efflux on the uptake and washout kinetics of [11C]6 from the rat brain. The two scanners used for this study have comparable resolution: the microPET-Focus-F220 (used for the control rat) has a spatial resolution of 1.5 mm and 3.4 % sensitivity at the center of the field of view and the microPET-Inveon MultiModality scanner (used for the CycA rat) has 1.5 mm spatial resolution and a sensitivity of 10% at the center of the field of view. The radiotracer injections were performed within 5 minutes of each other from a single radiosynthesis to eliminate any variability due to specific activity. Because of the low density of the D3 receptor, only 36 fmol/mg protein in the rat striatum [7], and low tracer uptake in control rats for the biodistribution study, a region of interest for each rat was drawn over the entire brain. As can be seen from the summed images over the 90 min acquisition period shown in Fig. 4A, no tracer accumulation is seen in the brain of the control rat. However, there is an increase in the accumulation of [11C]6 in the CycA treated rat. The normalized time activity curve in Fig. 4A demonstrates comparable initial uptake in the two rats, with a very early peak and rapid washout in the control animal. The CycA treated rat demonstrates slower washout kinetics with the radioactive peak evident ~ 5 min post injection and an increased retention of the tracer over the duration of the scan.
4. Discussion
In recent years, a number of potent and selective dopamine D3 ligands labeled with either carbon-11 or fluorine-18 have been reported and evaluated in animal models to determine if they possess the pharmacological properties needed to function as an in vivo imaging agent (Fig. 1). Unfortunately, all D3-selective radiotracers evaluated to date have yielded disappointing results with respect to their ability to cross the blood-brain barrier and selectively label D3 receptors in vivo. This observation is somewhat perplexing since some of the radiotracers shown in Fig. 1 possess a log P value well within the range needed to enable crossing the blood-brain barrier, with D3 receptor affinity and selectivity which should lead to the labeling of D3 receptors in vivo.
Over the past 5 years, our group has synthesized a number of conformationally-flexible benzamide analogs based on lead compound 5 and measured their affinity for dopamine D2, D3, and D4 receptors, and their intrinsic activity at D2 and D3 receptors [40, 41]. This effort led to the identification of four potential radiotracers for imaging the D3 receptor (Fig. 2) which could be radiolabeled with either carbon-11 or fluorine-18 using conventional radiolabeling procedures. Initial brain uptake studies of [11C]6, and [18F]7–9 conducted in Sprague-Dawley rats revealed low brain uptake 5 min post- injection. This was unexpected since the calculated log P values of these analogs (Table 1) indicate that they should be able to cross the blood-brain and label D3 receptors in vivo. In addition, compounds 6 (WC-10) and 8 (WC-44) were found to be behaviorally active in rat models of L-DOPA-induced dyskinesia [26] and prepulse inhibition [44] in a dose range of 1.0 to 10 mg/kg, which initially seemed inconsistent with our initial rodent brain uptake studies using [11C]6 and [18F]8. This unanticipated result suggested that some other mechanism may be operational at the tracer level which led to a low brain uptake. One possible explanation was the involvement of ABC transporter proteins. This hypothesis was suggested by previous studies demonstrating that the serotonin 5-HT1A PET ligands, [11C](R)-(-)RWAY, [18F]MPPF and [carbonyl-11C]WAY100635, which also contain an N-2-methoxyphenyl piperazine group, are substrates for P-glycoprotein in rats [49,54,59,60]. Although ABC proteins are expressed in many species, some studies have shown differences in ABC transporter-mediated efflux of PET radiotracers in rodents versus nonhuman primates and humans [58]. For example, [11C](R)-(-)RWAY is a substrate for P-gp in rodent brain but not a substrate for P-gp in nonhuman primate [61] and human brain [62].
