

Abstract
BRCA1-associated protein-1 (BAP1) is a 729 residue, nuclear-localized deubiquitinating enzyme (DUB) that displays tumor suppressor properties in the BAP1-null NCI-H226 lung carcinoma cell line. Studies that have altered BAP1 cellular levels or enzymatic activity have reported defects in cell cycle progression, notably at the G1/S transition. Recently BAP1 was shown to associate with the transcriptional regulator host cell factor 1 (HCF-1). The BAP1/HCF-1 interaction is mediated by the HCF-1 Kelch domain and an HCF-1 binding motif (HBM) within BAP1. HCF-1 is modified with ubiquitin in vivo, and ectopic studies suggest BAP1 deubiquitinates HCF-1. HCF-1 is a chromatin-associated protein thought to both activate and repress transcription by linking appropriate histone-modifying enzymes to a subset of transcription factors. One known role of HCF-1 is to promote cell cycle progression at the G1/S boundary by recruiting H3K4 histone methyltransferases to the E2F1 transcription factor so that genes required for S-phase can be transcribed. Given the robust associations between BAP1/HCF-1 and HCF-1/E2Fs, it is reasonable to speculate that BAP1 influences cell proliferation at G1/S by co-regulating transcription from HCF-1/E2F-governed promoters
Keywords: BAP1, Ubiquitin C-terminal hydrolase, Protease, Deubiquitylase
Introduction
BRCA1-associated protein-1 (BAP1) is a nuclear-localized deubiquitinating enzyme (DUB) that displays tumor suppressor properties in the NCI-H226 lung carcinoma cell line [1]. This enzyme was first identified as a BRCA1 binding protein in a yeast two hybrid screen and subsequently shown to have a mild synergistic effect on BRCA1-mediated growth suppression [2]. NCI-H226 cells carry a naturally occurring homozygous deletion of the BAP1 gene (BAP1−/−) [2], and lentiviral-based restoration of BAP1 in these cells suppresses both tumor formation in mice and cell growth in monolayer cultures [1]. Cell cycle analysis in NCI-H226, HeLa, and MCF10a cells suggest BAP1 is involved in promoting the G1/S transition [1, 3, 4]. Recent studies have found a strong link between BAP1 and the transcriptional regulator host cell factor 1 (HCF-1) [4–6]. HCF-1 is an abundant nuclear protein believed to regulate transcription by binding to transcription factors that have an HCF-1 binding motif (HBM) and recruiting activating or repressing histone modifying enzymes to these transcription complexes. HCF-1 was shown to co-regulate promoters bound by the E2F family of transcription factors [7, 8]. The E2Fs are known to promote cell cycle progression by activating genes required for S-phase, and E2F1 required HCF-1 for Set1 and MLL (histone K4-H3 methyl transferases) localization to E2F-regulated promoters. Given the strong links between BAP1/HCF-1 and between HCF-1/E2Fs, it very possible that cell cycle alterations observed in BAP1 studies are a product of misregulated HCF-1/E2F promoters. This review summarizes these findings and proposes models for BAP1’s role at HCF-1/E2F complexes.
DUBs and the Ubiquitin Pathway
Post-translational modification by ubiquitin (Ub) influences nearly all eukaryotic cellular processes. The 76-residue ubiquitin protein is conjugated to lysine side chains of protein substrates through sequential enzymatic reactions performed by E1, E2, and E3 enzymes (reviewed in [9, 10]). Ubiquitination occurs in many forms and the type of modification often dictates the cellular fate of the substrate. For example, monoubiquitination of membrane-associated proteins signals their internalization and degradation via the endocytic pathway [11, 12]. In contrast, ubiquitination can lead to the assembly of a polyubiquitin chain, a polymer of ubiquitin molecules tethered via isopeptide bonds between the lysine of one Ub and the C-terminus of a another. Because Ub has seven lysine residues, polyUb chains can be very complex (homogenous/heterogeneous, straight/forked, reviewed in [13]) and synthesis of specific chains can be directed by certain E2/E3 pairs [14]. Homogenous K48-linked chains are well known to target substrates to the 26S proteasome for degradation, but the consequences of attaching other polyUb chains are varied and serve to regulate substrates through proteolytic and non-proteolytic roles [9, 10, 15].
Like other post-translational modifications, ubiquitination is a reversible process and deubiquitinating enzymes (DUBs) like BAP1 are the proteases that perform this role. The human genome encodes 95 putative DUBs that can be divided into five classes based on their conserved catalytic domain; most DUBs are cysteine proteases, but a few belong to a family of metalloproteases (reviewed in [16–19]). DUBs are usually modular enzymes containing additional domains thought to be responsible for localization, autoregulating DUB activity, and binding to specific substrates or polyUb chains [16]. In addition to reversing substrate ubiquitination, DUBs also recycle monomeric Ub from polyUb chains and process newly synthesized ubiquitin from its proprotein forms.
The UCH Family of DUBs
BAP1 is a 729 residue protein that belongs to the UCH family of DUBs. There are four UCH family members in humans and a single ortholog in yeast (Fig. 1). The smaller UCH enzymes (UCH-L1 and UCH-L3) are single domain proteins with a papain-like fold and activity as ubiquitin-specific thiol proteases. They are likely involved in post/co-translationally hydrolyzing linear peptide bonds of proubiquitin precursors [20]. These DUBs are active in cleaving small peptides from the C-terminal glycine of ubiquitin (G76), but show little activity towards larger, structured fusions or towards isopeptide linked di-Ub [16, 20–22]. UCH37 (also known as UCH-L5), the third member of the UCH gene family, contains a short C-terminal extension and associates with the proteasome and the INO80 chromatin remodeling complex [23–27]. In the proteasome UCH37- acts to trim polyubiquitin chains as the substrate is degraded by this multicatalytic protease complex. The largest UCH family DUB, BAP1, contains a 500 amino acid extension C-terminal to the catalytic domain and is the subject of this review.
Fig. 1.
