

Abstract
Although membrane proteins make up 30% of the proteome and are a common target for therapeutic drugs, determination of their atomic structure remains a technical challenge. Electron crystallography represents an alternative to the conventional methods of X-ray diffraction and NMR and relies on the formation of two-dimensional crystals. These crystals are produced by reconstituting purified, detergent-solubilized membrane proteins back into the native environment of a lipid bilayer. This chapter reviews methods for producing two-dimensional crystals and for screening them by negative stain electron microscopy. In addition, we show examples of the different morphologies that are commonly obtained and describe basic image analysis procedures that can be used to evaluate their promise for structure determination by cryoelectron microsopy.
Keywords: membrane protein structure, electron crystallography, two-dimensional crystals, electron microscopy, crystal screening
Introduction
Electron crystallography is a method of membrane protein structure determination that involves imaging two-dimensional (2D) crystals by cryo-electron microscopy (cryo-EM). Such 2D crystals are produced when membrane proteins adopt a regular array within the plane of a lipid bilayer. Methods for electron crystallography were originally pioneered in the 1970s by Henderson and Unwin in their studies of bacteriorhodopsin (1). Intense efforts over the next two decades led to developments both in electron microscope design and in software for analyzing the resulting images, thus producing the first atomic resolution structure of a membrane protein in its native membranous environment (2). Since then, electron crystallography has developed into a powerful tool to elucidate the 3D structure of a wide range of membrane proteins (3–6), thus offering a plausible alternative to X-ray crystallography.
Similar to X-ray crystallography, the bottleneck preventing a more generalized use of electron crystallography is the preparation of well-ordered crystals. In the case of membrane proteins, there are three predominant morphologies adopted by 2D crystals: (i) flattened lipid vesicles with two, overlapping 2D lattices; (ii) tubular lipid vesicles which retain a cylindrical shape and comprise a helical array of membrane proteins; and (iii) a planar lipid bilayer with a single, coherent 2D array of proteins. Historically, 2D crystallization of membrane proteins has involved either in situ crystallization or in vitro reconstitution (7,8). The advantage of in situ crystallization is that the membrane protein is never removed from its native membrane. However, this approach requires a high concentration of the relevant protein in the native membrane and is not therefore generally applicable. More generally, 2D crystals have been grown by reconstitution of purified, detergent-solubilized membrane proteins into lipid bilayers under defined conditions (7,9). Reconstitution involves the controlled removal of detergent, either by dialysis or by adsorption to a hydrophobic resin, in the presence of exogenous lipid. Under favorable conditions the membrane proteins assemble into an ordered 2D array within the resulting lipid bilayers (7). In this chapter we will describe protocols and procedures to obtain 2D crystals of membrane proteins and to evaluate their quality and potential for structure determination by electron crystallography.
2. Materials
2.1. Analysis of Lipid
Solid phase: 20×20 cm thin layer chromatography (TLC) plates with silica gel 60 F254 coating (Merck & Co., Whitehouse Station, NJ).
Glass cutter.
Glass chromatography tank (25 cm × 27 cm × 10 cm).
Graduated disposable 5 μl glass capillaries (CAMAG, Muttenz, Switzerland).
Mobile phase: mixture of chloroform/methanol/25% aqueous ammonia (65: 25: 5, by volume).
Compressed nitrogen supply.
Kontes Chromatography TLC Reagent Sprayer with Standard Ground Joint Sprayer (Fisher Scientific, Pittsburgh PA).
A 0.1% solution in water of 8-anilino-1-naphtalene sulfonic acid (ANSA) (Sigma, St. Louis, MO).
E. coli polar phospholipid extract (Avanti Polar Lipids, Alabaster, AL) solubilized at 1mg/ml in chloroform.
2.2. Biobead Reconstitution
SM2 Biobeads (BioRad, Hercules, CA), washed with ethanol and then with water and stored under water at 4°C with 1mM NaN3. Care should be taken to prevent the Biobeads from drying out. Prior to use, bulk liquid should be removed by aspiration with a pipette and they should be kept covered and cold until water can be added back.
Dry N2 gas.
Lyophilizer or vacuum dessicator
50 mM Tris pH 7.3
Lipid stock solutions purchased in chloroform (Avanti Polar Lipids): e.g., egg yolk phosphatidyl choline, egg yolk phosphatidyl ethanolamine, egg yolk phosphatidic acid, or dioleoylphosphatidylcholine, dioleoylphosphatidyl ethanolamine, palmitoyl-oleoylphosphatidic acid.
50% sucrose stock solution in 50 mM Tris pH 7.3
Detergent stock solution 10mg/ml (e.g. C12E8, Anatrace, Maumee, OH) and stored at 4°C.
2.3. Detergent Removal by Dialysis
50–200 μl microdialysis buttons and o-rings (Hampton Research, Aliso Viejo, CA)
40 mm dialysis membrane 12000–14000 MW cutoff (Spectra/Pore, VWR International, West Chester, PA). Prepare square pieces by cutting all four sides of the strip and soak in a beaker of water. Beaker may be stored in the refrigerator.
2.4. Solid Carbon Films
Carbon rod, either 1/8″ or 1/4″ depending on the source provided with your vacuum evaporator. Alternatively, a carbon thread can also be used (Electron Microscopy Sciences, Hatfield, PA).