P-gp is a 170 kDa protein with low substrate specificity and widespread tissue distribution belonging to the ABC transporters which are involved in multidrug resistance [45,53]. P-gp is expressed in the capillary endothelial cells which comprise the blood-brain barrier, and protect the brain from the accumulation of toxic substances. In order to determine if the radiolabeled benzamides described in this report are substrates for P-gp, the brain uptake studies in Sprague-Dawley rats were repeated following a 30 min pretreatment with the P-gp inhibitor, cyclosporine A (CycA). In this case, the administration of CycA resulted in a 8.8 fold increase in brain uptake of [11C]6 at 30 min post injection. Similarly, CycA administration resulted in a 6.3-fold, 5.0-fold 6.1-fold increase in brain uptake of [18F]7–9 at 5 min, and a 2.7-fold, 5.4-fold and 5.3-fold increase at 30 min for the three 18F-labeled tracers respectively. Furthermore, microPET imaging studies of [11C]6 under control and CycA-treated conditions clearly demonstrate a dramatic increase in brain uptake. This effect on PET imaging agents is often described as P-gp modulation, however, it must be noted that CycA is also an inhibitor of MDR1 and MDR2. Therefore, our data are consistent with the hypothesis that [11C]6 and the three 18F-labeled radiotracers [18F]7, [18F]8 and [18F]9 are probable substrates of one or more ABC transporter proteins in rat brain since the uptake of these radiotracers was increased by pre-treatment with CycA.
While this work was being conducted, Mason and colleagues reported that PG 01037, an N-(2,3-dichlorophenyl)piperazine analog structurally similar to compound 5, is a substrate for P-gp expressed in Madin-Darby canine kidney (MDCK)-MDR1 cells, and that efflux was blocked with verapamil, a known P-gp inhibitor [63]. These data are consistent with the tracer experiments of [11C]6 and [18F]7–9, and may explain why relatively high doses of compounds 6 (WC-10), 8 (WC-44) and other structurally-related benzamide analogs are needed in rodent behavioral assays, despite the fact that these compounds have high (i.e., nanomolar to subnanomolar) affinity for D3 receptors in vitro. While the low dose administration of radiolabeled imaging agents [11C]6 and [18F]7–9 likely enabled efficient removal from the CNS by P-gp, MDR1, and/or MDR2, these transporter proteins were likely to have been saturated at the higher doses administered to rats in the behavioral pharmacology studies.
In summary, the data described in this paper show that the radiolabeled benzaminde analogs [11C]6 and [18F]7–9 are capable of crossing the rat blood brain barrier provided that CycA is administered to prevent efflux by ABC transporter proteins. Our data plus the earlier studies with [11C](R)-(-)RWAY, [18F]MPPF and [carbonyl-11C]WAY100635, which contain either the N-2-methoxyphenylpiperazine or N-2-(2-fluoroethoxy)phenylpiperazine pharmacophore, indicate that these compounds are substrates for one or more ABC transporter proteins in rodent brain. This mechanism may also be responsible for the low brain uptake reported for the D3 receptor PET radiotracers shown in Figure 1 since most contain the N-phenylpiperazine pharmacophore; these tracers were originally thought to not cross the blood-brain barrier because of the relatively high log P values. MicroPET imaging studies in nonhuman primates are currently ongoing to determine if the [11C]6 and [18F] 7–9 are capable of serving as radiotracers for imaging D3 receptors in vivo with PET in a nonrodent species.
