

Abstract
Electron crystallography relies on electron cryomicroscopy of two-dimensional (2D) crystals and is particularly well suited for studying the structure of membrane proteins in their native lipid bilayer environment. To obtain 2D crystals from purified membrane proteins, the detergent in a protein-lipid-detergent ternary mixture must be removed, generally by dialysis, under conditions favoring reconstitution into proteoliposomes and formation of well-ordered lattices. To identify these conditions a wide range of parameters such as pH, lipid composition, lipid-to-protein ratio, ionic strength and ligands must be screened in a procedure involving four steps: crystallization, specimen preparation for electron microscopy, image acquisition, and evaluation. Traditionally, these steps have been carried out manually and, as a result, the scope of 2D crystallization trials has been limited. We have therefore developed an automated pipeline to screen the formation of 2D crystals. We employed a 96-well dialysis block for reconstitution of the target protein over a wide range of conditions designed to promote crystallization. A 96-position magnetic platform and a liquid handling robot were used to prepare negatively stained specimens in parallel. Robotic grid insertion into the electron microscope and computerized image acquisition ensures rapid evaluation of the crystallization screen. To date, 38 2D crystallization screens have been conducted for 15 different membrane proteins, totaling over 3000 individual crystallization experiments. Three of these proteins have yielded diffracting 2D crystals. Our automated pipeline outperforms traditional 2D crystallization methods in terms of throughput and reproducibility.
Keywords: Automation, dialysis, electron crystallography, high-throughput, membrane protein, two-dimensional (2D) crystals
Introduction
Biological membranes surround all cells and many internal organelles. A 50–60Å thick bilayer [1, 2] of lipid constitutes the matrix and membrane proteins are either embedded in this bilayer or are associated with its surface. These proteins mediate a wide variety of cellular functions, including nutrient transport, electrolyte balance, signal transduction and energy conversion. Specifically, membrane proteins act as receptors, enzymes, channels, transporters, pumps, structural proteins and cell-cell adhesion molecules. When considered on a genome-wide scale, membrane proteins constitute approximately 30% of the transcriptome in all phyla [3, 4] and defective membrane proteins give rise to a broad spectrum of human diseases including cystic fibrosis [5, 6], cancer [7], Alzheimer’s disease [8], and various cardiomyopathies [9–12]. As a result, ~60% of the therapeutic drugs currently used in the United States target membrane proteins and their assemblies [13, 14].
Despite their tremendous relevance to basic cell biology and to therapeutic medicine, our understanding of membrane proteins from a structural perspective is limited. Certainly, there has recently been significant progress in solving the structures of detergent-solubilized membrane proteins, but visualizing them within their native membrane environment has proven extremely challenging. This is largely due to the difficulty in applying X-ray crystallography and solution NMR spectroscopy to membrane proteins embedded in the two-dimensional (2D) plane of the lipid bilayer. Despite recent advances in solid state NMR [15], electron crystallography is the most generally applicable method. Electron crystallography involves the application of electron cryomicroscopy on 2D crystalline lattices, and was pioneered in the 1970s by Henderson and Unwin through their studies of bacteriorhodopsin [16]. Electron crystallography is routinely used to produce structures at 5–8 Å resolution and has produced atomic structures of bacteriorhodopsin [17], plant light-harvesting complex [18], human red cell aquaporin-1 [19], eye lens aquaporin-0 [20], rat aquaporin-4 [21], glutathione transferase [22], and acetylcholine receptor [23].
Well ordered, 2D crystals of a given membrane protein generally grow within a relatively narrow window of conditions. To find this window, the most common factors, such as lipid, detergent, lipid-to-protein ratio (LPR), pH, salt and temperature [24, 25], need to be comprehensively screened. In X-ray crystallography, automated high-throughput screens encompass a large number of factors and represent a standard approach for producing crystals that diffract to high-resolution [26–29]. This screening is facilitated by the use of 24- or 96-well crystallization trays for vapor diffusion, which can be rapidly evaluated by optical microscopy. In contrast, 2D crystallography has generally been limited to smaller, manual screens, due to difficult logistics in parallelizing and automating both the crystallization and evaluation steps. Early efforts in automating 2D crystallization include a device for dialyzing up to 30 samples in parallel [30]. More recently, we designed a 96-well crystallization block [31] and Iacovache et al. have presented a robot for dispensing cyclodextrin as an alternative to remove detergent and effect crystallization [32]. Evaluation of these crystal trials represents another serious bottleneck resulting from the laborious nature of specimen preparation and imaging by transmission electron microscopy (EM).
To address these hurdles we have developed a pipeline that automates the key steps involved in setting up and screening 2D crystallization trials. Our dialysis device [31] allows 96 unique samples to be dialyzed against 96 different buffers. Following dialysis, a 96-position magnetic platform is employed for automated negative staining of these crystallization trials, which is required for EM. Robotic grid insertion into the electron microscope and computerized image acquisition thereafter facilitate the rapid evaluation of the crystallization screen [33]. A matrix of 2D crystallization conditions has also been developed to systematically evaluate the effect of select parameters such as lipids, pH, LPRs, and cations. The screen has been applied to 15 different prokaryotic membrane proteins, thus producing more than 3000 crystallization experiments that have been analyzed by EM. The results from the outcomes have thereafter been scored based both on the quality and quantity of objects found in each sample. From three of these membrane proteins, 2D crystals have been produced.
Materials and methods
Chemicals
Lyophilized lipids (DMPC, DOPC, DOPG, POPC, DOPE and E.coli polar lipid extract) were purchased from Avanti Polar Lipids (Alabaster, AL). Lipid stocks, at a final lipid concentration of 20mg/ml, were prepared in a mixture of 96% chloroform and 4% methanol and stored in Teflon-capped vessels under N2 gas at −80°C. Stock solutions of butylatedhydroxytoluene (BHT) were prepared at 2mg/ml in ethanol. All detergents were purchased from Anatrace Inc. (Maumee, OH) and were stored desiccated at −20°C prior to making stock solutions.