Mica sheets (Electron Microscopy Sciences)
Crystallizing dish (190×100mm) (VWR International)
Small test tube rack, tygon tubing, 5–10 ml glass pipette, hemostats, plastic tray, 2 pennies
90 mm plastic petri dishes, 90mm Grade 4 Whatman filter paper
300 mesh Cu EM grids (Ted Pella, Redding, CA)
Stainless steel forceps (e.g. #5 Dumont)
Vacuum evaporator (e.g. Edwards Auto306, Edwards Vacuum, Wilmington, MA)
2.5. Holey Carbon Films
Glass slides: standard 1×4″ slides for light microscopy cleaned by soaking in acetone overnight in glass staining dish (20–30 slide capacity, Electron Microscopy Sciences or Ted Pella).
2–3 Coplin staining jars (Electron Microscopy Sciences or Ted Pella).
Stock solution of 0.4% Formvar in chloroform (Electron Microscopy Sciences). Store in glass-stoppered flask at room temperature.
Stock solution of 50% glycerol in water.
Plastic boxes for storing glass slides (Electron Microscopy Sciences).
Crystallizing dish (190×100mm) (VWR International).
EM grids, carbon rod/thread and vacuum evaporator as above.
Methanol, ethylene dichloride, 50% hydrofluoric acid.
2.6. Negative Staining
-
1
Solid carbon films (prepared in step 3.5).
-
2
Stainless steel forceps, either reverse grip or with o-ring to hold them closed.
-
3
Glow discharge apparatus (e.g. Edwards Auto306, Edwards Vacuum).
-
4
Stock solution of 1% uranyl acetate in water.
-
5
Grade 1 Whatman filter paper, torn into strips.
-
7
Plastic boxes for storage of EM grids (Electron Microscopy Sciences)
2.7. Screening of Samples by Electron Microscopy
Electron microscope: 80 kV or higher accelerating voltage (e.g., JEOL, USA, Peabody MA)
Stainless steel forceps (e.g. #5 Dumont, Ted Pella)
ImageJ computer software for calculating Fourier transform of images (10).
3. Methods
The following methods focus on the reconstitution, crystallization and screening of membrane proteins. Prior to applying these methods, we assume that the membrane protein of interest has been purified in a detergent-solubilized state, either from natural sources or from an cell expression system. In either case, it is generally useful to know the lipid composition of the purified protein, so the first set of methods describes a suitable analysis using thin layer chromatography. After that, we describe two alternative methods for 2D crystallization. Conceptually, crystallization involves two steps: reconstitution of the membrane protein into a lipid bilayer and ordering of molecules within that bilayer to form a 2D array. When dialysis is used, detergent is removed slowly (up to 2 weeks) and these two processes occur simultaneously. Detergent removal by hydrophobic resins is two orders of magnitude faster (3–4 hours) and in this case crystallization is often carried out as a second, subsequent step. The conditions for 2D crystallization generally require a screen involving a number of different factors--such as pH, salt concentration, lipid composition, detergent composition--and the resulting samples must be assessed by EM. In particular, EM is used to evaluate the morphology of the resulting proteoliposomes and to look for the presence of a regular lattice. Therefore, we describe methods for preparing two different kinds of carbon-coated EM grids and for preparing negatively stained samples. Furthermore, examples are given of different morphologies produced by reconstitution and of two different lattice assemblies that can be produced. For structure determination by electron crystallography, cryoelectron microscopy is required to image the 2D crystals. Although these methods are beyond the scope of this chapter, we also include a protocol for producing holey carbon films, which are useful for preparing frozen-hydrated samples of 2D crystals.
3.1. Analysis of Lipid Species Co-Purifying with Membrane Proteins
Using a graduated glass cylinder, add 100 ml chloroform/methanol/25% aqueous ammonia (65:25:5, by volume) to a glass chromatography tank that is lined with filter paper. Cover tightly and allow vapors in the tank to equilibrate for 10 min.
Use a glasscutter to divide TLC plates into 5×10 cm pieces. Use gloves to avoid contamination of the Si-coated surface.
Using a pencil, draw a very light line across the plate 1.5 cm from the bottom. Be careful not to scratch the Si coating.
Using glass capillaries, spot 2–4 μl of sample at 5mm intervals along the pencil line. Standard samples should be included so that lipid species associated with the protein can be identified. Amount of lipid added in each spot should be 1–5 μg.
Dry sample using a stream of N2 gas.
Using pair of long forceps, grasp the top of the plate and place it into the tank such that the spots of sample lie just above the level of the solvent. Allow the top edge of the plate to lean against the back wall of the developing chamber.
Cover the tank tightly and allow the solvent to progress up the plate. Leave undisturbed until the ascending solvent front reaches the top of the plate (approx. 30–45 minutes).
Remove the TLC plate with forceps and use a stream of N2 gas in chemical fume hood to dry the plate thoroughly.
Once dry, spray the plate briefly with 0.1% ANSA solution in the chemical fume hood.
Illuminate plate with an UV lamp either from above or from below and use a digital camera to record a picture. If the staining is weak, step 9 may be repeated.