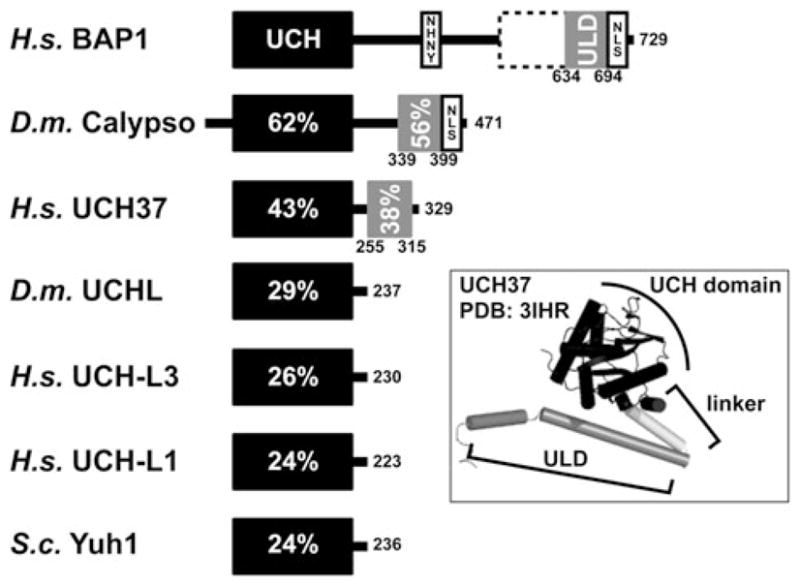
Domain architecture and sequence conservation of human, fruit fly, and yeast UCH DUBs. Human BAP1 is composed of an N-terminal UCH domain (res. 1–250), an HBM (NHNY, res. 363–366), a region that mediates association with BRCA1 (res. 596–721), a 60 residue helical motif that shares conservation with UCH37 (ULD for UCH37-like domain, res. 634–694), and a bona fide nuclear localization signal (NLS, RRRKGR, res. 717–722). The UCH domains of other human, yeast, and fruit fly DUBs are depicted by black bars and percentages reflect the sequence identity compared to BAP1. Calypso and UCH37 also contain ULDs (gray bars) and Calypso contains a putative NLS at its C-terminus (RRRKGR, res. 459–464). The inset maps the UCH and ULD domains of UCH37 onto its crystal structure. The UCH domain, shown in black, is connected to the helical ULD, shown in gray, by a short helical linker
Domain Structure of BAP1
Like other UCH family DUBs, BAP1 has an N-terminal catalytic domain containing the ubiquitin-binding site and the catalytic triad responsible for cleavage of the ubiquitin isopeptide bond. Notably, two of the human UCH-containing DUBs have C-terminal extensions; the BAP1 and UCH37 proteins exhibit 38% identity over 60 residues at the C-terminus, designated as the ULD (UCH37-like domain, Fig. 1). UCH37 uses a portion of its helical ULD to bind Rpn13, a component of the 19S regulatory subunit of the proteasome [24–26] and perhaps to bind some component of the INO80 chromatin remodeling complex [23]. What is the role of this ULD? The UCH37/Rpn13 association stimulates UCH37 activity, in part by relieving autoinhibition, and recruits UCH37 to proteasomes where it hydrolyzes distal subunits from polyUb chains (reviewed in [16]). The UCH37/Rpn13 interaction is dependent on a short KEKE-motif within the ULD of UCH37 [26]. The BAP1 ULD lacks this motif and BAP1 is not known to associate with proteasomes or Rpn13. The Drosophila homolog of BAP1, Calypso has a very similar domain architecture sharing high sequence identity, a ULD, and a putative NLS. As discussed in the following section and shown in Fig. 1, the BAP1 C-terminal extension mediates other protein–protein interactions and contains a functional nuclear localization signal.
Protein Partners of BAP1
Initially identified in a yeast two-hybrid screen using the RING-finger domain of the breast cancer type 1 susceptibility protein BRCA1 as bait, BAP1 was shown to enhance BRCA1-mediated growth suppression in MCF7 cells [2]. The BAP1 C-terminal domain mediates binding to the N-terminal RING of BRCA1. Since the BRCA1/BARD1 heterodimer is a ubiquitin ligase that forms polyubiquitin linkages through K6 and K29 on ubiquitin [28–30], it is tempting to speculate that BAP1 acts on autoubiquitinated BRCA1 and/or its putative ubiquitin ligase substrates, including core histones [31, 32], RNA pol II [33], FANCD2 [34], nucleophosmin/B23 [35], and estrogen receptor-α [36]. However, direct tests of this hypothesis by assessing BAP1’s DUB activity on these putative substrates are lacking. In one case, BAP and its Drosophila homolog Calypso, were able to deubiquitinate histone H2A in vitro [37]. Other studies have shown that growth suppression is independent of BRCA1 [1] and that BAP1 disrupts the BRCA1/BARD1 heterodimer, but cannot reverse its autoubiquitination [3, 32]. Thus, a cellular role for BAP1 in BRCA1-mediated functions remains speculative.
The BAP1/HCF-1 interaction was first identified by mass spectrometry in immunoprecipitates of overexpressed BAP1 and the determinants were subsequently mapped to the Kelch domain of HCF-1 and an HBM within BAP1 (residues 363–366, NHNY) [6]. The association has since been observed by two other groups [4, 5]. Mutation of the BAP1 HBM to alanine (BAP1-ΔHBM) abolishes its ability to bind HCF-1 [4, 6] but does not impact BAP1’s DUB activity when measured with Ub-AMC, a fluorescent DUB substrate [6].
Two groups have identified proteins that co-immunoprecipitate with overexpressed BAP1 using mass spectrometry [4, 5]. Several identified proteins appear with high confidence in both reports and are discussed below. Intriguingly, BAP1 primarily associates with proteins involved in chromatin modifications and transcriptional processes. As discussed below, HCF-1 localizes to chromatin and associates with several transcription factors as well as proteins involved in different chromatin modifying activities. BAP1 pulls-down both FoxK1 and FoxK2, members of the Forkhead transcription factors (>40 in humans [38]). FoxK1 is expressed in myogenic progenitor cells where it regulates cell cycle progression [39–41]. ASXL1 and ASXL2 are putative polycomb group (PcG) proteins and recently BAP1 and ASXL1 (as wells as the Drosophila homologs Calypso and ASX) were shown to form a complex called PR-DUB (polycomb repressive-DUB) that can reverse ubiquitination of histone H2A in vitro [37]. In Drosophila, PR-DUB localizes to PcG target genes where it is thought to actively repress HOX gene expression by regulating histone H2A ubiquitination [37]. Transcriptional repression of a PcG target gene required Calypso’s active site cysteine (C131) [37]. BAP1 also appears to interact with E2O, a poorly understood and non-canonical ubiquitin conjugating enzyme (E2). E2 enzymes contain a core UBC domain of ~150 amino acids and some E2s have short N- or C-terminal domains or extensions [9, 17]. E2O is very unique in that it is a 1292 residue protein with a ~1,000 residue N-terminal extension which shares little homology to other proteins. The E2O/BAP1 interaction is intriguing as the two proteins promote opposing enzymatic activities and may act upon similar substrates antagonizing one another.