Proteins for 2D crystallization screens
We conducted 2D crystallization trials on fifteen different membrane proteins (Table 1). The Semliki forest virus E1 protein and the β1-adrenergic receptor were produced by our collaborators following procedures described by Sanchez-San Martin et al. [34] and Warne et al. [35], respectively. The remaining protein targets were produced in our own laboratory or in collaborating laboratories associated with the New York Consortium on Membrane Protein Structure (NYCOMPS; www.nycomps.com). These protein targets were expressed and purified according to a standard protocol routinely employed by NYCOMPS. Prokaryotic membrane protein targets, carrying either N- or C-terminal His10-tags to allow purification by metal affinity chromatography, were expressed in E. coli BL21(DE3)pLysS-T1R cells (Sigma-Aldrich, Inc.) from IPTG-inducible T7 vectors. Prior to large-scale production, the detergent preference of each membrane protein target was pre-screened using size exclusion chromatography [36]. To this end each target was first produced on a mid-scale. A 500ml of transformed cells were grown at 37°C to an OD600 of 0.6–1, induced with 0.4 μM IPTG and harvested 3h later. Following isolation of cytoplasmic E. coli membranes by ultracentrifugation at 100,000xg, the membrane protein target was extracted in buffer containing 30 mM imidazole and 1% dodecylmaltoside (DDM) for 4h. The solubilizate was then bound to 0.1 ml of Ni-NTA beads (Qiagen, Valencia, CA) and eluted in DDM at twice its critical micelle concentration (CMC). Aliquots (5μl) of the eluted protein were thereafter injected on a silica based size exclusion column (TSKgel Super SW3000, 4.6mm by 30cm, Tosoh Biosciences, Montgomeryville, PA) at a flow rate of 0.25ml/min using a HPLC system (Agilent Technologies, Santa Clara, CA). This column had been pre-equilibrated with a standard mobile phase of aqueous buffer containing decylmaltoside (DM), DDM, octylglucoside (OG), or nonylglucoside (NG) at twice their respective CMC’s, and protein elution was monitored by UV absorbance. Membrane proteins yielding elution profiles characteristic of monodisperse preparations (i.e., a single sharp peak) were classified as stable in the corresponding detergent. As assessed by SDS-PAGE, using bovine serum albumin as a concentration standard, the yield of purified membrane protein target was on average >0.5mg per liter of E. coli culture.
Table 1. Summary of 2D crystallization trials.
| Protein | Organism | (Putative) Function | MW (kDa) | pI | TMD | N-term | % TM | Screens | Best Outcome |
|---|---|---|---|---|---|---|---|---|---|
| E2P1 | M.voltae | Protease | 25.9 | 8.7 | 4 | Inside | 45 | 4 | Sheets, Tubes |
| E2P2 | M.maripaludis | Protease | 25.9 | 5.0 | 4 | Inside | 39 | 3 | Sheets, Tubes, |
| E2P3 | M.marisnigri | Protease | 31.8 | 5.3 | 7 | Inside | 59 | 2 | Sheets, Tubes |
| E2P4 | M.hungatei | Protease | 32.9 | 7.8 | 9 | Inside | 61 | 2 | Sheets, Tubes |
| P2A3 | S.oneidensis | Cation efflux family protein | 32.5 | 5.4 | 5 | Inside | 36 | 5 | Helical crystals |
| Rhomboid PA3086 | P.aeruginosa | Intramembrane protease | 31.8 | 9.9 | 6 | Inside | 45 | 4 | Lipidic structures |
| YkgB-D332 | E.coli | Unknown | 21.9 | 6.0 | 3 | Inside | 38 | 1 | Tubes |
| YkgB-D36 | E.coli | Unknown | 21.8 | 5.7 | 3 | Inside | 37 | 4 | Helical crystals |
| β1-adrenergic receptor | M.gallopavo | G-protein coupled receptor | 54.1 | 9.3 | 7 | Outside | 33 | 3 | Sheets, Tubes |
| Rhomboid GlpG | E.coli | Intramembrane protease | 31.3 | 9.2 | 6 | Inside | 46 | 1 | Aggregates |
| Cytochrome b561 | P.aeruginosa | Electron carrier activity | 20.6 | 9.6 | 4 | Inside | 47 | 1 | Lipidic structures |
| E1 protein | Semliki Forest Virus | Inducer of membrane fusion | 47.4 | 7.6 | 0 (1) | Outside | 5 | 4 | Sheet crystals |
| P40B7 | B.subtilis | Sporulation kinase C | 48.8 | 6.3 | 2 | Inside | 10 | 1 | Vesicles, Sheets |
| Kdp-ATPase (4 subunits) | E.coli | High-affinity K-pump | 159.2 | 5.2–9.4 | 17 | 3 in, 1out | 31 | 1 | Sheets, Tubes |
| P39H10 | K. pneumoniae | Diguanylate cyclase | 45.7 | 6.5 | 2 | Inside | 11 | 1 | Sheets, Tubes |
PI: isoelectric point; TMD: transmembrane domain; N-Term: location of N-teminus; %TM: % of transmembrane sequence ; Screens: Number of screens
Crystallization was conducted at a protein concentration of 0.5mg/ml; therefore, ~2.5mg of protein were needed to set up a complete 96-condition screen. Thus following identification of the optimal detergent, the expression of the target membrane protein was scaled up to 6 liters. The cytoplasmic membranes were isolated by ultracentrifugation at 100,000xg and the membrane protein target was extracted in buffer containing 30 mM imidazole and the optimal detergent at a concentration of 5–10 times its CMC for 4h. Following solubilization, the protein was bound to 0.5 ml of Ni-NTA beads and eluted with 250 mM Imidazole in the same detergent (at 2–4 times its CMC). After elution, the linker region between the tag and the target protein was removed by proteolysis with either TEV or thrombin. The last step in the purification of a target protein involved size exclusion chromatography over a HR Superdex 200 (GE Healthcare) column pre-equilibrated with aqueous buffer containing detergent at twice its CMC. Following purification, each target membrane protein was concentrated to >1mg/ml and clarified by centrifugation at 200,000xg for 30 min. Protein purity and concentration was routinely assessed by SDS-PAGE.
Lipid solubilization
The minimum detergent concentration needed to solubilize each lipid species used for the 2D crystallization screens was determined by turbidity [37]. Using a Hamilton syringe, 0.75μg of the lipid stock solution and 4μg of BHT were transferred to individual glass test tubes. A thin lipid film was produced by vigorously vortexing the tube while evaporating the solvent with N2 gas. To ensure complete solvent removal, the test tube was placed in a vacuum (~10−3 mbar) for 1h. The dry lipid films were resuspended in 0.5ml of aqueous solution, which contained small amounts of detergent: DDM (0.05mg/ml), Triton X-100 (0.05mg/ml), OG (1mg/ml), OTG (1mg/ml) and C8E5 (1mg/ml). To these samples, increasing detergent amounts were added and the absorbance at 500nm was monitored. With limited amounts of detergent, lipid vesicles are present and scattering of 500nm light (OD500) is relatively high. As more detergent is added, and the lipid vesicles become solubilized, the OD500 decreases, eventually falling to a stable baseline corresponding to fully solubilized lipid as lipid-detergent mixed micelles. For crystallization, lipid stocks contained 1.5mg/ml of lipid, a sufficient amount of detergent to ensure full solubilization (Table 2), and 10mM MES at pH 6.5. These stocks were stored for up to four days at 4°C, or frozen at −80°C for long-term storage.