3.2. 2D Crystallization using Biobeads
Prepare thin film of dried lipid by pipetting 500 μg of lipid stock solution into a glass test tube (7cm × 1cm) or small round bottom flask (25 ml) (see Notes 1 and 2). Rotate the flask rapidly to swirl the solution around to bottom while blowing N2 gas across the solution to evaporate the chloroform. This procedure should produce a thin, even film of dry lipid on the bottom of the tube. The film should appear white and not contain any thick, clear areas. If the film appears too thick, dissolve it with more chloroform and try again. Once satisfied, place tube into a lyophilizer or vacuum desiccator for 1 hr to remove residual chloroform. Then add 100 μl of detergent stock solution (1 mg) and vortex to dissolve the lipid. This represents the lipid stock solution to be used the same day for reconstitution.
Prepare solutions with 1mg/ml protein concentration and with lipid-to-protein ratios ranging from 0.25 to 2.0 by weight. The final detergent concentration should be at least double the lipid concentration to ensure complete solubilization. The buffer conditions should be tailored to the particular protein under investigation, initially to prevent aggregation and eventually to induce 2D crystallization.
Add 100 μl of these protein-detergent-lipid solutions to a test tube. Add a small stir bar and place on a magnetic stirrer at room temperature. Several test tubes can be placed in a small beaker and stirred simultaneously.
Over a 3–4 hr period, add Biobeads at a Biobead-to-detergent mass ratio of 16:1 g/mg for C12E8 (see Note 3). This includes detergent that accompanies both the protein and lipid stock solutions. There are two strategies for adding Biobeads, which often produce different results. Fast reconstitution involves simply adding all the Biobeads at the beginning of the incubation period (3 hours). Slow reconstitution involves four consecutive additions at 30 min intervals of a 1 g:1 mg Biobeads:detergent mass ratio, a 4:1 ratio for a 1 hr incubation, followed by a final addition of the remaining 8:1 ratio followed by 1 hr incubation, all at room temperature (see Note 4).
Remove the reconstituted protein solution and layer on top of a discontinuous sucrose gradient composed of 3 layers (10%, 25%, and 50%) of 0.7 ml each in a 2ml centrifuge tube. Place the centrifuge tube in a swinging bucket rotor and centrifuge at 35,000 rpm for at least 3 hr.
Collect the protein band from the sucrose gradient and dilute 10–20 fold in buffer (see Note 5). Centrifuge either in the same swinging bucket rotor or in a refrigerated benchtop centrifuge at 15,000–20,000 rpm for 30 min. Repeat this wash step at least once to remove sucrose.
Freeze-thaw cycles are often effective in increasing the size of proteoliposomes and promoting crystallization. For this procedure, do not resuspend the final pellet from the previous step. Instead, remove the supernatant and add the buffer that you wish to use for crystallization to produce a final protein concentration of ~1 mg/ml. Without resuspending the pellet, immerse the tube into liquid N2. Then thaw the pellet between thumb and forefinger. Repeat 3–4 times. Finally, resuspend any remaining pellet with a pipette. Crystallization often requires incubation for 1–7 days. This final solution can also be dialyzed against a crystallization buffer if desired (see below).
3.3. 2D Crystallization by Detergent Removal
Prepare lipid stock solutions and protein solutions as described in steps 1 and 2 of section 3.2.
Prepare dialysis solutions with a range of pH, salts and other additives that could be effective in promoting 2D crystal formation and aliquot into 100 ml beakers.
Add lipid/protein/detergent solutions to microdialysis buttons with a volume that exceeds their stated capacity by at least 10% such that the meniscus extends above the surface of the button.
Place o-ring on top of a 3×3 cm2 piece of dialysis membrane. In one motion, place dialysis membrane on top of the button and press o-ring into groove. The goal is to prevent the introduction of air bubbles into the dialysis button (Fig. 1a).
Place 2–4 dialysis buttons on the bottom of each 100ml beaker with dialysis membrane oriented upwards (Fig. 1b). Only the detergent and buffer components will exchange with the dialysis buffer, so buttons with different lipid-to-protein ratios or different lipid species can be mixed within one beaker. In this case, a permanent marker can be used to label the bottom of individual buttons.
Allow dialysis to continue for two weeks, changing the buffer daily. Solutions can be kept refrigerated or at room temperature or cycled between different temperatures as part of the crystallization strategy. Stirring is not generally required.
To harvest samples, remove buttons from the beaker and place on a piece of parafilm. Use a scalpel to cut an “X” in the dialysis membrane. Then use a micropipette to remove the solution and store in an microfuge tube.
Fig. 1.

Dialysis buttons for removal of detergent. (A) Individual dialysis button is shown with dialysis membrane held in place by a black o-ring. After securing the dialysis membrane in this way, the excess can be cut away with scissors. (B) Dialysis buttons in the bottom of a 100 ml beaker filled with dialysis solution. Stirring is not necessary, though the solution should be changed at least once per day to promote efficient detergent removal.
3.4. Preparation of Solid Carbon Film For Electron Microscopy
Sharpen carbon rod, remove dust remnants and mount in vacuum evaporator. Handle the carbon rod with gloves to prevent contamination with body oil.
Cut 4×2 cm2 piece of mica with scissors. Cleave in half longitudinally using your fingernail and place on filter paper in a petri dish with freshly cleaved surface facing upwards. Cut triangular piece of filter paper, fold in half such that one vertex overlaps the opposite side of the triangle and place in petri dish alongside the mica (see Fig. 2). Place petri dish into vacuum evaporator and obtain a vacuum of at least 1×10−6 torr.
If available, use retractable shield to cover mica sheets. Increase current through carbon rod until vacuum begins to degrade. Reduce current until vacuum recovers. Repeat until degradation is minimal. This step is designed to burn contamination off the carbon rod prior to evaporation.