Enzymatic Activity of BAP1
Recent studies on BAP1 have primarily focused on the in vivo properties of BAP1 and its role in cell cycle regulation (see below). From a biochemical perspective, very little is known regarding its catalytic properties and substrate selectivity. BAP1 has been shown to cleave small derivatives from the C-terminus of ubiquitin [2, 6], but its in vitro activity towards protein substrates or polyUb has not been thoroughly examined. It does not act upon autoubiquitinated BRCA1 [3, 32] and has only been tested against two BRCA1/BARD1 ubiquitin ligase substrates, histone H2A and H2B, where BAP1/ASXL1 complexes selectively disassembled mono-ubiquitinated histone H2A [37]. As discussed below, HCF-1 is the other known substrate of BAP1, and in vivo studies using overexpressed and epitope-tagged Ub, HCF-1, and BAP1 found that BAP1 prefers to cleave K48-polyUb over K63-polyUb [4, 6]. We will first turn our attention to the biological effects of BAP1 and then speculate on the underlying biochemical mechanisms that may be involved.
BAP1 is a Cancer-Related DUB
The BAP1 gene is mutated (including rearrangements, homozygous deletions, and missennse mutations) in lung and breast carcinomas [2, 42]. BAP1 is located on chromosome 3 (3p21.3), in a region (3p) that harbors deletions, sometimes including BAP1, in >80% of breast carcinomas, >90% of lung carcinomas, and almost all renal carcinomas [2, 43]. The NCI-H226 non-small cell lung carcinoma line carries a homozygous BAP1 deletion (BAP1−/−) [2], and recently it was shown that lentiviral-based restoration of BAP1 in this cell line suppresses both tumor formation in mice and growth in monolayer cultures [1]. These tumor suppressor properties are dependent on localization of BAP1 to the nucleus and its active site cysteine residue. Cancer-associated point mutations (A95D, G178V) were also shown to abolish BAP1’s DUB activity in vitro [1].
BAP1 Regulates the G1/S Transition
Several groups have reported alterations in cell proliferation and cell cycle distribution when BAP1 concentrations are enhanced, depleted, or restored. These findings are summarized in Table 1. In the study by Ventii et al. [1] BAP1 was restored in the NCI-H226 cell line (BAP1−/−) by lentiviral infection resulting in complete growth suppression in and an increase in the number of cells in S and sub-G1 phases. Thus, BAP1 reexpression in NCI-H226 kills by apoptosis and necrosis. More direct evidence for the involvement of BAP1 in regulating the G1/S transition is provided by knocking down the expression of BAP1. Transient expression of a shRNA against BAP1 in cells synchronized at G1/S showed a delay in progressing thru the G1/S transition [3]. In MCF10A cells that express endogenous BAP1, overexpression of BAP1 had minimal effects on cell growth, but knockdown of endogenous BAP1 led to reduced cell growth [4]. The differences observed between the two cell lines may reflect their genetic backgrounds; for example, NCI-H226 cells have probably upregulated other pathways to efficiently transition thru G1/S in response to the loss of BAP1. Intriguingly, overexpression of BAP1-C91S in MCF10A cells severely reduced cell growth, and growth suppression was dependent on the HBM of BAP1 [4]. While it is possible that BAP1 uses its HBM to interact with other proteins, these findings strongly suggest that growth inhibition is mediated thru HCF-1.
Table 1.
Summary of studies on BAP1’s role in cell proliferation
| Reference | Cell line | Alteration | Measurement | Effect |
|---|---|---|---|---|
| Jensen et al. [2] | MCF7 | Transfection of BRCA1 and/or BAP1 and BAP1-ΔUCH | Cell growth after 21–28 days | Overexpression of BAP1 or BRCA1 alone suppresses cell growth. Co-expression enhances suppression and this is dependent on presence of UCH domain |
| Ventii et al. [1] | NCI-H226 | Lentiviral restoration of BAP1 and BAP1 C91A and ΔNLS in cultured cells | Survival of infected cells | BAP1 expression suppresses growth and effects are dependent on C91A and nuclear localization |
| Lentiviral restoration of BAP1 and BAP1 C91A and ΔNLS in tumors in mice | Tumor growth rate and tumor volume | BAP1 expression suppresses growth and effects are dependent on C91A and nuclear localization | ||
| Lentiviral restoration of BAP1 and BAP1 C91A and ΔNLS in cultured cells | Cell cycle by PI staining and flow cytometry | BAP1 expression results in more cells in S and sub-G1 phases. C91A and ΔNLS mutants have distributions like control cells | ||
| Lentiviral restoration of BAP1 and BAP1 C91A and ΔNLS in cultured cells | Apoptosis by PI/Annexin V-FITC staining and flow cytometry | Cells expressing BAP1 undergo apoptosis and necrosis. C91A and ΔNLS mutants have distributions more like control cells | ||
| Misaghi et al. [6] | HeLa | Knockdown of endogenous BAP1 with transfected siRNA | Cell cycle by PI staining and flow cytometry | Depletion of BAP1 results in fewer cells in G1 and more cells in G2/M and sub-G1 phases suggesting a slower transition through S phase |