Acknowledgments
This research was supported by the NIH grant: DA 16181, DA29840, and NS48056. We would like to thank the Small Animal Imaging Facility for assistance with the microPET imaging study. The microPET-Inveon MultiModality scanner was acquired under an NIH-NCRR HEI grant (S10-025097 PI. Richard Laforest).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sibley DR, Monsma FJ., Jr Molecular biology of dopamine receptors. Trends Pharmacol Sci. 1992;13:61–9. doi: 10.1016/0165-6147(92)90025-2. [DOI] [PubMed] [Google Scholar]
- 2.Luedtke RR, Mach RH. Progress in developing D3 dopamine receptor ligands as potential therapeutic agents for neurological and neuropsychiatric disorders. Curr Pharm Des. 2003;9:643–71. doi: 10.2174/1381612033391199. [DOI] [PubMed] [Google Scholar]
- 3.Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- 4.Reavill C, Taylor SG, Wood MD, Ashmeade T, Austin NE, Avenell KY, et al. Pharmacological actions of a novel, high-affinity, and selective human dopamine D3 receptor antagonist, SB-277011-A. J Pharmacol Exp Ther. 2000;294:1154–65. [PubMed] [Google Scholar]
- 5.Joyce JN, Gurevich EV. D3 Receptors and the actions of neuroleptics in the ventral striatopallidal system of schizophrenics. Annals of the New York Academy of Sciences. 1999;877:595–613. doi: 10.1111/j.1749-6632.1999.tb09291.x. [DOI] [PubMed] [Google Scholar]
- 6.Stanwood GD, Artymyshyn RP, Kung MP, Kung HF, Lucki I, McGonigle P. Quantitative autoradiographic mapping of rat brain dopamine D3 binding with [125I]7-OH-PIPAT: evidence for the presence of D3 receptors on dopaminergic and nondopaminergic cell bodies and terminals. J Pharmacol Exp Ther. 2000;295:1223–31. [PubMed] [Google Scholar]
- 7.Bancroft GN, Morgan KA, Flietstra RJ, Levant B. Binding of [3H]PD 128907, a putatively selective ligand for the D3 dopamine receptor, in rat brain: a receptor binding and quantitative autoradiographic study. Neuropsychopharmacology. 1998;18:305–16. doi: 10.1016/S0893-133X(97)00162-0. [DOI] [PubMed] [Google Scholar]
- 8.Murray AM, Ryoo HL, Gurevich E, Joyce JN. Localization of dopamine D3 receptors to mesolimbic and D2 receptors to mesostriatal regions of human forebrain. Proc Natl Acad Sci U S A. 1994;91:11271–5. doi: 10.1073/pnas.91.23.11271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levant B. Differential distribution of D3 dopamine receptors in the brains of several mammalian species. Brain Res. 1998;800:269–74. doi: 10.1016/s0006-8993(98)00529-0. [DOI] [PubMed] [Google Scholar]
- 10.Joyce JN. Dopamine D3 receptor as a therapeutic target for antipsychotic and antiparkinsonian drugs. Pharmacol Ther. 2001;90:231–59. doi: 10.1016/s0163-7258(01)00139-5. [DOI] [PubMed] [Google Scholar]
- 11.Griffon N, Pilon C, Sautel F, Schwartz JC, Sokoloff P. Two intracellular signaling pathways for the dopamine D3 receptor: opposite and synergistic interactions with cyclic AMP. J Neurochem. 1997;68:1–9. doi: 10.1046/j.1471-4159.1997.68010001.x. [DOI] [PubMed] [Google Scholar]
- 12.Guigoni C, Aubert I, Li Q, Gurevich VV, Benovic JL, Ferry S, et al. Pathogenesis of levodopa-induced dyskinesia: focus on D1 and D3 dopamine receptors. Parkinsonism Relat Disord. 2005;11 (Suppl 1):S25–9. doi: 10.1016/j.parkreldis.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 13.Schwartz JC, Levesque D, Martres MP, Sokoloff P. Dopamine D3 receptor: basic and clinical aspects. Clin Neuropharmacol. 1993;16:295–314. doi: 10.1097/00002826-199308000-00002. [DOI] [PubMed] [Google Scholar]