Retract shield to expose mica sheets. Gradually increase current until carbon begins to evaporate. Maintain lowest possible current during evaporation to prevent sparking. After 15–30 sec, turn down current and inspect shadow produced by depositing carbon on the folded triangle of filter paper. Repeat evaporation as required to obtain a light, but distinct gray shadow. (see Note 6 and Fig. 2)
Vent vacuum evaporator, remove petri dishes and store covered at room temperature (see Note 7) until ready to coat EM grids.
Fill crystallization dish with water and place on a tray. Place test tube rack under water. Cut 3×5 cm2 piece of filter paper and place on top of the test tube rack. Weigh down the filter paper by placing pennies on the edges.
Individually place 300 mesh EM grids on top of the filter paper with forceps in a close packed array that matches the size of the mica. (see Notes 8 and 9).
Carefully add water until crystallization dish is almost overflowing. Wipe surface with 10ml glass pipette to remove dust from the surface. Insert 40–50 cm length of tygon tubing into bottom of crystallization dish and suck on end to create siphon. Clamp tygon tubing to stop siphon until later.
Pick up mica with forceps. “Huff” on the mica to humidify the carbon-coated surface (don’t blow with pursed lips; say “hot” without pronouncing the “t”). Hold mica at 10–20° relative to the water surface and gently lower it into the water. The carbon should float off of the mica and remain at the surface. Once the carbon is completely separated, drop the mica into the bottom of the crystallization dish.
Use the glass pipette and/or the forceps to maneuver the carbon over the filter paper. Release the hemostats to allow the siphon to remove water gradually from the crystallization tray. The carbon should land on top of the grids as the water recedes below the filter paper. The siphon can be paused just before the carbon contacts the filter paper in order to perfect the positioning of the carbon over the grids.
Lift the filter paper together with the carbon-coated grids and place on a piece of dry filter paper in a plastic petri dish. Store at room temperature until ready for use.
Fig. 2.
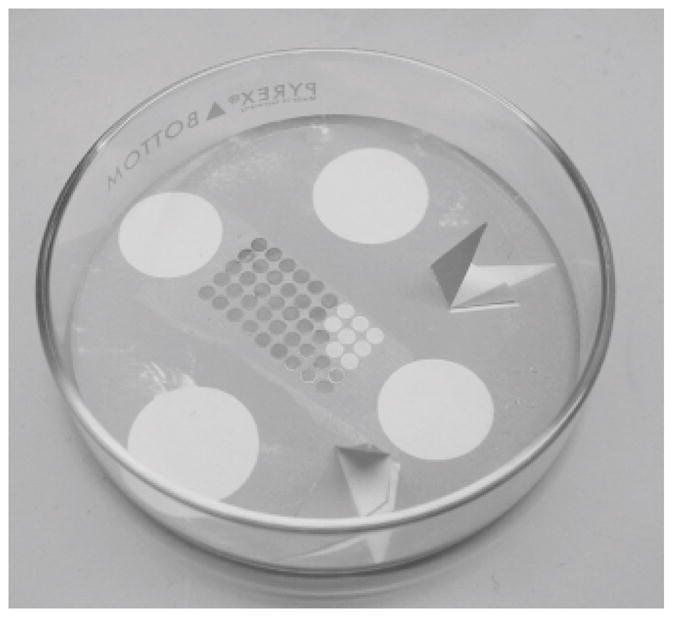
Carbon coating of EM grids. A group of EM grids lie on a piece of white filter paper in the bottom of a petri dish. Two triangular pieces of filter paper have been folded over to cast a shadow during carbon evaporation. Four pennies were placed on the filter paper to hold it down during the evaporation process, but have been removed to reveal their shadow. An array of EM grids have been placed in the center of the Petri dish; eight of these grids have been removed from the right side of the array. The contrast of these shadows is a good measure of the thickness of the carbon that has been deposited either, as in this case, on the plastic coated grids, or on the freshly cleaved mica.
3.5. Preparation of Holey Carbon Films
Mix 50 ml of the Formvar solution with 10–50 drops of the glycerol solution. Sonicate with a probe sonicator for 2–5 min, and wait for 0–10 min after the sonication. Conditions for sonication should be adjusted to obtain the desired size and distribution of holes in the plastic film (see Note 10 and Fig. 3).
Using forceps, hold a glass slide vertically, dip it into the sonicated solution, and slowly pull it out. Allow the slide to air dry and examine it in the light microscope using phase contrast optics. Once suitable plastic films have been obtained, they can be stored in a plastic box for years. It is helpful to make notes with a permanent marker on the top of the slides for later reference.
To transfer the holey plastic film to EM grids, it must be floated onto the surface of water as follows. Use a razor blade to scrape the edges of the glass slide and also to scrape away the upper and lower margins of the plastic film to provide a distinct edge. Fill the crystallizing dish with water. In order to facilitate release of the plastic film released from the glass slide, wave the slide (plastic film down) over an open bottle of hydrofluoric acid to loosen the association. Holding the glass slide with hemostats at a 10–20° angle relative to the surface of the water, lower it gradually such that the plastic film floats off.
Place EM grids directly on top of the plastic film with the polished side down. Gently lay either a piece of parafilm or its backing paper on top of the grids and plastic film. Allow water to saturate the region between the plastic and the parafilm. Gently lift the parafilm/backing paper, which should also lift the plastic film and associated grids off the water surface. Place this sandwich onto filter paper in a plastic petri dish and allow it to dry.