| Nishikawa et al. [3] | HeLa | Stable expression of shRNA towards BAP1 followed by synchronization at G1/S | Cell cycle by PI staining and flow cytometry | BAP1 depleted cells arrest earlier in G1/S and require more time to transition into S-phase |
| Machida et al. [4] | MCF10A | Retroviral expression of shRNA towards BAP1 | Cell number after 10 days | BAP1 depletion inhibits growth. 60–70% reduction in cell number after 10 days |
| Retroviral expression of BAP1 and BAP1-C91S | Cell number after 10 days | BAP1 overexpression has little effect on cell number but C91S inhibits growth by 80% | ||
| Retroviral expression of BAP1 and BAP1-C91S, BAP1-ΔHBM, and C91S-ΔHBM | Cell number after 11 days | C91S growth repression is relieved in the C91S-ΔHBM mutant. The ΔHBM mutant alone has a minor effect on cell number |
HCF-1 is a Transcriptional Regulator
HCF-1 is a 2035 residue protein that localizes to the nucleus and associates with chromatin [44, 45]. Full-length HCF-1 is proteolytically processed into mature N- and C-terminal fragments that remain stably associated [45, 46]. HCF-1 is known to be involved in cell proliferation as early studies on HCF-1 identified a P134S mutation in a temperature-sensitive hamster cell line (tsBN67) [47]. The P134S mutation prevents HCF-1 from associating with chromatin [44], and when grown at the non-permissive temperature, tsBN37 cells arrest in G0/G1 phase and show defects in proper cytokinesis [47, 48]. Later it was shown that the N-terminal fragment is necessary for the G1/S transition while the C-terminal fragment is needed for proper cytokinesis [49]. The N-terminal fragment contains a Kelch domain that binds to (D/E/N)-H–x-Y motifs found in BAP1 [4, 6], the viral transactivator VP16 [50], several transcription factors (including LZIP, Krox20, E2F1, and E2F4 [7, 50–52]), and the Set1 and MLL histone H3-lysine 4 (H3K4) methyltransferases (HMTs) [53, 54]. HCF-1 was shown to simultaneously bind VP16 and Set1 [54], suggesting the Kelch domain can accommodate binding to two HBMs. Also within the N-terminal fragment of HCF-1 is a basic region that binds other transcription factors, Sp1 and GABPβ [55, 56] as well as histone deacetylase complexes (HDACs) via Sin3a [54]. HCF-1 complexes containing Set1, MLL, or Sin3a also coprecipitate other members of HMT and HDAC complexes, and experiments using ChIP and activity assays indicate that these complexes exhibit histone modifying activities [7, 54, 57]. The C-terminal fragment of HCF-1 has been studied less extensively, but has been shown to associate with protein phosphatase 1 and PDCD2 [58, 59] and play a role in mitosis, ensuring proper chromosome alignment and segregation and cytokinesis presumably by regulating PR-Set7, a H4K20 HMT, and the levels of mono versus dimethyl marks [49, 60].
Substrates of BAP1 DUB Activity Coimmunoprecipitate with HCF1
It has been suggested that HCF-1 is a substrate of BAP1 DUB activity. Two groups have shown that anti-HCF-1 immunoprecipitates contain polymeric ubiquitin (at least some in the form of Ub-HCF-1) when HA-Ub and tagged-HCF-1 are co-overexpressed [4, 6]. The ubiquitination sites on HCF-1 have been mapped to specific lysines using mass spectrometry [4, 6]. In the Misaghi et al. [6] study, V5-tagged HCF-1 was found to be ubiquitinated in its C-terminal fragment (Lys 1807/1808) whereas immunoprecipitation of the Flag-tagged Kelch domain demonstrated that it was modified at four sites (Lys 105, 163, 244, and 363) [4]. Co-overexpression studies that include WT-BAP1 showed a reduced amount of Ub/Ub-HCF-1 in HCF-1 immunoprecipitates [4, 6]. Analysis of the ubiquitin chain linkages using ubiquitin lysine mutants showed that both K63 and K48-linked chains are immunoprecipitated with HCF-1, but that BAP1 prefers cleaving K48-linked chains [4, 6]. In both studies, co-overexpression of catalytically dead BAP1 (C91S or C91A) resulted in the recovery of more polymeric ubiquitin in HCF-1 immunoprecipitates and in one case resulted in the inhibition of cell growth [4, 6]. The interpretation is that overexpressed, inactive BAP1 has a dominant negative effect by saturating HCF-1 binding sites and preventing endogenous BAP1 from deubiquitinating its substrates (see Fig. 2). This was tested by disrupting the BAP1/HCF-1 interaction using the BAP1-ΔHBM mutant, and consistent with this model, this mutant was unable to reduce ubiquitin levels in HCF-1 immunoprecipitates and had a mild effect on proliferation [4]. Thus, overexpressed BAP1-C91S-ΔHBM mutant does not exhibit the dominant negative effects of overexpressed C91S-BAP1 [4]. While it is not known if BAP1 uses its HBM to interact with other proteins, these findings strongly suggest that an HCF-1 dependent interaction results in growth inhibition. These results are consistent with HCF-1 or proteins associated with HCF-1 being substrates for BAP1. Interestingly, neither of these two groups observed significant changes to HCF-1 levels when BAP1 concentrations were modulated via overexpression or RNAi [4, 6] suggesting that the stability of HCF-1 is not dependent on the presence of BAP1 activity.
Fig. 2.