- 14.Sokoloff P, Giros B, Martres MP, Bouthenet ML, Schwartz JC. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature. 1990;347:146–51. doi: 10.1038/347146a0. [DOI] [PubMed] [Google Scholar]
- 15.Suzuki M, Hurd YL, Sokoloff P, Schwartz JC, Sedvall G. D3 dopamine receptor mRNA is widely expressed in the human brain. Brain Res. 1998;779:58–74. doi: 10.1016/s0006-8993(97)01078-0. [DOI] [PubMed] [Google Scholar]
- 16.Hackling AE, Stark H. Dopamine D3 receptor ligands with antagonist properties. Chembiochem. 2002;3:946–61. doi: 10.1002/1439-7633(20021004)3:10<946::AID-CBIC946>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 17.Millan MJ, Dekeyne A, Rivet JM, Dubuffet T, Lavielle G, Brocco M. S33084, a novel, potent, selective, and competitive antagonist at dopamine D3-receptors: II. Functional and behavioral profile compared with GR218,231 and L741,626. J Pharmacol Exp Ther. 2000;293:1063–73. [PubMed] [Google Scholar]
- 18.Heidbreder CA, Gardner EL, Xi Z-X, Thanos PK, Mugnaini M, Hagan JJ, et al. The role of central dopamine D3 receptors in drug addiction: a review of pharmacological evidence. Brain Research Reviews. 2005;49:77–105. doi: 10.1016/j.brainresrev.2004.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Higley AE, Spiller K, Grundt P, Newman AH, Kiefer SW, Xi Z-Z, et al. PG01037, a novel dopamine D3 receptor antagonist, inhibits the effects of methamphetamine in rats. Journal of Psychopharmacology. doi: 10.1177/0269881109358201. published online Feb 8, 2010 before print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sokoloff P, Le Foll B, Perachon S, Bordet R, Ridray S, Schwartz JC. The dopamine D3 receptor and drug addiction. Neurotox Res. 2001;3:433–41. doi: 10.1007/BF03033202. [DOI] [PubMed] [Google Scholar]
- 21.Newman AH, Grundt P, Nader MA. Dopamine D3 receptor partial agonists and antagonists as potential drug abuse therapeutic agents. J Med Chem. 2005;48:3663–79. doi: 10.1021/jm040190e. [DOI] [PubMed] [Google Scholar]
- 22.Bordet R, Ridray S, Carboni S, Diaz J, Sokoloff P, Schwartz JC. Induction of dopamine D3 receptor expression as a mechanism of behavioral sensitization to levodopa. Proc Natl Acad Sci U S A. 1997;94:3363–7. doi: 10.1073/pnas.94.7.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bordet R, Ridray S, Schwartz JC, Sokoloff P. Involvement of the direct striatonigral pathway in levodopa-induced sensitization in 6-hydroxydopamine-lesioned rats. Eur J Neurosci. 2000;12:2117–23. doi: 10.1046/j.1460-9568.2000.00089.x. [DOI] [PubMed] [Google Scholar]
- 24.Visanji NP, Fox SH, Johnston T, Reyes G, Millan MJ, Brotchie JM. Dopamine D3 receptor stimulation underlies the development of L-DOPA-induced dyskinesia in animal models of Parkinson's disease. Neurobiology of Disease. 2009;35:184–92. doi: 10.1016/j.nbd.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 25.Kumar R, Riddle L, Griffin SA, Grundt P, Newman AH, Luedtke RR. Evaluation of the D3 dopamine receptor selective antagonist PG01037 on L-dopa-dependent abnormal involuntary movements in rats. Neuropharmacology. 2009;56:944–55. doi: 10.1016/j.neuropharm.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar R, Riddle LR, Griffin SA, Chu W, Vangveravong S, Neisewander J, et al. Evaluation of D2 and D3 dopamine receptor selective compounds on L-dopa-dependent abnormal involuntary movements in rats. Neuropharmacology. 2009;56:956–69. doi: 10.1016/j.neuropharm.2009.