Table 2. Standard screening matrix used for systematic 2D crystallization screens.
|
Lipids: DMPC, DOPC, POPC, DOPG, E.coli polar lipid extract pH: 6.0 (20mM MES), 7.0 (20mM TES), 8.0 (20mM TES) LPRs: 0.25, 0.75, 1.5 Cations (mM): Na+ (100), Mg2+ (10) |
||||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
| A | X | DOPC 0.25 |
POPC 0.75 |
DOPG 1.5 |
X | DOPC 0.25 |
POPC 0.75 |
DOPG 1.5 |
X | DOPC 0.25 |
POPC 0.75 |
DOPG 1.5 |
| B | DMPC 0.25 |
DOPC 0.75 |
POPC 1.5 |
E.coli 0.25 |
DMPC 0.25 |
DOPC 0.75 |
POPC 1.5 |
E.coli 0.25 |
DMPC 0.25 |
DOPC 0.75 |
POPC 1.5 |
E.coli 0.25 |
| C | DMPC 0.75 |
DOPC 1.5 |
DOPG 0.25 |
E.coli 0.75 |
DMPC 0.75 |
DOPC 1.5 |
DOPG 0.25 |
E.coli 0.75 |
DMPC 0.75 |
DOPC 1.5 |
DOPG 0.25 |
E.coli 0.75 |
| D | DMPC 1.5 |
POPC 0.25 |
DOPG 0.75 |
E.coli 1.5 |
DMPC 1.5 |
POPC 0.25 |
DOPG 0.75 |
E.coli 1.5 |
DMPC 1.5 |
POPC 0.25 |
DOPG 0.75 |
E.coli 1.5 |
| E | X | DOPC 0.25 |
POPC 0.75 |
DOPG 1.5 |
X | DOPC 0.25 |
POPC 0.75 |
DOPG 1.5 |
X | DOPC 0.25 |
POPC 0.75 |
DOPG 1.5 |
| F | DMPC 0.25 |
DOPC 0.75 |
POPC 1.5 |
E.coli 0.25 |
DMPC 0.25 |
DOPC 0.75 |
POPC 1.5 |
E.coli 0.25 |
DMPC 0.25 |
DOPC 0.75 |
POPC 1.5 |
E.coli 0.25 |
| G | DMPC 0.75 |
DOPC 1.5 |
DOPG 0.25 |
E.coli 0.75 |
DMPC 0.75 |
DOPC 1.5 |
DOPG 0.25 |
E.coli 0.75 |
DMPC 0.75 |
DOPC 1.5 |
DOPG 0.25 |
E.coli 0.75 |
| H | DMPC 1.5 |
POPC 0.25 |
DOPG 0.75 |
E.coli 1.5 |
DMPC 1.5 |
POPC 0.25 |
DOPG 0.75 |
E.coli 1.5 |
DMPC 1.5 |
POPC 0.25 |
DOPG 0.75 |
E.coli 1.5 |
pH6.0,
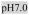 ,
,
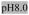
Rows A-D contain 100mM Na+; Rows E-H contain 10mM Mg2+
Set up of the 96-well crystallization block
Aliquots of a purified membrane protein target were mixed with detergent-solubilized lipids in selected LPRs and the protein-lipid-detergent ternary mixtures were incubated for 1h at room temperature in 96-well disposable microtiter plates. Thereafter, 53μl of each sample was transferred to individual sample wells of the crystallization block [31]. Sheets of dialysis membrane (MWCO 12,000Da, Spectrum Laboratories, Rancho Dominguez, CA) were boiled in 5mM EDTA at pH 8.0 and cut to match the dimensions of the crystallization block (~12cm × 8cm). After securing the top and bottom halves of the block together, 950μl of dialysis buffer was added to each of the buffer chambers and the block was placed in an incubator at either 4, 20 or 37°C. Buffer was exchanged twice daily for 2 weeks. To recover each crystallization trial, the dialysis membrane was punctured with a pipette tip, and the dialysate was transferred to corresponding positions in a 96-well disposable microtiter plate.
Automated staining protocol
Batches of carbon-coated EM grids were prepared as described in Vink et al. 2007 [31]. For negative staining, the EM grids were manually transferred onto the magnetic support platform (SPRI plate 384 Post Magnet Plate, Agencourt, Beverly MA) that was thereafter placed in a liquid-handling robot (Biomek FX, Beckman Coulter, Fullerton, CA). Using the 96-pipette head, the robot applied 2μl of each crystallization condition to the grids for 30s, followed by 8μl of stain solution (0.25% uranyl acetate in water). An 8μl aliquot was immediately aspirated from the grids and replaced with 8μl of fresh stain solution. This step was repeated with an 8s incubation and then again with a 16s incubation. Finally, an 8μl aliquot of stain was added to each grid, the magnetic platform removed from the robot, excess stain blotted away (one row of 12 grids at a time) using long strips of filter paper (figure 3b), and the grids were left to air-dry. The grids were then manually transferred to a custom-made storage block for later evaluation by TEM. The stain solution was filtered immediately before the protocol to remove any precipitate.
Figure 3. Setup for preparation of EM specimens by negative staining.

a) The magnetic plate used for the automated negative staining protocol. Each post on the plate is made from an individual rare earth magnet. A carbon coated, glow-discharged Ni-grid is manually transferred to the plate by tweezers. b) Following completion of the staining protocol, the last aliquot of stain is left on the grids and the magnetic plate is removed from the liquid-handling robot. Long strips of filter paper are used to blot away the remaining stain from the grids, one row of grids at a time.
Results
Overview of the 2D crystallization screening pipeline
The screening of 2D crystallization trials can be divided into four key steps: (1) crystallization, (2) preparation of specimens for EM, (3) image acquisition, and (4) data analysis and evaluation. All these steps have been incorporated into an automated pipeline (Fig. 1) that, to date, has been used to carry out 38 2D crystallization screens on 15 different membrane protein targets. Thus, over 3000 unique crystallizations (90 samples in initial screens and at least 60 in follow up screens) have been evaluated so far. For 2D crystallization, reconstitution of a purified target protein into a lipid bilayer is achieved by using dialysis to remove the detergent. To dialyze multiple samples in parallel, a previously designed crystallization block was employed [31]. This block was built with standard 96-well SBS microplate dimensions and permits the dialysis of 96 unique samples against 96 different buffers. Our dialysis block accommodates sample and buffer volumes of 50μl and 950μl respectively, thus effectively producing a 20-fold dilution with each buffer exchange. Following detergent dialysis for periods of up to two weeks, EM specimens are prepared by negative staining in batches of up to 96: a liquid handling robot transfers 2μl of each dialysate onto a carbon-coated EM-grid and then washes the grids with stain. Specimen insertion into the electron microscope and image acquisition has been fully automated. The image-acquisition software (Leginon; [38, 39]) coordinates insertion of the specimens by a custom made robot and records a set of images from each grid in an unattended manner [33]. Currently, an experienced microscopist performs data analysis and evaluation, but efforts to automate these steps using algorithms for crystal detection and automated collection of diffraction data are underway.
Figure 1. Pipeline to screen 2D crystallization trials of membrane proteins.

Target membrane proteins are purified in detergent micelles in a stable and monodisperse form. Following the addition of lipids to form protein-lipid-detergent ternary mixtures, excess detergent is removed by dialysis using the 96-well crystallization block. Upon completion of a 2D crystallization experiment the 96 conditions are harvested, transferred to EM grids and negative stained specimens made in parallel using a liquid-handling robot. The EM grids are robotically inserted into the electron microscope, and images of the crystallization outcomes are recorded automatically by image-acquisition software. Finally, an experienced microscopist performs data analysis and evaluation. A negatively stained image of a 2D crystal of YkgB-D36, in which the lattice is clearly visible, is shown as an example of a successful outcome.