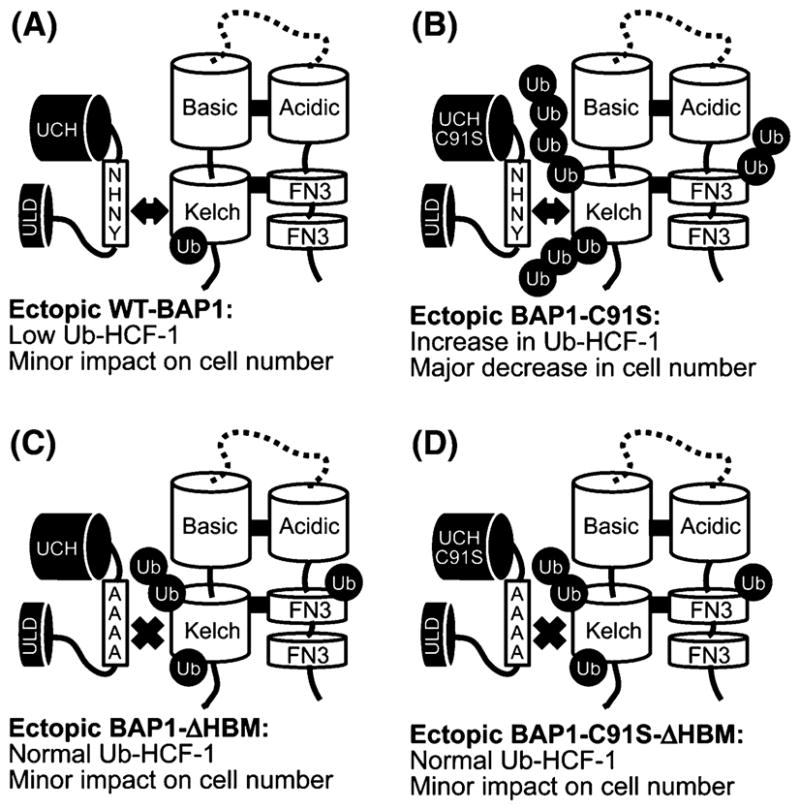
Model summarizing data from BAP1 overexpression studies. The report by Machida et al. [4] showed that knockdown of BAP1 levels by siRNA and overexpression of catalytically inactive BAP1 (C91S) both resulted in growth inhibition of MCF10a cells. To investigate the effects of BAP1 overexpression on the ubiquitination status of HCF-1, the authors co-overexpressed Flag-Kelch domain and HA-Ubiquitin and used anti-Flag immunoprecipitations to isolate the Kelch domain and any associated ubiquitin. Immunoblots revealed that WT BAP1 reduced the amount of ubiquitin that copurified with Flag-Kelch compared to cells lacking exogenous BAP1 (a). Expression of BAP1-C91S resulted in an increase in ubiquitin/Ub-Kelch (b) and thus the dominant negative effect on cell growth could be a product of inactive BAP1 saturating HCF-1 binding sites preventing endogenous, active BAP1 from deubiquitinating HCF-1. This model was tested by disrupting the BAP1/HCF-1 association, done by incorporating the ΔHBM alanine mutations with inactive BAP1 (BAP1-C91S-ΔHBM). When overexpressed the BAP1-C91S-ΔHBM protein resulted in a reduced amount of ubiquitin/Ub-Kelch that copurifies compared to BAP1-C91S (d). The BAP1-C91S-ΔHBM protein also rescued growth defects caused by overexpression of BAP1-C91S
HCF-1 Regulates Transcription from E2F-Responsive Promoters
The emerging model for the function of HCF-1 is that it regulates the transcription of various genes by recruiting chromatin remodeling complexes to transcription factors. In one such role, Herr and colleagues show that HCF-1 regulates cell cycle progression via association with the E2F family of transcription factors [7]. There are at least 8 E2F family members in humans (E2F1-8), and E2F family members can have overlapping and distinct transcriptional targets and regulate the expression of genes involved in DNA replication, DNA damage, and cell cycle progression, and apoptosis [61–64]. Five of the E2Fs (E2F1-5) contain domains that mediate interactions with the inhibitory ‘pocket proteins’ (Rb, p107, and p130) and control cell cycle progression by activating or repressing genes that promote the G1/S transition [62, 64]. Activating E2F proteins (E2F1-3a) occupy E2F-responsive promoters during the G1/S transition and S phase, while repressive E2Fs (E2F3b-5) bind these promoters during late S through early G1 phases. In one study, ectopic HCF-1 was shown to immunoprecipitate E2F1, E2F3a, and E2F4, and the associations were dependent on HBMs within these E2Fs [7]. HCF-1 preferentially associated with the activator E2F1 during the G1/S transition and thru S phase, but with the repressive E2F4 during late S through early G1 phases [7]. Remarkably, HCF-1 selectively associates with HMTs (Set1 and MLL) when bound to E2F1, but with HDAC activity (via Sin3a) when bound to E2F4 [7]. RNAi depletion of HCF-1 did not have an effect on E2F1 promoter binding, but drastically diminished transcription and the presence HMTs and H3K4 trimethylation at these promoters [7]. A similar result was obtained when investigating the varicella zoster virus (VZV) immediate-early (IE) promoter where knockdown of HCF-1 severely reduces Set1 and MLL promoter occupancy, H3K4 trimethylation, and transcription [57].
A Model for BAP1 and HCF-1 in Cell Proliferation
As discussed above, it is believed that BAP1 is involved in promoting the G1/S transition. Overexpression of the dominant negative C91S-BAP1mutant inhibits cell cycle progression and inhibition is dependent on the BAP1 HBM [4]. The dominant negative effect of C91A/S-BAP1 expression leads to the accumulation of polymeric ubiquitin in HCF-1 immunoprecipitates and growth inhibition [4, 6]. RNAi depletion of BAP1 causes growth inhibition [4] and a delayed progression into S-phase [3]. Because HCF-1 concentrations remain largely unchanged when BAP1 is overexpressed or depleted, it is possible that either ubiquitination of HCF-1 regulates a non-proteolytic function of HCF-1 or that the polymeric ubiquitin present in BAP-1 immunoprecipitates is conjugated to HCF-1-associated proteins.
HCF-1 promotes cell proliferation at the G1/S transition by recruiting H3K4 HMTs to E2F1 and activating transcription of E2F-responsive promoters [7]. Given the strong links between BAP1/HCF-1 and HCF-1/E2Fs, which are described above, the most logical conclusion is that ubiquitination of HCF-1 (or a HCF-1-associated protein) prevents activation of E2F-responsive promoters. Deubiquitination by BAP1 would relieve this inhibitory effect and promote cell proliferation. Consistent with this model, we have found that endogenous E2F4 and E2F1 are able to co-IP BAP1 from HeLa cell lysates (ZME and KDW, unpublished data). E2F1 protein levels are known to be regulated in a cell cycle dependent manner as E2F1 level peak in G1/early S and disappear in late S/G2/M phases. Ubiquitination is thought to trigger E2F1 destruction in late S phase, and indeed cullins E3 ligases complexes that promote E2F1 ubiquitination have been identified (Cul1Skp2 in humans [65], Cul4Cdt2 in Drosophila [66]). It remains unknown whether BAP1 also acts upon ubiquitinated E2F1.