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Kampen JM, Eckman CB. Dopamine D3 receptor agonist delivery to a model of Parkinson's disease restores the nigrostriatal pathway and improves locomotor behavior. J Neurosci. 2006;26:7272–80. doi: 10.1523/JNEUROSCI.0837-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghosh B, Antonio T, Zhen J, Kharkar P, Reith MEA, Dutta AK. Development of (S)-N6-(2-(4-(isoquinolin-1-yl)piperazin-1-yl) ethyl)-N6-propyl-4,5,6,7-tetrahydrobenzo[d]-thiazole-2,6-diamine and its analogue as a D3 receptor preferring agonist: Potent in vivo activity in Parkinson's disease animal models. J. Med. Chem. 2010;53:1023–37. doi: 10.1021/jm901184n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pilla M, Perachon S, Sautel F, Garrido F, Mann A, Wermuth CG, et al. Selective inhibition of cocaine-seeking behaviour by a partial dopamine D3 receptor agonist. Nature. 1999;400:371–5. doi: 10.1038/22560. [DOI] [PubMed] [Google Scholar]
- 30.Khaled MATM, Farid Araki K, Li B, Coen KM, Marinelli PW, Varga J, et al. The selective dopamine D3 receptor antagonist SB 277011-A, but not the partial agonist BP 897, blocks cue-induced reinstatement of nicotine-seeking. Int J Neuropsychopharmacol. 2009:1–10. doi: 10.1017/S1461145709991064. [DOI] [PubMed] [Google Scholar]
- 31.Garcia-Ladona FJ, Cox BF. BP 897, a selective dopamine D3 receptor ligand with therapeutic potential for the treatment of cocaine-addiction. CNS Drug Rev. 2003;9:141–58. doi: 10.1111/j.1527-3458.2003.tb00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carr KD, Yamamoto N, Omura M, Cabeza de Vaca S, Krahne L. Effects of the D3 dopamine receptor antagonist, U99194A, on brain stimulation and d-amphetamine reward, motor activity, and c-fos expression in ad libitum fed and food-restricted rats. Psychopharmacology (Berl) 2002;163:76–84. doi: 10.1007/s00213-002-1132-0. [DOI] [PubMed] [Google Scholar]
- 33.Witkin JM, Levant B, Zapata A, Kaminski R, Gasior M. The dopamine D3/D2 agonist (+)-PD-128,907 [(R-(+)-trans-3,4a,10b-tetrahydro-4-propyl-2H,5H-[1]benzopyrano[4,3-b]-1,4-oxazin-9-ol)] protects against acute and cocaine-kindled seizures in mice: Further evidence for the involvement of D3 receptors. Journal of Pharmacology and Experimental Therapeutics. 2008;326:930–8. doi: 10.1124/jpet.108.139212. [DOI] [PubMed] [Google Scholar]
- 34.Hocke C, Prante O, Salama I, Hubner H, Lober S, Kuwert T, et al. 18F-Labeled FAUC 346 and BP 897 derivatives as subtype-selective potential PET radioligands for the dopamine D3 receptor. ChemMedChem. 2008;3:788–93. doi: 10.1002/cmdc.200700327. [DOI] [PubMed] [Google Scholar]
- 35.Sovago J, Farde L, Halldin C, Langer O, Laszlovszky I, Kiss B, et al. Positron emission tomographic evaluation of the putative dopamine-D3 receptor ligand, [11C]RGH-1756 in the monkey brain. Neurochem Int. 2004;45:609–17. doi: 10.1016/j.neuint.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 36.Kuhnast B, Valette H, Besret L, Demphel S, Coulon C, Ottaviani M, et al. Synthesis and radiolabeling of N-[4-[4-(2-[11C]methoxyphenyl)piperazin-1-yl]butyl]benzo[b]thiophene-2-carboxamide - a potential radiotracer for D3 receptor imaging with PET. Nucl Med Biol. 2006;33:785–95. doi: 10.1016/j.nucmedbio.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 37.Bennacef I, Salinas CA, Bonasera TA, Gunn RN, Audrain H, Jakobsen S, et al. Dopamine D3 receptor antagonists: the quest for a potentially selective PET ligand. Part 3: Radiosynthesis and in vivo studies. Bioorg Med Chem Lett. 2009;19:5056–9. doi: 10.1016/j.bmcl.2009.07.055. [DOI] [PubMed] [Google Scholar]