2D crystallization matrix
Defining a matrix of conditions to effectively sample the key parameters influencing 2D crystallization is essential. These parameters include lipid composition, LPR, protein concentration, pH, temperature, detergent type, ions, inhibitors and ligands. To conduct a truly comprehensive screen, each parameter should be varied independently, but this becomes impractical when a large number of parameters are included and the number of conditions grows exponentially. We therefore selected a subset of parameters, based on previous experience and test screens, to be evaluated in initial screens of novel membrane protein targets. In particular, we chose to vary pH (6.0, 7.0, 8.0), lipid type (DMPC, DOPC, POPC, DOPG, E. coli polar lipid extract) LPR (0.25, 0.75, 1.5), and cation (10mM Mg2+ or 100mM Na+), thus producing a total of 90 conditions (3 (pH) × 5 (lipid) × 3 (LPR) × 2 (cation)), a number that can be accommodated in a single 96-well crystallization block. All solutions also contained 5mM NaN3. Table 2 lists the initial parameters sampled by our screening matrix.
Choice of detergent is essential for protein stability during purification. However, since detergent is removed during reconstitution, we reasoned that it is of secondary importance for crystal formation relative to the lipids that ultimately form the crystalline environment. We therefore used the purification detergent, at least for initial 2D crystallization screens. Although temperature is also an important parameter, initial screens were incubated at room temperature, and the effect of temperature was instead evaluated in follow-up screens, where either constant incubation at 4, 20 and 37°C, or cycling between these temperatures at 12-hour intervals was performed. Other parameters evaluated in follow up screens included lipid mixtures, sampling of LPRs at smaller intervals, and cationic species and concentration.
Proteins, detergents, and lipids
The membrane proteins that have been channeled through the 2D crystallization pipeline represent either our own research interests or those of NYCOMPS and additional collaborators. All target proteins are postulated α-helical integral membrane proteins, containing at least one putative α-helical transmembrane domain (TMD). The E1-fusion protein from the Semliki Forest Virus is a special case, since it presents its hydrophobic fusion peptide only upon exposure to low pH [40], and can be treated as a soluble protein until setup for crystallization. All proteins are also monomeric, except the high-affinity potassium pump Kdp that is composed of 4 different subunits [41]. Although all the proteins screened to date, except the β1-adrenergic receptor, are from prokaryotic sources, we intend to shift much of our focus to eukaryotic targets in the future.
The detergent can play an important role in the stability and monodispersity of a membrane protein. To identify the optimal detergent for purification we routinely prescreened the detergent preference of our target proteins by size exclusion chromatography (Fig. 2a), and used elution as a single sharp peak, following prolonged incubation in a given detergent, as indicative of stability and monodispersity [36]. For each target protein, the different steps of solubilization, metal affinity purification, and size exclusion chromatography were all carried out in this optimal detergent. This general strategy allowed us to produce sufficient amounts (~2.5mg of purified protein needed per 90 conditions; 0.5mg/ml protein × 53 μl per well × 90 wells) of each target protein, and with enough purity (Fig. 2b) and stability as required to set up a full 96-well 2D crystallization trial.
Figure 2. Production of target membrane proteins and lipid solubilization.

a) Membrane protein stability screen in different detergents. In this case, the protein YkgB-D332 was purified in DDM and run on a silica based size exclusion chromatography column equilibrated with an aqueous mobile phase containing the detergents DM, DDM, OG and NG at a concentration of twice the CMC. The elution profiles (rate of 0.25 ml/min) are shown. The color codes for the traces are red=DM, blue=DDM, green=OG and pink=NG. b) As evaluated by SDS-PAGE the proteins used in the 2D crystallization trials were at least 90% pure. Note that the acryl amide concentration used in the gels varies. The Kdp protein is composed of four different subunits, of which one is only 3kDa and appears as a smear at the bottom of the gel. 1: E2P1, 2: E2P2, 3: E2P3, 4: E2P4, 5: P2A3, 6: Rhomboid PA3086, 7: YkgB-D332, 8: YkgB-D36, 9: β1-adrenergic receptor, 10: Rhomboid GlpG, 11: Cytochrome b561, 12: E1 protein, 13: P40B7, 14: Kdp-ATPase, 15: P39H10. c) Turbidity measurements were employed to assess the solubilization of lipid by detergent. The initial turbidity of the solution was normalized to 100%. The lipid concentration was kept constant at 1.5mg/ml throughout the experiment. Increasing amounts of highly concentrated OG were added to the mixture and the UV absorbance was recorded at 500nm following equilibration. As detergent is added, and the lipid vesicles become solubilized as lipid-detergent mixed micelles, the OD500 decreases, eventually falling to a stable baseline corresponding to fully solubilized lipid. All lipids became fully solubilized at an OG concentration of 10mg/ml.
The detergent used for protein purification was also used to solubilize the lipids included in the crystallization screen. To find an optimal detergent concentration for complete lipid solubilization we studied the solubilization process using turbidity measurements [37]. The five most commonly used detergents in protein purification (Triton X-100, DDM, C8E5, OG and OTG) and the five lipids used in our 2D crystallization screens (DMPC, DOPC, POPC, DOPG, and E.coli polar lipid extract) were evaluated (Fig. 2c, Table 3). The detergent concentration required to solubilize a particular lipid varied considerably among the different detergents. Triton X-100 (CMC = 0.15mg/ml) was clearly the most efficient, requiring <3mg/ml to solubilize all the lipids tested. The other low-CMC detergent, DDM (CMC = 0.087mg/ml), required considerably higher concentrations (up to 14mg/ml) to achieve complete solubilization. Detergents with higher CMC’s - OTG (CMC = 2.8mg/ml), C8E5 (CMC = 2.5mg/ml) and OG (CMC = 5.3mg/ml) - laid somewhere in between. For 2D crystallization, the detergent concentration was kept constant across all the screening conditions, thus ensuring comparable kinetics for reconstitution/crystallization. Therefore, the detergent concentration required for complete solubilization of the most challenging lipid was used for all lipid stock. For example, a concentration of 4mg/ml of Triton X-100 was employed for preparing all the Triton-containing lipid stocks. In all cases, an incubation period of 1h was used to allow the protein-lipid-detergent ternary mixture to equilibrate prior to initiating detergent removal.
Table 3. Lipid solubilization by detergent.
The numbers indicate the amount of a particular detergent (in mg/ml) needed to solubilize 1.5mg/ml of lipid, as assessed by turbidity measurements at 500nm.
| DDM | Triton X-100 | OTG | OG | C8E5 | |
|---|---|---|---|---|---|
| DMPC | 2 | 1 | 7* | 6 | 4 |
| DOPC | 14* | 3* | 7* | 8 | 7* |
| DOPG | 4 | 2 | 4 | 4 | 4 |
| POPC | 13 | 3* | 5 | 8 | 5 |
| E.coli polar lipids | 7 | 3* | 5 | 10* | 5 |
Conditions used for preparing lipid/detergent stocks for 2D crystallization.