Etch the plastic films to remove “pseudo-holes” as follows. Place a piece of filter paper in a glass petri dish. Add enough methanol to saturate the filter paper as well as a residual amount of liquid in the bottom of the dish. Individually place the grids onto the filter paper plastic-side down. Cover and allow to incubate for 10–15 min. Remove the grids and place them on a dry piece of filter paper.
Select a group of grids by examining them individually in a light microscope using phase contrast optics and a 40x objective. Place the grids on a glass slide plastic-side up into the vacuum evaporator. Include a folded triangular piece of filter paper as described above for carbon evaporation. Prepare carbon rod/thread and evaporate carbon as described above (see Fig. 2). The carbon coated grids can be stored at room temperature, though they should be used within a few days/weeks of the evaporation.
Remove the plastic films using ethylene dichloride. Similar to step 5 above, place 2–3 pieces of filter paper into a small glass petri dish and saturate them with ethylene dichloride. Place the grids on the filter paper carbon side facing upwards. Cover and incubate for 10–30 min. Examine the grids with a light microscope using phase contrast optics and the 40x objective to ensure that the carbon film remains intact.
Fig. 3.

Holey carbon films. The density and size of holey carbon films can be controlled by the concentration and sonication of the glycerol/chloroform mixture (see text). (A) Closely packed holes are useful for cryoelectron microscopy of helical crystals, where one wishes to image the crystal as it lays across the hole suspended in vitreous ice. (B) Widely spaced holes are useful for cryoelectron microscopy of planar 2D crystals that require support by the carbon. These holes allow the grids to be blotted from behind, thus concentrating crystals on the carbon surface. This is a useful strategy for 2D crystals that fail to adsorb efficiently to solid carbon supports. The size of holes in (A) is ~ 2 μm.
3.6. Preparation of Negatively Stained Samples
Place solid carbon grids on a clean glass slide with the carbon facing upwards. Place slide inside glow discharge apparatus (see Note 11) and evacuate to ~250 mTorr. Turn on electrical discharge and observe a faint blue/purple glow throughout chamber (more visible with room light turned down). Allow discharge to proceed for ~30 seconds. If sparking is observed or grids are jumping, the discharge is too strong and the carbon film will break. If in doubt, examine one or two grids by phase-contrast light microscopy to ensure that the carbon film was not broken.
Grasp grid by edge using reverse-grip forceps with carbon coat facing up. Pipette 5 μl of reconstituted protein sample onto surface of the grid. If glow discharge was effective, sample should spread readily across grid. Pooling of sample indicates that grid is still too hydrophobic.
Incubate ~30 sec, then blot excess sample from grid by touching torn edge of filter paper to edge of the grid. Do not scrape filter paper across top of grid.
Immediately pipette 5 μl of 1% uranyl acetate solution onto grid. Do not allow grid to dry before adding this solution. It is easiest if a pipette is pre-loaded with stain before prior to the blotting step. Incubate 15–30 sec and blot stain from side as in step 3. Depending on the sample, this staining step may be repeated 2–3 times. After the final application, thoroughly remove all stain from grid and forceps by touching torn filter paper to side of the grid and to the side of the forceps where stain may have wicked. Try to remove as much liquid as possible.
Allow grid to air-dry and store in plastic grid box. Uranyl acetate stained grids should last for several years. Other stains may be more volatile (see Note 12).
3.7. Screening of 2D Crystallization Trials by Electron Microscopy
Insert stained grid into electron microscopy according to manufacturer’s instructions.
Start with microscope at low magnification (150x – 300x). Be sure that objective aperture has been removed as it will limit the field of view in this mode. Observe overall appearance of grid. Most squares should be light grey, possibly with some dark flecks, whereas broken squares will appear bright (see Fig. 4). Too much sample and/or stain will result in very dark, possibly black squares. If too many grid squares are broken or black, see Note 13.
Choose an area of the grid with several good squares. Increase magnification to ~3000x, insert objective aperture to increase contrast and adjust sample height to the eucentric position. Adjust focus to achieve 5–10 μm underfocus
-
Scan grid square noting features and recording images. Look for the following basic morphologies (see Fig. 4):
Vesicles with poor contrast and irregular shape, which are likely to be pure lipid.
Large aggregates of protein.
Round vesicles with good contrast, which are likely to be proteoliposomes.
Rod-shaped vesicles, which are possibly a sign of helical crystals.
Large sheets, which are possibly a sign of 2D crystals.
When rod-shaped vesicles or large sheets are found, increase the magnification to ~50,000X and defocus to 1–2 um underfocus (see Note 14). At this magnification, you may be able to see a lattice if sample is crystalline, depending on how much of the protein is outside of the membrane where negative stain is most likely to produce contrast. Fig. 5 shows examples of helical and 2D crystals.
After recording an image (preferably on a CCD camera, see Note 15), calculate a Fourier transform. A 2D crystal will produce discrete diffraction spots, and a helical crystal will produce layer lines (Fig. 5). In addition, Thon rings may be visible in the background and can be used to judge defocus and astigmatism.
Continue screening other grid squares in areas all over grid. Stain is inherently variable, and some areas will be better stained than others. Signs of suitable staining include good contrast, a halo of stain around object and low, featureless background.