- 38.Turolla EA, Matarrese M, Belloli S, Moresco RM, Simonelli P, Todde S, et al. 11C-labeling of N-[4-[4-(2,3-dichlorophenyl)piperazin-1-yl]butyl]arylcarboxamide derivatives and evaluation as potential radioligands for PET imaging of dopamine D3 receptors. J Med Chem. 2005;48:7018–23. doi: 10.1021/jm050171k. [DOI] [PubMed] [Google Scholar]
- 39.Mach RH, Huang Y, Freeman RA, Wu L, Vangveravong S, Luedtke RR. Conformationally-flexible benzamide analogues as dopamine D3 and sigma 2 receptor ligands. Bioorg Med Chem Lett. 2004;14:195–202. doi: 10.1016/j.bmcl.2003.09.083. [DOI] [PubMed] [Google Scholar]
- 40.Chu W, Tu Z, McElveen E, Xu J, Taylor M, Luedtke RR, et al. Synthesis and in vitro binding of N-phenyl piperazine analogs as potential dopamine D3 receptor ligands. Bioorg Med Chem. 2005;13:77–87. doi: 10.1016/j.bmc.2004.09.054. [DOI] [PubMed] [Google Scholar]
- 41.Tu Z, Li S, Cui J, Xu J, Taylor M, Ho D, et al. Synthesis and pharmacological evaluation of fluorine containing D3 dopamine receptor selective analogues. J Med Chem. doi: 10.1021/jm101323b. under review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu J, Chu W, Tu Z, Jones LA, Luedtke RR, Perlmutter JS, et al. [3H]4-(Dimethylamino)-N-[4-(4-(2-methoxyphenyl)piperazin- 1-yl)butyl]benzamide, a selective radioligand for dopamine D3 receptors. I. In vitro characterization. Synapse. 2009;63:717–28. doi: 10.1002/syn.20652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu J, Hassanzadeh B, Chu W, Tu Z, Jones LA, Luedtke RR, et al. [3H]4-(dimethylamino)-N-(4-(4-(2-methoxyphenyl)piperazin-1-yl) butyl)benzamide: a selective radioligand for dopamine D3 receptors. II. Quantitative analysis of dopamine D3 and D2 receptor density ratio in the caudate-putamen. Synapse. 2010;64:449–59. doi: 10.1002/syn.20748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weber M, Chang WL, Durbin JP, Park PE, Luedtke RR, Mach RH, et al. Using prepulse inhibition to detect functional D3 receptor antagonism: effects of WC10 and WC44. Pharmacol Biochem Behav. 2009;93:141–7. doi: 10.1016/j.pbb.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loscher W, Potschka H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx. 2005;2:86–98. doi: 10.1602/neurorx.2.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luedtke RR, Freeman RA, Boundy VA, Martin MW, Huang Y, Mach RH. Characterization of 125I-IABN, a novel azabicyclononane benzamide selective for D2-like dopamine receptors. Synapse. 2000;38:438–49. doi: 10.1002/1098-2396(20001215)38:4<438::AID-SYN9>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 47.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 48.Shimizu H, Daly JW, Creveling CR. A radioisotopic method for measuring the formation of adenosine 3',5'-cyclic monophosphate in incubated slices of brain. J Neurochem. 1969;16:1609–19. doi: 10.1111/j.1471-4159.1969.tb10360.x. [DOI] [PubMed] [Google Scholar]
- 49.Ishiwata K, Kawamura K, Yanai K, Hendrikse NH. In vivo evaluation of P-glycoprotein modulation of 8 PET radioligands used clinically. J Nucl Med. 2007;48:81–7. [PubMed] [Google Scholar]
- 50.Elsinga PH, Hendrikse NH, Bart J, van Waarde A, Vaalburg W. Positron emission tomography studies on binding of central nervous system drugs and P-glycoprotein function in the rodent brain. Mol Imaging Biol. 2005;7:37–44. doi: 10.1007/s11307-005-0951-x. [DOI] [PubMed] [Google Scholar]