Specimen preparation for EM
EM specimens were prepared by negative staining. The basic procedure for batch grid preparation has been described previously [31], but the protocol for negative staining has been significantly improved to increase both throughput and reproducibility. The previous strategy was based on a non-wicking support treated with adhesive to prevent capillary action from inadvertently displacing the grids. However, this adhesive provided inadequate support and made the grids sticky, thus resulting in deformed grids and uneven staining. Our updated procedure employs a commercially available magnetic platform (Agencourt SPRI plate 384 Post Magnet Plate) with 96 magnetic posts with a diameter of 3.3mm (Fig. 3). Standard EM grids (3.05mm diameter) made of Ni are held securely at the center of each post throughout the pipetting steps required for negative staining (Fig. 3a).
For negative staining of EM grids, controlled removal of the last drop of stain solution is critical to achieve optimal specimen preservation and uniform contrast. In conventional manual protocols, 2–5μl of sample is pipetted onto a glow-discharged grid and allowed to adsorb to the carbon film for ~30 seconds. The grid is then washed with one or more drops of negative stain (e.g., 5μl of 1% uranyl acetate). Excess solution is removed by gently blotting the grid from one side with filter paper and, after the final drop, the grid is allowed to air dry. With our earlier adhesive-dependent staining protocol, the liquid-handling robot aspirated as much liquid as possible from the grids at the end of the washing steps. This procedure sometimes left behind small puddles of stain solution and therefore produced uneven staining and large stain precipitates on some of the samples. Because the magnetic platform more securely holds the grids, it is now possible to leave the final drop of stain on the grids, and to blot an entire row of twelve grids simultaneously with long strips of filter paper (Fig. 3b). Our final automated staining procedure yields high-quality staining that is very reproducible (~95% success rate). The remaining 5% of the grids generally suffer from broken carbon, which is likely caused by defects in the plastic film, or by problems during carbon evaporation and glow discharge.
Scoring system: abundance and quality
To evaluate and catalog the outcomes from parallel 2D crystallization screens, individual trials should be scored in a comprehensive and systematic manner. A sound scoring system should allow important crystallization parameters to be identified for follow-up screens, and provide numerical data for statistical analyses. Also, to avoid wasting effort, proteins not suitable for 2D crystallization should be recognized early. Finally, the scoring should reflect both the crystalline qualities of the objects found, as well as their abundance. Abundance is particularly important for electron crystallography, since structure determination requires merging data from a considerable number of crystals at different orientations. Furthermore, crystal quality is generally not uniform throughout a specimen and the efficiency of recording high quality images is relatively low. Efforts at optimization must therefore include attempts to maximize the number as well as the quality of crystals produced.
We developed a system with six quality grades ranging from A–F, and a measure of abundance ranging from 1–4. The quality grades comprise the microscopic categories A) crystal lattice, B) planar sheets and tubular vesicles, C) vesicles and proteoliposomes, D) protein aggregates and E) lipidic structures, together with the macroscopic category F) macroscopic precipitation observed by light microscopy. Representative images of the different categories are shown in Fig. 4a. These categories not only distinguish between crystalline and non-crystalline objects, but also describe the nature of the non-crystalline objects to guide future attempts at refinement. For example, amorphous shapes with low contrast characterize non-ordered structures made up predominantly of lipids, and their occurrence suggests the presence of non-bilayer structures or other lipid polymorphism. In contrast, dense protein-rich aggregates indicate substantial protein precipitation and a general failure at reconstituting the lipid bilayer. Vesicles/Proteoliposomes signify successful reconstitution, but indicate that the protein is not packed optimally within the bilayer, or that conditions are not favorable for crystallization. Tubular or sheet-like morphologies indicate that protein density and organization is high enough to alter the normally spherical shape of lipid vesicles; these morphologies are often precursors to crystallization. The macroscopic category addresses any visible precipitation observed by light microscopy after detergent removal, which can range from amorphous aggregates to well-defined 3D-shapes (e.g. crystalline needles). The presence of macroscopic precipitates indicates that some of the protein has been removed from solution and is therefore unavailable for reconstitution and 2D crystal formation, thereby altering the effective LPR. Also, since evaluation of individual crystallization experiments by EM is time-consuming, elimination of trials with excessive macroscopic precipitation has the potential to increase overall throughput. Results from a screen are illustrated graphically in Fig. 4b, where the color corresponds to the scoring category and the height of each bar corresponds to abundance. These results show that the putative cation-transporter P2A3 crystallizes readily in DOPG; however, it is notable that objects belonging to several different categories are present in all of the individual crystallizations experiments.
Figure 4. Scoring system and outcome evaluation.

a) Representative images of the six-grade quality scoring system of the 2D crystallization outcomes. b) Outcome from a 2D crystallization screen of the protein P2A3 is displayed in the form of a histogram. For clarity, the histogram displays only results obtained at pH 7.0. The depth axis corresponds to the quality score described in a. In addition, the y-axis reflects the abundance of the observed objects; 1: up to 2 objects/grid square, 2: 3–10 objects/grid square, 3: 11–20 objects/grid square, and 4: more than 20 objects/grid square.
Screening results
To date, 38 2D crystallization screens have been performed on 15 different target proteins (Table 1). This corresponds to more than 3000 individual crystallization experiments evaluated by EM. Most of these proteins were subjected to an initial broad screen, followed by more limited optimization screens. Three protein targets have yielded diffracting crystals: P2A3, ykgB-D36 and Semliki Forest Virus E1 fusion protein (Fig. 5). In all three successful cases, crystals were observed in the initial screens and subsequent screens were therefore designed to improve their quality and abundance. Several others targets have yielded promising outcomes such as tubes, large vesicles and sheets; in these cases, however, subsequent screens have not produced crystalline order.
Figure 5. Gallery of negatively stained images of diffracting crystals obtained using the 2D crystallization pipeline.

a) P2A3 and b) YkgB-D36 yielded narrow tubular crystals, whereas c) SFV fusion protein produced flat, sheet-like crystals. The scale bars are 50nm.