-
Computer analysis of images require considerable expertise, but preliminary evaluation of ordered arrays can be done as follows.
Open 50K image in ImageJ software (10).
Box out an area of interest.
Under “Process” menu, choose to calculate Fourier transform
Adjust brightness/contrast of Fourier transform until diffraction spots or layer lines are visible.
Record resolution of diffraction spots closest to the origin as well as maximum resolution of spots furthest from the origin by positioning mouse over individual diffraction spots. ImageJ will report resolution of any given location in pixels (edge of the Fourier transform corresponds to the Nyquist limit, which is 2 pixel resolution). Multiply values reported by ImageJ by the calibrated pixel size to calculate the resolution in Angstroms. Example: ImageJ reports resolution of 33 pixels and the image was recorded at a magnification producing 2Å/pixel, so resolution of spot is 33 × 2 = 66 Å.
Calculate a filtered image by using the circle tool to encircle each of the visible spots in the Fourier transform. Choose to clear each spot and then calculate an inverse Fourier transform (under the “Process” menu).
-
To consult with a potential collaborator about prospects for structure determination, assemble the following data:
Images.
Sample Fourier transform showing diffraction spots or layer lines.
Estimate of highest-resolution visible in the Fourier transform.
Estimate of lattice size for the 2D crystal (i.e., resolution of the diffraction spots closest to the origin of the Fourier transform)..
Fig. 4.

Screening of negatively stained crystal trials. (A) Low magnification image showing multiple grid squares coated with continuous solid film. Although a few grid squares are broken on the lower right, the film is mostly intact. (B) Low magnification image showing considerable precipitate on this grid and many broken grid squares on the right hand side. This grid is less likely to produce nice images. (C) Pure lipid vesicles generally form extended, amorphous structures. (D) Proteoliposomes are generally spherical, with a tendency to collapse and form wrinkles when dried in negative stain. (E) Elongated shapes can indicate that the protein is forming an ordered lattice within the bilayer, which after further screening could produce helical crystals. (F) Single-layered membrane sheets lie flat on the carbon support and offer the best opportunity for high resolution structure determination. Unlike the vesicles in (D), these sheets do not show wrinkles that result from the collapse of the top bilayer onto the lower bilayer. Scale bars are 500 μm in (A–B) and 100 nm in (C–F).
Fig. 5.
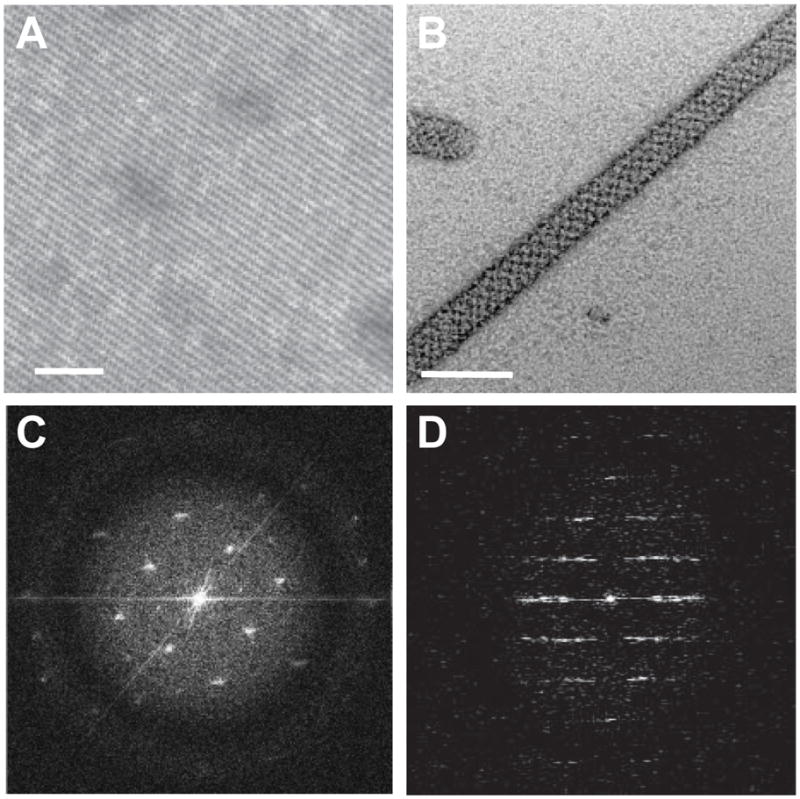
Negatively stained 2D crystals. (A) Planar 2D crystal shows a regular lattice. (B). A helical crystal also has a regular lattice, though a moire pattern is produced by overlap of lattices from the front and back sides of the cylindrical tube. (C) Fourier transform of a 2D crystal is characterized by discrete spots at regular intervals. A black circle surrounds the innermost spots, which is produced by the defocus used to record the image. Careful inspection will reveal additional spots appearing beyond this circle near the edge of the panel. (D) After rotating a helical crystal to align the long axis vertically, its Fourier transform is characterized by a regular series of horizontal “layer lines”. The pattern possesses mirror symmetry about both the horizontal and vertical axes. Scale bars in (A–B) correspond to 60 nm.