- 51.van Asperen J, Mayer U, van Tellingen O, Beijnen JH. The functional role of P-glycoprotein in the blood-brain barrier. J Pharm Sci. 1997;86:881–4. doi: 10.1021/js9701364. [DOI] [PubMed] [Google Scholar]
- 52.Abbott NJ, Romero IA. Transporting therapeutics across the blood-brain barrier. Mol Med Today. 1996;2:106–13. doi: 10.1016/1357-4310(96)88720-x. [DOI] [PubMed] [Google Scholar]
- 53.Pearce HL, Winter MA, Beck WT. Structural characteristics of compounds that modulate P-glycoprotein-associated multidrug resistance. Adv Enzyme Regul. 1990;30:357–73. doi: 10.1016/0065-2571(90)90026-x. [DOI] [PubMed] [Google Scholar]
- 54.Passchier J, van Waarde A, Doze P, Elsinga PH, Vaalburg W. Influence of P-glycoprotein on brain uptake of [18F]MPPF in rats. Eur J Pharmacol. 2000;407:273–80. doi: 10.1016/s0014-2999(00)00752-4. [DOI] [PubMed] [Google Scholar]
- 55.Hendrikse NH, de Vries EG, Franssen EJ, Vaalburg W, van der Graaf WT. In vivo measurement of [11C]verapamil kinetics in human tissues. Eur J Clin Pharmacol. 2001;56:827–9. doi: 10.1007/s002280000253. [DOI] [PubMed] [Google Scholar]
- 56.de Vries EF, Kortekaas R, van Waarde A, Dijkstra D, Elsinga PH, Vaalburg W. Synthesis and evaluation of dopamine D3 receptor antagonist 11C- GR218231 as PET tracer for P-glycoprotein. J Nucl Med. 2005;46:1384–92. [PubMed] [Google Scholar]
- 57.Doze P, Van Waarde A, Elsinga PH, Hendrikse NH, Vaalburg W. Enhanced cerebral uptake of receptor ligands by modulation of P-glycoprotein function in the blood-brain barrier. Synapse. 2000;36:66–74. doi: 10.1002/(SICI)1098-2396(200004)36:1<66::AID-SYN7>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 58.Syvänen S, Lindhe Ö, Palner M, Kornum BR, Rahman O, Långström B, et al. Species Differences in Blood-Brain Barrier Transport of Three Positron Emission Tomography Radioligands with Emphasis on P-Glycoprotein Transport. Drug Metabolism and Disposition. 2009;37:635–43. doi: 10.1124/dmd.108.024745. [DOI] [PubMed] [Google Scholar]
- 59.Liow JS, Lu S, McCarron JA, Hong J, Musachio JL, Pike VW, et al. Effect of a P-glycoprotein inhibitor, Cyclosporin A, on the disposition in rodent brain and blood of the 5-HT1A receptor radioligand, [11C](R)-(-)-RWAY. Synapse. 2007;61:96–105. doi: 10.1002/syn.20348. [DOI] [PubMed] [Google Scholar]
- 60.Lacan G, Plenevaux A, Rubins DJ, Way BM, Defraiteur C, Lemaire C, et al. Cyclosporine, a P-glycoprotein modulator, increases [18F]MPPF uptake in rat brain and peripheral tissues: microPET and ex vivo studies. Eur J Nucl Med Mol Imaging. 2008;35:2256–66. doi: 10.1007/s00259-008-0832-z. [DOI] [PubMed] [Google Scholar]
- 61.Yasuno F, Zoghbi SS, McCarron JA, Hong J, Ichise M, Brown AK, et al. Quantification of serotonin 5-HT1A receptors in monkey brain with [11C](R)-(-)-RWAY. Synapse. 2006;60:510–20. doi: 10.1002/syn.20327. [DOI] [PubMed] [Google Scholar]
- 62.Zhang XY, Yasuno F, Zoghbi SS, Liow JS, Hong J, McCarron JA, et al. Quantification of serotonin 5-HT1A receptors in humans with [11C](R)-(-)-RWAY: radiometabolite(s) likely confound brain measurements. Synapse. 2007;61:469–77. doi: 10.1002/syn.20392. [DOI] [PubMed] [Google Scholar]
- 63.Mason CW, Hassan HE, Kim KP, Cao J, Eddington ND, Newman AH, et al. Characterization of the transport, metabolism, and pharmacokinetics of the dopamine D3 receptor-selective fluorenyl- and 2-pyridylphenyl amides developed for treatment of psychostimulant abuse. J Pharmacol Exp Ther. 2010;333:854–64. doi: 10.1124/jpet.109.165084. [DOI] [PMC free article] [PubMed] [Google Scholar]