In the case of P2A3 and ykgB-D36, we obtained narrow tubular crystals, whereas SFV fusion protein produced flat, planar crystals (Fig. 5). The helical symmetry underlying the tubular crystals offers an advantage for structure determination, given that a full 3D dataset can be derived from each individual crystal. However, more extensive planar crystals produce a higher signal-to-noise ratio, and thus have a better chance of extending to high resolution. We therefore designed screens to perturb the environment of P2A3 in order to generate planar crystals, specifically by altering the lipid head group and the ionic conditions of the aqueous phase. Despite these efforts, we were unsuccessful in converting the tubular crystals into a planar morphology, suggesting that the molecular interactions stabilizing the crystal lattice, are stable and favor the high curvature of the reconstituted membranes. We also compared P2A3 crystallization outcomes from the 96-well crystallization block with results obtained from individual microdialysis buttons. The sample volume (50μl) for the two devices is the same, whereas the buffer volume used with dialysis buttons (50ml) is much larger than for the crystallization block (1ml). Even though more frequent buffer exchanges for the crystallization block (twice daily compared to once daily for the buttons) is designed to compensate for the smaller volumes, the kinetics of detergent removal are likely different for the two devices. Indeed, although we could easily obtain tubular crystals of P2A3 from both devices, there were differences in the morphologies and molecular packing of the resulting crystals, most clearly evident by a differing diameter of tubes, and different patterns of layer lines in the corresponding Fourier transforms (not shown). Since this diameter reflects the underlying molecular interactions and thus, the helical symmetries of the crystals, it is apparent that even a subtle change in the kinetics of detergent removal can alter the reconstitution process and affect the crystallization process.
Discussion
We have conducted 38 2D crystallization screens for 15 different membrane proteins, totaling over 3000 individual crystallization experiments. Compared to manual 2D crystallization methods, our automated pipeline allowed us to cover a wider range of 2D crystallization space on a larger number of membrane protein targets. The outcomes from our 2D crystallization trials range from diffracting crystals to amorphous aggregates (Table 1). By analyzing only the primary or predicted secondary structures of the screened proteins, it is not easy to foresee their crystallization propensity. For example, the three proteins that, so far, have yielded crystals have different topologies (1–5 transmembrane helices), hydrophobicities (5–37% of amino acid sequence being transmembrane), and isoelectric points (5.4–7.6), as predicted through www.cbs.dtu.dk/services/TMHMM/ and au.expasy.org/tools/protparam.html. Thus, additional proteins need to be screened, and more ambitious bioinformatics studies conducted, in order to draw more generalized conclusions. Nevertheless, our results reflect the expected range of outcomes and also shed light onto the mechanisms of 2D crystallization.
Reconstitution into a lipid bilayer is a first and critical step towards 2D crystallization, and was not achieved for those membrane protein targets that yielded mainly protein precipitates and/or lipidic structures. One possible reason for this failure is that the membrane protein is unstable in the initial ternary complex with detergent and lipid. Certainly, the pre-screening of protein stability in a variety of detergents should rule out a negative effect of pure detergent, but mixed micelles of detergent and lipid do not behave like micelles of pure detergent [42] and either lipid-protein or lipid-detergent interactions are capable of affecting both protein stability and the reconstitution process.
Specifically, Rigaud and colleagues described alternative pathways for reconstitution (reviewed in [43]) that depend critically on the rate of detergent removal and on the interactions between detergent, lipid and protein. The prevalence of each pathway depends on the relative kinetics of detergent removal from two co-existing populations of mixed micelles, namely ternary micelles (protein-detergent-lipid) and binary micelles (detergent-lipid). For rapid detergent removal, over a period of 1–2 hours (e.g. with Biobeads), both species break down at a similar rate and coalesce into a homogeneous population of proteoliposomes. For the slower detergent removal achieved by dialysis, the formation of detergent saturated liposomes and of protein-detergent-lipid aggregates as intermediate species, was postulated. These protein-detergent-lipid aggregates are distinct from the initial ternary micelles, because the limiting number of detergent molecules can drive protein-protein interactions that are not present in the fully solubilized state, where each membrane protein molecule is likely to exist within its own micelle. These detergent-depleted aggregates hold the key to reconstitution and to 2D crystallization. If the protein is unstable in this state, precipitation is the likely outcome. On the other hand, reconstitution can be achieved if the remaining detergent is able to facilitate incorporation of the protein into the existing liposomes. Finally, if fusion of the aggregates is favored over their incorporation into detergent-saturated liposomes, two populations of vesicles, one lipid-rich and the other protein-rich, will be produced [44].
Specific inter-molecular associations within the detergent depleted protein-lipid aggregates may govern the resulting crystal symmetry and morphology. These interactions may occur between TMDs of adjacent proteins, between the hydrophilic portions of neighboring proteins, and also between proteins and lipids (Protein Data Bank (www.rcsb.org/pdb)). In many cases, a given protein will form both tubular and planar crystals [45], indicating that there are alternative sets of crystal contacts that are differentially selected by the conditions during reconstitution and crystal maturation. Among the proteins screened in our pipeline, P2A3 regularly yielded thin tubular crystals composed of dimeric protein units inserted asymmetrically into the membrane. Despite wide-ranging lipid and buffer screens, we were unable to induce major changes in the curvature of these tubular crystals, suggesting that the intermolecular contacts between protein monomers are very strong, and constrain their incorporation into proteoliposomes. In analogy to the Ca-ATPase [44], the high curvature of these crystals likely reflects a strong protein-protein interaction that orients the proteins within the micelles and as they are incorporated into the lipid bilayer, thus preventing molecules from being inserted in an “upside-down” orientation. Differences in the distance of the protein-protein contacts on either side of the membrane are presumably responsible for the curvature. In contrast, proteins that have a two-fold symmetry axis oriented parallel to the membrane plane are symmetric across the membrane and inevitably produce extended, planar crystals (e.g., eye lens aquaporin-0 [20], light harvesting complex II [46], cytochrome oxidase [47], glutathione transferase [22]).
Even though P2A3 consistently produced thin tubular crystals in the presence of DOPG, crystal morphology and helical symmetry was influenced by relatively subtle differences in the kinetics of detergent removal. This behavior underlines the importance of detergent removal rates for 2D crystallization, at least during the crystal optimization phase. Because it is difficult to control the rate of detergent removal by dialysis, it is important to explore alternative reconstitution techniques such as dilution [48], adsorption by Biobeads [49] or complexation by cyclodextrins [50]. We therefore envision our dialysis block mainly as a screening device and foresee that crystal optimization will require alternative approaches and devices for controlling detergent removal.
Conclusion
The automated pipeline presented in this communication, significantly speeds up the screening of 2D crystallization conditions and offers a more general approach to electron crystallography. In addition to throughput, automation has the potential to increase the reproducibility of the crystallization process, which is particularly important for electron crystallography as it relies on data collection from a large number of individual crystals. Given the amount of effort required for structure determination, an increased level of automation not only in crystallization, but in imaging and data analysis will be essential to consolidate electron crystallography as a viable tool for routine structure determination of membrane proteins.
Acknowledgments
This paper is dedicated to Russ Hinchliffe, who was indispensable in the design and manufacture of the dialysis block and other components used in our pipeline. We are grateful to Ms. Kumiko Sugawara for producing the proteins YkgB-D332 and YkgB-D36 used for crystallization. We also wish to thank Ms. KD Derr, Dr. Ruben Diaz-Avalos and Dr. William Rice at The New York Structural Biology Center for fruitful discussions and members of the New York Consortium on Membrane Protein Structure for their supply of both proteins and ideas. Funding for this work was provided by grant R01-GM081817 from the National Institutes of Health and grant MCB-0546087 from the National Science Foundation.