Perspective
Though much progress has been made in the 2 years since the first report of a BAP1/HCF-1 association, little is known about the precise consequences of BAP1’s DUB activity on HCF-1 complexes. It does not appear that BAP1 grossly alters HCF-1 levels [4, 6], suggesting a non-proteolytic outcome of HCF-1 ubiquitination. Given the large number of proteins that associate with HCF-1, and the complexity in regulating E2F promoters, many regulatory functions can be envisioned. For example, ubiquitination of HCF-1 could regulate the associations with transcription factors (e.g., E2F1 vs. E2F4) or with activating or repressing chromatin modifying complexes (e.g. HMTs vs. HDACs). Alternatively, ubiquitination could regulate association with chromatin or have effects dependent on the cell cycle stages. In the big picture, BAP1/HCF-1 could regulate transcription at many promoters and influence several different cellular pathways. Effects observed from BAP1 overexpression and depletion studies may be a culmination of misregulating several genes. It is also plausible that other proteins, in addition to HCF-1, are deubiquitinated at BAP1/HCF-1 complexes. Certain transcription factors and histones H2A and H2B are known to be modified with ubiquitin, and BAP1 was shown to deubiquitinate H2A when complexed with ASXL1 [37]. Interestingly, the Drosophila homologs of BAP1 and ASXL1, Calypso and ASX, also act upon H2A [37], but Calypso lacks an HBM suggesting it may not function with Drosophila HCF-1. This also suggests that in humans BAP1 may have multiple cellular roles. Dissecting the precise mechanism of BAP1-mediated growth inhibition will require a better understanding of BAP1/HCF-1/transcription factor complexes and the number and types of promoters co-regulated by BAP1/HCF-1.
Contributor Information
Ziad M. Eletr, Department of Biochemistry, Emory University, Atlanta, GA 30322, USA
Keith D. Wilkinson, Email: genekdw@emory.edu, Department of Biochemistry, Emory University, Atlanta, GA 30322, USA
References
- 1.Ventii KH, Devi NS, Friedrich KL, Chernova TA, Tighiouart M, Van Meir EG, et al. BRCA1-associated protein-1 is a tumor suppressor that requires deubiquitinating activity and nuclear localization. Cancer Research. 2008;68:6953–6962. doi: 10.1158/0008-5472.CAN-08-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jensen DE, Proctor M, Marquis ST, Gardner HP, Ha SI, Chodosh LA, et al. BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene. 1998;16:1097–1112. doi: 10.1038/sj.onc.1201861. [DOI] [PubMed] [Google Scholar]
- 3.Nishikawa H, Wu W, Koike A, Kojima R, Gomi H, Fukuda M, et al. BRCA1-associated protein 1 interferes with BRCA1/BARD1 RING heterodimer activity. Cancer Research. 2009;69:111–119. doi: 10.1158/0008-5472.CAN-08-3355. [DOI] [PubMed] [Google Scholar]
- 4.Machida YJ, Machida Y, Vashisht AA, Wohlschlegel JA, Dutta A. The deubiquitinating enzyme BAP1 regulates cell growth via interaction with HCF-1. Journal of Biological Chemistry. 2009;284:34179–34188. doi: 10.1074/jbc.M109.046755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Misaghi S, Ottosen S, Izrael-Tomasevic A, Arnott D, Lamkanfi M, Lee J, et al. Association of C-terminal ubiquitin hydrolase BRCA1-associated protein 1 with cell cycle regulator host cell factor 1. Molecular and Cellular Biology. 2009;29:2181–2192. doi: 10.1128/MCB.01517-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tyagi S, Chabes AL, Wysocka J, Herr W. E2F activation of S phase promoters via association with HCF-1 and the MLL family of histone H3K4 methyltransferases. Mol Cell. 2007;27:107–119. doi: 10.1016/j.molcel.2007.05.030. [DOI] [PubMed] [Google Scholar]
- 8.Tyagi S, Herr W. E2F1 mediates DNA damage and apoptosis through HCF-1 and the MLL family of histone methyltransferases. EMBO Journal. 2009;28:3185–3195. doi: 10.1038/emboj.2009.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pickart CM. Mechanisms underlying ubiquitination. Annual Review of Biochemistry. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 10.Hershko A, Ciechanover A. The ubiquitin system. Annual Review of Biochemistry. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 11.Hicke L. Protein regulation by monoubiquitin. Nature Reviews Molecular Cell Biology. 2001;2:195–201. doi: 10.1038/35056583. [DOI] [PubMed] [Google Scholar]
- 12.Schwartz AL, Ciechanover A. The ubiquitin-proteasome pathway and pathogenesis of human diseases. Annual Review of Medicine. 1999;50:57–74. doi: 10.1146/annurev.med.50.1.57. [DOI] [PubMed] [Google Scholar]
- 13.Komander D. The emerging complexity of protein ubiquitination. Biochemical Society Transactions. 2009;37:937–953. doi: 10.1042/BST0370937. [DOI] [PubMed] [Google Scholar]
- 14.Kim HT, Kim KP, Lledias F, Kisselev AF, Scaglione KM, Skowyra D, et al. Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. Journal of Biological Chemistry. 2007;282:17375–17386. doi: 10.1074/jbc.M609659200. [DOI] [PubMed] [Google Scholar]
- 15.Bernassola F, Karin M, Ciechanover A, Melino G. The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer Cell. 2008;14:10–21. doi: 10.1016/j.ccr.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 16.Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annual Review of Biochemistry. 2009;78:363–397. doi: 10.1146/annurev.biochem.78.082307.091526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123:773–786. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Amerik AY, Hochstrasser M. Mechanism and function of deubiquitinating enzymes. Biochimica et Biophysica Acta. 2004;1695:189–207. doi: 10.1016/j.bbamcr.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 19.Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nature Reviews Molecular Cell Biology. 2009;10:550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- 20.Larsen CN, Krantz BA, Wilkinson KD. Substrate specificity of deubiquitinating enzymes: ubiquitin C-terminal hydrolases. Biochemistry. 1998;37:3358–3368. doi: 10.1021/bi972274d. [DOI] [PubMed] [Google Scholar]