3.8. Conclusion
Although the methods for growing 2D crystals are relatively straight forward, it is unfortunately not possible to predict which conditions to use for a particular membrane protein. As with X-ray crystallography, a wide range of conditions must therefore be screened in order to identify a general set of parameters that maintain protein stability and that produce suitable membrane morphologies. Promising conditions are then subjected to subsequent screening designed to optimize size of membrane assemblies and order of any resulting 2D crystals. Unlike soluble proteins, membrane proteins generally require detergent solubilization, which complicates the physical chemistry of the crystallization solution and represents an additional parameter that must be included in the screen. For 2D crystallization, the situation is even more complex, as the composition of added lipid and the procedures used for reconstitution are also crucial to successful crystallization. As a result, a high-throughput approach is necessary for sampling a sufficient number of crystallization conditions. Although liquid-handling robots, multi-well crystallization plates and automated imaging are standard for X-ray crystallography, 2D crystallization has, until recently, been done largely by hand. This is due to the lack of suitable apparatus for multi-well dialysis and to the cumbersome nature of sample preparation and imaging by EM. Recently, some progress has been made on both these fronts. Strategies for automated imaging of samples in the electron microscope have been reported, using either a special 100-grid holder (11) or a sample-loading robot (12). Also, devices for microdialysis and negative staining have been developed on a 96-well format (13). By integrating all of these pieces, it is now possible to construct a pipeline for high-throughput screening of 2D crystallization trials. Thus, electron crystallography is becoming a plausible strategy not only for structure determination of membrane proteins, but for participation in the genome-wide effort being taken by the NIH sponsored Protein Structure Initiative.
Acknowledgments
This work was supported by NIH grant R01 GM81817.
Footnotes
2 μg of butylatedhydroxytoluene (BHT) can be added to the lipid solution prior to drying to minimize oxidation.
Lipid composition has a significant influence on the results of reconstitution and crystallization. To start, try a mixture of three lipids in the following ratio: 80% phosphatidylcholine as the bulk lipid, 10% phosphatidylethanolamine which tends to destabilize bilayer structures and thus induce fusion into larger sheets, 10% phosphatidic acid or phosphatidylserine which carries a negative charge and minimizes aggregation of vesicles.
Biobeads have different capacities for different detergents. According to Rigaud and colleagues (14), the following absorptive capacities can be expected in mg detergent/g Biobeads: Triton X100 -185, C12E8 -190, dodecylmaltoside - 105, Cholate - 80, CHAPS - 85, octylglucoside - 117. An excess of Biobeads relative to these values is always used due to the slow kinetics of binding.
Fast reconstitution often produces a more homogeneous preparation of proteoliposomes with an even distribution of randomly inserted protein molecules. Slow reconstitution can produce a heterogeneous preparation of vesicles with varying lipid-to-protein ratios and an asymmetric insertion of proteins into the bilayer (15). Presumably, this heterogeneity and preferential sidedness is a result of protein-protein interactions during bilayer formation, which depends on the kinetics of the procedure.
The protein should be visible as a white band at the interface of the 25% and 50% sucrose layers. However, there may be multiple bands if a heterogeneous population of proteoliposomes were produced. At first, it may therefore be useful to collect samples systematically throughout the sucrose gradient and assay them for lipid and protein content to characterize the results of the reconstitution (15).
Some report that multiple evaporations of carbon produces a stronger, flatter and more conductive carbon film. Some prefer graphite rods rather than amorphous carbon, but in each case the rods should be the highest possible purity.
Storage conditions affect the ability of the carbon film to separate from the mica in subsequent step 9. Generally, the carbon will not separate immediately after evaporation and should be stored for at least a couple of days. Separation may be helped by equilibrating the mica in a humid chamber immediately before floating.
Grids can be cleaned if desired by bath sonication in a small beaker filled with acetone. This removes traces of machine oil and other contaminants that may be present. After sonication, decant bulk acetone, then turn the beaker upside down on a piece of filter paper. As the grids dry, they tend to drop onto the filter paper, especially after tapping the bottom of the beaker.
Grids should be placed on the filter paper in a consistent way, usually with the polished (shiny) side up. This removes ambiguity about which side the carbon is on. Grids can be purchased with a Rh coating on one side, which makes it easy to distinguish the two different sides.
Two parameters should be explored to obtain the desired size and distribution of holes: amount of glycerol solution in Formvar/chloroform, and incubation time between sonication and dipping of the glass slide. More glycerol solution and longer incubation times produce larger holes. If very few holes separated by a large distance is desired, use lower amounts of glycerol and longer incubation times (e.g., Fig. 3b). This method was previously described by Baumeister and Seredynski (16). An alternative method for producing holey films is described by Fukami and Adachi (17), though the materials are no longer generally available.
Glow discharge apparatuses vary greatly among institutions. You should check with local EM staff for detailed operating instructions. For negatively charged proteins, it may be helpful to have a small amount of amylamine vapour inside the glow discharge apparatus. This will tend to make the carbon film positively charged. Glow discharged grids are good for about one day, after which they will become hydrophobic again.
There are many possible stains and a book by Robin Harris provides an excellent reference (18):
• Uranyl acetate: Most commonly used stain. pH, generally not adjusted, is 4.5. Stock solution lasts for months.
• Uranyl formate: A finer grain stain than uranyl acetate, but must be freshly prepared.
• Sodium phosphotungstate: Can be titrated to neutral or basic pH.
• Ammonium molybdate: pH 6.0–7.0. Much less contrast than heavy metal salts.