List of Abbreviations
- BHT
Butylated hydroxytoluene
- C8E4
Tetraethylene glycol monooctyl ether
- C8E5
Pentaethylene glycol monooctyl ether
- DM
n-Decyl-β-D-maltopyranoside
- DDM
n-Dodecyl-β-D-maltopyranoside
- DMPC
1,2-Dimyristoyl-sn-glycero-3-phosphocholine
- DOPC
1,2-Dioleoyl-sn-glycero-3-phosphocholine
- DOPE
1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine
- DOPG
1,2-Dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)
- LDAO
Lauryldimethylamine-oxide
- OG
n-Octyl-β-D-glucopyranoside
- NG
n-Nonyl-β-D-glucopyranoside
- OTG
n-Octyl-β-D-thioglucopyranoside
- POPC
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- EM
Electron microscopy
- 2D
Two-dimensional
References
- 1.Engelman DM. Lipid bilayer structure in the membrane of Mycoplasma laidlawii. J Mol Biol. 1971;58:153–65. doi: 10.1016/0022-2836(71)90238-5. [DOI] [PubMed] [Google Scholar]
- 2.Mitra K, Ubarretxena-Belandia I, Taguchi T, Warren G, Engelman DM. Modulation of the bilayer thickness of exocytic pathway membranes by membrane proteins rather than cholesterol. Proc Natl Acad Sci U S A. 2004;101:4083–8. doi: 10.1073/pnas.0307332101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wallin E, von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998;7:1029–38. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stevens TJ, Arkin IT. Do more complex organisms have a greater proportion of membrane proteins in their genomes? Proteins. 2000;39:417–20. doi: 10.1002/(sici)1097-0134(20000601)39:4<417::aid-prot140>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 5.Planells-Cases R, Jentsch TJ. Chloride channelopathies. Biochim Biophys Acta. 2009;1792:173–89. doi: 10.1016/j.bbadis.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 6.Verkman AS, Galietta LJ. Chloride channels as drug targets. Nat Rev Drug Discov. 2009;8:153–71. doi: 10.1038/nrd2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Croce CM. Oncogenes and cancer. N Engl J Med. 2008;358:502–11. doi: 10.1056/NEJMra072367. [DOI] [PubMed] [Google Scholar]
- 8.Evin G, Sernee MF, Masters CL. Inhibition of gamma-secretase as a therapeutic intervention for Alzheimer’s disease: prospects, limitations and strategies. CNS Drugs. 2006;20:351–72. doi: 10.2165/00023210-200620050-00002. [DOI] [PubMed] [Google Scholar]
- 9.Herren T, Gerber PA, Duru F. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: a not so rare “disease of the desmosome” with multiple clinical presentations. Clin Res Cardiol. 2009;98:141–58. doi: 10.1007/s00392-009-0751-4. [DOI] [PubMed] [Google Scholar]
- 10.Rac ME, Safranow K, Poncyljusz W. Molecular basis of human CD36 gene mutations. Mol Med. 2007;13:288–96. doi: 10.2119/2006-00088.Rac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gazzerro E, Sotgia F, Bruno C, Lisanti MP, Minetti C. Caveolinopathies: from the biology of caveolin-3 to human diseases. Eur J Hum Genet. 2009 doi: 10.1038/ejhg.2009.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehnart SE, Ackerman MJ, Benson DW, Jr, Brugada R, Clancy CE, Donahue JK, George AL, Jr, Grant AO, Groft SC, January CT, Lathrop DA, Lederer WJ, Makielski JC, Mohler PJ, Moss A, Nerbonne JM, Olson TM, Przywara DA, Towbin JA, Wang LH, Marks AR. Inherited arrhythmias: a National Heart, Lung, and Blood Institute and Office of Rare Diseases workshop consensus report about the diagnosis, phenotyping, molecular mechanisms, and therapeutic approaches for primary cardiomyopathies of gene mutations affecting ion channel function. Circulation. 2007;116:2325–45. doi: 10.1161/CIRCULATIONAHA.107.711689. [DOI] [PubMed] [Google Scholar]
- 13.Drews J. Drug discovery: a historical perspective. Science. 2000;287:1960–4. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- 14.Imming P, Sinning C, Meyer A. Drugs, their targets and the nature and number of drug targets. Nat Rev Drug Discov. 2006;5:821–34. doi: 10.1038/nrd2132. [DOI] [PubMed] [Google Scholar]
- 15.Hong M. Oligomeric structure, dynamics, and orientation of membrane proteins from solid-state NMR. Structure. 2006;14:1731–40. doi: 10.1016/j.str.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Henderson R, Unwin PNT. Three-dimensional model of purple membrane obtained from electron microscopy. Nature. 1975;257:28–32. doi: 10.1038/257028a0. [DOI] [PubMed] [Google Scholar]
- 17.Kimura Y, Vassylyev DG, Miyazawa A, Kidera A, Matsushima M, Mitsuoka K, Murata K, Hirai T, Fujiyoshi Y. Surface of bacteriorhodopsin revealed by high-resolution electron crystallography. Nature. 1997;389:206–11. doi: 10.1038/38323. [DOI] [PubMed] [Google Scholar]
- 18.Kühlbrandt W, Wang DN, Fujiyoshi Y. Atomic model of plant light-harvesting complex by electron crystallography. Nature. 1994;367:614–21. doi: 10.1038/367614a0. [DOI] [PubMed] [Google Scholar]
- 19.Murata K, Mitsuoka K, Hirai T, Walz T, Agre P, Heymann JB, Engel A, Fujiyoshi Y. Structural determinants of water permeation through aquaporin-1. Nature. 2000;407:599–605. doi: 10.1038/35036519. [DOI] [PubMed] [Google Scholar]
- 20.Gonen T, Cheng Y, Sliz P, Hiroaki Y, Fujiyoshi Y, Harrison SC, Walz T. Lipid-protein interactions in double-layered two-dimensional AQP0 crystals. Nature. 2005;438:633–8. doi: 10.1038/nature04321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tani K, Mitsuma T, Hiroaki Y, Kamegawa A, Nishikawa K, Tanimura Y, Fujiyoshi Y. Mechanism of aquaporin-4’s fast and highly selective water conduction and proton exclusion. J Mol Biol. 2009;389:694–706. doi: 10.1016/j.jmb.2009.04.049. [DOI] [PubMed] [Google Scholar]
- 22.Holm PJ, Bhakat P, Jegerschold C, Gyobu N, Mitsuoka K, Fujiyoshi Y, Morgenstern R, Hebert H. Structural basis for detoxification and oxidative stress protection in membranes. J Mol Biol. 2006;360:934–45. doi: 10.1016/j.jmb.2006.05.056. [DOI] [PubMed] [Google Scholar]
- 23.Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol. 2005;346:967–89. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 24.Kühlbrandt W. Two-dimensional crystallization of membrane proteins. Q Rev Biophys. 1992;25:1–49. doi: 10.1017/s0033583500004716. [DOI] [PubMed] [Google Scholar]