- 21.Johnston SC, Riddle SM, Cohen RE, Hill CP. Structural basis for the specificity of ubiquitin C-terminal hydrolases. EMBO Journal. 1999;18:3877–3887. doi: 10.1093/emboj/18.14.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Komander D, Reyes-Turcu F, Licchesi JD, Odenwaelder P, Wilkinson KD, Barford D. Molecular discrimination of structurally equivalent Lys 63-linked and linear poly-ubiquitin chains. EMBO Report. 2009;10:466–473. doi: 10.1038/embor.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao T, Song L, Jin J, Cai Y, Takahashi H, Swanson SK, et al. Distinct modes of regulation of the Uch37 de-ubiquitinating enzyme in the proteasome and in the Ino80 chromatin-remodeling complex. Molecular Cell. 2008;31:909–917. doi: 10.1016/j.molcel.2008.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yao T, Song L, Xu W, DeMartino GN, Florens L, Swanson SK, et al. Proteasome recruitment and activation of the Uch37 deubiquitinating enzyme by Adrm1. Nature Cell Biology. 2006;8:994–1002. doi: 10.1038/ncb1460. [DOI] [PubMed] [Google Scholar]
- 25.Qiu XB, Ouyang SY, Li CJ, Miao S, Wang L, Goldberg AL. hRpn13/ADRM1/GP110 is a novel proteasome subunit that binds the deubiquitinating enzyme, UCH37. EMBO Journal. 2006;25:5742–5753. doi: 10.1038/sj.emboj.7601450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamazaki J, Iemura S, Natsume T, Yashiroda H, Tanaka K, Murata S. A novel proteasome interacting protein recruits the deubiquitinating enzyme UCH37 to 26S proteasomes. EMBO Journal. 2006;25:4524–4536. doi: 10.1038/sj.emboj.7601338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lam YA, Xu W, DeMartino GN, Cohen RE. Editing of ubiquitin conjugates by an isopeptidase in the 26S proteasome. Nature. 1997;385:737–740. doi: 10.1038/385737a0. [DOI] [PubMed] [Google Scholar]
- 28.Nishikawa H, Ooka S, Sato K, Arima K, Okamoto J, Klevit RE, et al. Mass spectrometric and mutational analyses reveal Lys-6-linked polyubiquitin chains catalyzed by BRCA1-BARD1 ubiquitin ligase. Journal of Biological Chemistry. 2004;279:3916–3924. doi: 10.1074/jbc.M308540200. [DOI] [PubMed] [Google Scholar]
- 29.Morris JR, Solomon E. BRCA1:BARD1 induces the formation of conjugated ubiquitin structures, dependent on K6 of ubiquitin, in cells during DNA replication and repair. Human Molecular Genetics. 2004;13:807–817. doi: 10.1093/hmg/ddh095. [DOI] [PubMed] [Google Scholar]
- 30.Wu-Baer F, Lagrazon K, Yuan W, Baer R. The BRCA1/BARD1 heterodimer assembles polyubiquitin chains through an unconventional linkage involving lysine residue K6 of ubiquitin. Journal of Biological Chemistry. 2003;278:34743–34746. doi: 10.1074/jbc.C300249200. [DOI] [PubMed] [Google Scholar]
- 31.Chen A, Kleiman FE, Manley JL, Ouchi T, Pan ZQ. Autoubiquitination of the BRCA1*BARD1 RING ubiquitin ligase. Journal of Biological Chemistry. 2002;277:22085–22092. doi: 10.1074/jbc.M201252200. [DOI] [PubMed] [Google Scholar]
- 32.Mallery DL, Vandenberg CJ, Hiom K. Activation of the E3 ligase function of the BRCA1/BARD1 complex by polyubiquitin chains. EMBO Journal. 2002;21:6755–6762. doi: 10.1093/emboj/cdf691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Starita LM, Horwitz AA, Keogh MC, Ishioka C, Parvin JD, Chiba N. BRCA1/BARD1 ubiquitinate phosphorylated RNA polymerase II. Journal of Biological Chemistry. 2005;280:24498–24505. doi: 10.1074/jbc.M414020200. [DOI] [PubMed] [Google Scholar]
- 34.Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, et al. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Molecular Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 35.Sato K, Hayami R, Wu W, Nishikawa T, Nishikawa H, Okuda Y, et al. Nucleophosmin/B23 is a candidate substrate for the BRCA1-BARD1 ubiquitin ligase. Journal of Biological Chemistry. 2004;279:30919–30922. doi: 10.1074/jbc.C400169200. [DOI] [PubMed] [Google Scholar]
- 36.Eakin CM, Maccoss MJ, Finney GL, Klevit RE. Estrogen receptor alpha is a putative substrate for the BRCA1 ubiquitin ligase. Proceedings of National Academic Science of United States of America. 2007;104:5794–5799. doi: 10.1073/pnas.0610887104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly-Hartig N, McGinty RK, Fraterman S, et al. Histone H2A deubiquitinase activity of the polycomb repressive complex PR-DUB. Nature. 2010;465:243–247. doi: 10.1038/nature08966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaestner KH, Knochel W, Martinez DE. Unified nomenclature for the winged helix/forkhead transcription factors. Genes and Development. 2000;14:142–146. [PubMed] [Google Scholar]
- 39.Shi X, Bowlin KM, Garry DJ. Fhl2 interacts with foxk1 and corepresses foxo4 activity in myogenic progenitors. Stem Cells. 2010;28:462–469. doi: 10.1002/stem.274. [DOI] [PubMed] [Google Scholar]
- 40.Meeson AP, Shi X, Alexander MS, Williams RS, Allen RE, Jiang N, et al. Sox15 and Fhl3 transcriptionally coactivate Foxk1 and regulate myogenic progenitor cells. EMBO Journal. 2007;26:1902–1912. doi: 10.1038/sj.emboj.7601635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hawke TJ, Jiang N, Garry DJ. Absence of p21CIP rescues myogenic progenitor cell proliferative and regenerative capacity in Foxk1 null mice. Journal of Biological Chemistry. 2003;278:4015–4020. doi: 10.1074/jbc.M209200200. [DOI] [PubMed] [Google Scholar]
- 42.Jensen DE, Rauscher FJ., 3rd Defining biochemical functions for the BRCA1 tumor suppressor protein: analysis of the BRCA1 binding protein BAP1. Cancer Letters. 1999;143(Suppl 1):S13–S17. doi: 10.1016/s0304-3835(99)90004-6. [DOI] [PubMed] [Google Scholar]