If all or most carbon squares are broken, then glow discharge may have been too aggressive. Check grids in light microscope before and after glow discharge to see if this was a problem. If not, poor sample handling, application of too much sample or of a sample composed of very coarse precipitate may result in broken carbon. If most squares are nearly or completely black, sample may be too concentrated and should be diluted. Alternatively, a wash step can be added before the first stain step. After removing sample, pipette 5ul of buffer onto grid, remove with filter paper and proceed with staining.
A low-dose software kit on the electron microscope is very helpful at this point. This kit will allow you to jump between screening magnification and imaging magnification. It also facilitates focusing on an area adjacent to the feature of interest, thus minimizing electron radiation and helping to preserve higher-resolution features.
CCD imaging is preferred for screening samples because 1) an unlimited number of images can be taken in any given session and 2) Fourier transforms can be calculated immediately, thus allowing rapid evaluation of image quality and crystalline order. A large format, high-sensitivity CCD is not needed for screening; a 1 mega pixel format is generally sufficient, allows for faster image readout and reduces data storage requirements.
References
- 1.Henderson R, Unwin PNT. Three-dimensional model of purple membrane obtained from electron microscopy. Nature. 1975;257:28–32. doi: 10.1038/257028a0. [DOI] [PubMed] [Google Scholar]
- 2.Henderson R, Baldwin JM, Ceska TA, Zemlin F, Beckmann E, Downing KH. Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J Mol Biol. 1990;213:899–929. doi: 10.1016/S0022-2836(05)80271-2. [DOI] [PubMed] [Google Scholar]
- 3.Subramaniam S, Hirai T, Henderson R. From structure to mechanism: electron crystallographic studies of bacteriorhodopsin. Philos Transact A Math Phys Eng Sci. 2002;360:859–74. doi: 10.1098/rsta.2001.0971. [DOI] [PubMed] [Google Scholar]
- 4.Stahlberg H, Fotiadis D, Scheuring S, Remigy H, Braun T, Mitsuoka K, Fujiyoshi Y, Engel A. Two-dimensional crystals: a powerful approach to assess structure, function and dynamics of membrane proteins. FEBS Lett. 2001;504:166–72. doi: 10.1016/s0014-5793(01)02746-6. [DOI] [PubMed] [Google Scholar]
- 5.Unger VM. Electron cryomicroscopy methods. Curr Opin Struct Biol. 2001;11:548–54. doi: 10.1016/s0959-440x(00)00260-8. [DOI] [PubMed] [Google Scholar]
- 6.Gonen T, Cheng Y, Sliz P, Hiroaki Y, Fujiyoshi Y, Harrison SC, Walz T. Lipid-protein interactions in double-layered two-dimensional AQP0 crystals. Nature. 2005;438:633–8. doi: 10.1038/nature04321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kühlbrandt W. Two-dimensional crystallization of membrane proteins. Quart Rev Biophys. 1992;25:1–49. doi: 10.1017/s0033583500004716. [DOI] [PubMed] [Google Scholar]
- 8.Yeager M, Unger VM, Mitra AK. Three-dimensional structure of membrane proteins determined by two-dimensional crystallization, electron cryomicroscopy, and image analysis. Methods Enzymol. 1999;294:135–80. doi: 10.1016/s0076-6879(99)94010-7. [DOI] [PubMed] [Google Scholar]
- 9.Jap BK, Zulauf M, Scheybani T, Hefti A, Baumeister W, Aebi U, Engel A. 2D crystallization: from art to science. Ultramicroscopy. 1992;46:45–84. doi: 10.1016/0304-3991(92)90007-7. [DOI] [PubMed] [Google Scholar]
- 10.Rasband W. ImageJ, US National Institutes of Health. 1997–2008 http://rsb.info.nih.gov/ij/
- 11.Lefman J, Morrison R, Subramaniam S. Automated 100-position specimen loader and image acquisition system for transmission electron microscopy. J Struct Biol. 2007;158:318–26. doi: 10.1016/j.jsb.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng A, Leung A, Fellmann D, Quispe J, Suloway C, Pulokas J, Abeyrathne PD, Lam JS, Carragher B, Potter CS. Towards automated screening of two-dimensional crystals. J Struct Biol. 2007;160:324–31. doi: 10.1016/j.jsb.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vink M, Derr K, Love J, Stokes DL, Ubarretxena-Belandia I. A high-throughput strategy to screen 2D crystallization trials of membrane proteins. J Struct Biol. 2007;160:295–304. doi: 10.1016/j.jsb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rigaud JL, Levy D, Mosser G, Lambert O. Detergent removal by non-polar polystyrene beads: applications to membrane protein reconstitution and two-dimensional crystallization. Eur Biophys J. 1998;27:305–19. [Google Scholar]
- 15.Young HS, Rigaud JL, Lacapere JJ, Reddy LG, Stokes DL. How to make tubular crystals by reconstitution of detergent-solubilized Ca2+-ATPase. Biophys J. 1997;72:2545–58. doi: 10.1016/S0006-3495(97)78898-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baumeister W, Seredynski J. Preparation of Perforated Films with Predeterminable Hole Size Distributions. Micron. 1976;7:49–54. [Google Scholar]
- 17.Fukami A, Adachi K. A New Method of Preparation of a Self-Perforated Micro Plastic Grid and its Application (1) J Electron Micros. 1965;14:112–8. [PubMed] [Google Scholar]
- 18.Harris JR. Negative Staining and Cryoelectron Microscopy. BIOS Scientific Publishers Limited; Oxford, UK: 1997. [Google Scholar]