- 25.Mosser G. Two-dimensional crystallogenesis of transmembrane proteins. Micron. 2001;32:517–40. doi: 10.1016/s0968-4328(00)00047-0. [DOI] [PubMed] [Google Scholar]
- 26.Stevens RC. High-throughput protein crystallization. Current Opinion in Structural Biology. 2000;10:558–63. doi: 10.1016/s0959-440x(00)00131-7. [DOI] [PubMed] [Google Scholar]
- 27.Hui R, Edwards A. High-throughput protein crystallization. J Struct Biol. 2003;142:154–61. doi: 10.1016/s1047-8477(03)00046-7. [DOI] [PubMed] [Google Scholar]
- 28.Joachimiak A. High-throughput crystallography for structural genomics. Curr Opin Struct Biol. 2009;19:573–84. doi: 10.1016/j.sbi.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chayen NE, Saridakis E. Protein crystallization: from purified protein to diffraction-quality crystal. Nat Methods. 2008;5:147–53. doi: 10.1038/nmeth.f.203. [DOI] [PubMed] [Google Scholar]
- 30.Ringler P, Heymann B, Engel A. Two-dimensional crystallization of membrane proteins. In: Baldwin S, editor. Membrane Transport. Oxford University Press; Oxford: 2000. pp. 229–268. [Google Scholar]
- 31.Vink M, Derr K, Love J, Stokes DL, Ubarretxena-Belandia I. A high-throughput strategy to screen 2D crystallization trials of membrane proteins. J Struct Biol. 2007;160:295–304. doi: 10.1016/j.jsb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iacovache I, Biasini M, Kowal J, Kukulski W, Chami M, Gisou Van der Goot F, Engel A, Rémigy H. The 2DX robot: a membrane protein 2D crystallization Swiss Army knife. Journal of Structural Biology. 2009 doi: 10.1016/j.jsb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 33.Hu M, Vink M, Kim C, Derr K, Koss J, D’Amico K, Cheng A, Pulokas J, Ubarretxena-Belandia I, Stokes D. Automated Electron Microscopy for Evaluating Two-dimensional Crystallization of Membrane Proteins. J Struct Biol. 2010 doi: 10.1016/j.jsb.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanchez-San Martin C, Sosa H, Kielian M. A stable prefusion intermediate of the alphavirus fusion protein reveals critical features of class II membrane fusion. Cell Host Microbe. 2008;4:600–8. doi: 10.1016/j.chom.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Warne T. Expression and purification of truncated, non-glycosylated turkey beta-adrenergic receptors for crystallization. Biochimica et Biophysica Acta (BBA) -Biomembranes. 2003;1610:133–140. doi: 10.1016/s0005-2736(02)00716-2. [DOI] [PubMed] [Google Scholar]
- 36.Kawate T, Gouaux E. Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure. 2006;14:673–681. doi: 10.1016/j.str.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 37.Paternostre MT, Roux M, Rigaud JL. Mechanisms of membrane protein insertion into liposomes during reconstitution procedures involving the use of detergents. 1. Solubilization of large unilamellar liposomes (prepared by reverse-phase evaporation) by triton X-100, octyl glucoside, and sodium cholate. Biochemistry. 1988;27:2668–77. doi: 10.1021/bi00408a006. [DOI] [PubMed] [Google Scholar]
- 38.Potter CS, Chu H, Frey B, Green C, Kisseberth N, Madden TJ, Miller KL, Nahrstedt K, Pulokas J, Reilein A, Tcheng D, Weber D, Carragher B. Leginon: a system for fully automated acquisition of 1000 electron micrographs a day. Ultramicroscopy. 1999;77:153–61. doi: 10.1016/s0304-3991(99)00043-1. [DOI] [PubMed] [Google Scholar]
- 39.Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B. Automated molecular microscopy: the new Leginon system. J Struct Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 40.Gibbons DL, Vaney MC, Roussel A, Vigouroux A, Reilly B, Lepault J, Kielian M, Rey FA. Conformational change and protein-protein interactions of the fusion protein of Semliki Forest virus. Nature. 2004;427:320–5. doi: 10.1038/nature02239. [DOI] [PubMed] [Google Scholar]
- 41.Gassel M, Siebers A, Epstein W, Altendorf K. Assembly of the Kdp complex, the multi-subunit K+-transport ATPase of Escherichia coli. Biochim Biophys Acta. 1998;1415:77–84. doi: 10.1016/s0005-2736(98)00179-5. [DOI] [PubMed] [Google Scholar]
- 42.Almgren M. Mixed micelles and other structures in the solubilization of bilayer lipid membranes by surfactants. Biochim Biophys Acta. 2000;1508:146–63. doi: 10.1016/s0005-2736(00)00309-6. [DOI] [PubMed] [Google Scholar]
- 43.Rigaud J-L, Lévy D. Reconstitution of membrane proteins into liposomes. Meth Enzymol. 2003;372:65–86. doi: 10.1016/S0076-6879(03)72004-7. [DOI] [PubMed] [Google Scholar]
- 44.Young HS, Rigaud JL, Lacapere JJ, Reddy LG, Stokes DL. How to make tubular crystals by reconstitution of detergent-solubilized Ca2(+)-ATPase. Biophys J. 1997;72:2545–58. doi: 10.1016/S0006-3495(97)78898-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jap BK, Zulauf M, Scheybani T, Hefti A, Baumeister W, Aebi U, Engel A. 2D crystallization: from art to science. Ultramicroscopy. 1992;46:45–84. doi: 10.1016/0304-3991(92)90007-7. [DOI] [PubMed] [Google Scholar]
- 46.Kühlbrandt W, Wang DN. Three-dimensional structure of plant light-harvesting complex determined by electron crystallography. Nature. 1991;350:130–4. doi: 10.1038/350130a0. [DOI] [PubMed] [Google Scholar]
- 47.Valpuesta JM, Henderson R, Frey TG. Electron cryo-microscopic analysis of crystalline cytochrome oxidase. J Mol Biol. 1990;214:237–51. doi: 10.1016/0022-2836(90)90158-I. [DOI] [PubMed] [Google Scholar]
- 48.Dolder M, Engel A, Zulauf M. The micelle to vesicle transition of lipids and detergents in the presence of a membrane protein: towards a rationale for 2D crystallization. FEBS Lett. 1996;382:203–8. doi: 10.1016/0014-5793(96)00180-9. [DOI] [PubMed] [Google Scholar]
- 49.Rigaud JL, Mosser G, Lacapere JJ, Olofsson A, Levy D, Ranck JL. Bio-Beads: an efficient strategy for two-dimensional crystallization of membrane proteins. J Struct Biol. 1997;118:226–35. doi: 10.1006/jsbi.1997.3848. [DOI] [PubMed] [Google Scholar]
- 50.Signorell GA, Kaufmann TC, Kukulski W, Engel A, Remigy HW. Controlled 2D crystallization of membrane proteins using methyl-beta-cyclodextrin. J Struct Biol. 2007;157:321–8. doi: 10.1016/j.jsb.2006.07.011. [DOI] [PubMed] [Google Scholar]