- 43.Angeloni D. Molecular analysis of deletions in human chromosome 3p21 and the role of resident cancer genes in disease. Briefings in functional genomics and proteomics. 2007;6:19–39. doi: 10.1093/bfgp/elm007. [DOI] [PubMed] [Google Scholar]
- 44.Wysocka J, Reilly PT, Herr W. Loss of HCF-1-chromatin association precedes temperature-induced growth arrest of tsBN67 cells. Molecular and Cellular Biology. 2001;21:3820–3829. doi: 10.1128/MCB.21.11.3820-3829.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilson AC, LaMarco K, Peterson MG, Herr W. The VP16 accessory protein HCF is a family of polypeptides processed from a large precursor protein. Cell. 1993;74:115–125. doi: 10.1016/0092-8674(93)90299-6. [DOI] [PubMed] [Google Scholar]
- 46.Wilson AC, Peterson MG, Herr W. The HCF repeat is an unusual proteolytic cleavage signal. Genes and Development. 1995;9:2445–2458. doi: 10.1101/gad.9.20.2445. [DOI] [PubMed] [Google Scholar]
- 47.Goto H, Motomura S, Wilson AC, Freiman RN, Nakabeppu Y, Fukushima K, et al. A single-point mutation in HCF causes temperature-sensitive cell-cycle arrest and disrupts VP16 function. Genes and Development. 1997;11:726–737. doi: 10.1101/gad.11.6.726. [DOI] [PubMed] [Google Scholar]
- 48.Reilly PT, Wysocka J, Herr W. Inactivation of the retinoblastoma protein family can bypass the HCF-1 defect in tsBN67 cell proliferation and cytokinesis. Molecular and Cellular Biology. 2002;22:6767–6778. doi: 10.1128/MCB.22.19.6767-6778.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Julien E, Herr W. Proteolytic processing is necessary to separate and ensure proper cell growth and cytokinesis functions of HCF-1. EMBO Journal. 2003;22:2360–2369. doi: 10.1093/emboj/cdg242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Freiman RN, Herr W. Viral mimicry: common mode of association with HCF by VP16 and the cellular protein LZIP. Genes and Development. 1997;11:3122–3127. doi: 10.1101/gad.11.23.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu R, Yang P, Padmakumar S, Misra V. The herpesvirus transactivator VP16 mimics a human basic domain leucine zipper protein, luman, in its interaction with HCF. Journal of Virology. 1998;72:6291–6297. doi: 10.1128/jvi.72.8.6291-6297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luciano RL, Wilson AC. HCF-1 functions as a coactivator for the zinc finger protein Krox20. Journal of Biological Chemistry. 2003;278:51116–51124. doi: 10.1074/jbc.M303470200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, et al. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Molecular and Cellular Biology. 2004;24:5639–5649. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wysocka J, Myers MP, Laherty CD, Eisenman RN, Herr W. Human Sin3 deacetylase and trithorax-related Set1/Ash2 histone H3–K4 methyltransferase are tethered together selectively by the cell-proliferation factor HCF-1. Genes and Development. 2003;17:896–911. doi: 10.1101/gad.252103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vogel JL, Kristie TM. The novel coactivator C1 (HCF) coordinates multiprotein enhancer formation and mediates transcription activation by GABP. EMBO Journal. 2000;19:683–690. doi: 10.1093/emboj/19.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gunther M, Laithier M, Brison O. A set of proteins interacting with transcription factor Sp1 identified in a two-hybrid screening. Molecular and Cellular Biochemistry. 2000;210:131–142. doi: 10.1023/a:1007177623283. [DOI] [PubMed] [Google Scholar]
- 57.Narayanan A, Ruyechan WT, Kristie TM. The coactivator host cell factor-1 mediates Set1 and MLL1 H3K4 trimethylation at herpesvirus immediate early promoters for initiation of infection. Proceedings of National Academic Science of United States of America. 2007;104:10835–10840. doi: 10.1073/pnas.0704351104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scarr RB, Sharp PA. PDCD2 is a negative regulator of HCF-1 (C1) Oncogene. 2002;21:5245–5254. doi: 10.1038/sj.onc.1205647. [DOI] [PubMed] [Google Scholar]
- 59.Ajuh PM, Browne GJ, Hawkes NA, Cohen PT, Roberts SG, Lamond AI. Association of a protein phosphatase 1 activity with the human factor C1 (HCF) complex. Nucleic Acids Research. 2000;28:678–686. doi: 10.1093/nar/28.3.678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Julien E, Herr W. A switch in mitotic histone H4 lysine 20 methylation status is linked to M phase defects upon loss of HCF-1. Molecular Cell. 2004;14:713–725. doi: 10.1016/j.molcel.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 61.DeGregori J, Johnson DG. Distinct and Overlapping Roles for E2F Family Members in Transcription, Proliferation and Apoptosis. Current Molecular Medicine. 2006;6:739–748. doi: 10.2174/1566524010606070739. [DOI] [PubMed] [Google Scholar]
- 62.Blais A, Dynlacht BD. Hitting their targets: an emerging picture of E2F and cell cycle control. Current Opinion in Genetics and Development. 2004;14:527–532. doi: 10.1016/j.gde.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 63.Cam H, Dynlacht BD. Emerging roles for E2F: beyond the G1/S transition and DNA replication. Cancer Cell. 2003;3:311–316. doi: 10.1016/s1535-6108(03)00080-1. [DOI] [PubMed] [Google Scholar]
- 64.Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nature Reviews Molecular Cell Biology. 2002;3:11–20. doi: 10.1038/nrm714. [DOI] [PubMed] [Google Scholar]
- 65.Marti A, Wirbelauer C, Scheffner M, Krek W. Interaction between ubiquitin-protein ligase SCFSKP2 and E2F–1 underlies the regulation of E2F-1 degradation. Nature Cell Biology. 1999;1:14–19. doi: 10.1038/8984. [DOI] [PubMed] [Google Scholar]
- 66.Shibutani ST, de la Cruz AF, Tran V, Turbyfill WJ, III, Reis T, Edgar BA, et al. Intrinsic negative cell cycle regulation provided by PIP box- and Cul4Cdt2-mediated destruction of E2f1 during S phase. Developmental Cell. 2008;15:890–900. doi: 10.1016/j.devcel.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]