

Abstract
Apoptosis is a process by which cells undergo a form of non-necrotic cellular suicide. Although it is a programmed process, apoptosis can be induced by various stressors. During sepsis, apoptosis has been regarded as an important cause of cell death in the immune system, leading to unresponsiveness to treatment. This study was designed to investigate how prior heat shock induction can influence the rate of apoptosis in animals that have experienced sepsis. Sprague-Dawley rats were used, and experimental sepsis was induced by cecal ligation and puncture (CLP). Animals in the heated group were anesthetized and received heat shock by whole-body hyperthermia. They were sacrificed 9 h and 18 h after CLP as early and late sepsis, respectively. Apoptosis was evaluated by “DNA ladder” detection in agarose electrophoresis and Tdt-mediated dUTP nick end-labeling (TUNEL) assay. Hsp72 was detected by Western blot analysis. The results showed that the DNA ladder was detected most clearly in the thymus at the late phase of sepsis with time course dependence, while it showed less clearly in heat shock treated animals. Histopathological study by TUNEL assay obtained similar results in the thymus, where the cortex was more susceptible to apoptosis than the medulla. The Western blot analysis showed that the heat shock induced Hsp72 concomitant with an increase in Bcl-2:Bax ratio. In conclusion, heat shock pretreatment prevents rats from sepsis-induced apoptosis that may account for the better outcome of experimental sepsis. An increase in the Bcl-2:Bax ratio may in part explain the molecular mechanism of the effect of heat shock pretreatment.
INTRODUCTION
Sepsis is regarded as a major initiator of multiple organ dysfunction syndromes and remains a leading cause of mortality in medical critical care (Parrillo 1993). The septic process triggers a series of as yet unknown events, even when the invading pathogen is successfully eradicated, and patients may continue to worsen with sequential loss of organ function. Despite advances in medical equipment and the administration of new drugs, mortality rates remain high. For decades, the pathogenesis of sepsis has been extensively investigated in animal studies, but our understanding of the subject remains limited.
In the last decade, a novel therapeutic strategy has been introduced to reduce the subsequent injury by using the protective function of the heat shock response. In response to various stresses, a series of highly conversed proteins named heat shock proteins (Hsps) can be induced. Amazingly, no Hsps are induced by cecal ligation and puncture (CLP)-induced sepsis or endotoxin-induced shock (Villar et al 1994; Yang et al 1998). In our previous study, we showed that heat shock pretreatment significantly reduced the mortality rate of rats with CLP-induced sepsis: 50% vs 0% at 18 h, 90% vs 0% at 24h, and 100% vs 50% 48 h after CLP operation in the control and preheated groups, respectively (Yang et al 1998). Similar results were also obtained from studies that involved the administration of sodium arsenite (Ribeiro et al 1994). However, it is unclear how a previous heat shock treatment protects subjects from sepsis.
In recent years, a possible cause of progressive organ failure in sepsis, called programmed cell death or apoptosis, has been suggested (Ayala et al 1995; Hiramatsu et al 1997). Apoptosis can be defined by either morphologic or biochemical criteria (Kerr et al 1972). The morphologic changes include condensation of nuclear chromatin, aggregation of cytoplasmic organelles, and blebbing of cell membranes. The most common biochemical feature of apoptosis is the fragmentation of DNA (DNA ladder formation) caused by endogenous endonuclease activity (Hale et al 1996). During the process of apoptosis, a progressively growing number of factors or proteins has been documented. The Bcl-2 family of proteins is considered to constitute a critical intracellular checkpoint of apoptosis within a common cell death pathway. Some proteins of this family, such as Bcl-2 and Bcl-χL, function as apoptosis suppressors. Other Bcl-2 homologues, including Bax and Bak, have a powerful apoptosis-promoting ability (Chao and Korsmeyer 1998; Wang and Reed 1998). The balance of expression of the Bcl-2 family contributes directly or indirectly to the induction of apoptosis. While the regulation of apoptosis has been extensively studied, little is known about the mechanisms of cell survival or death during sepsis. However, it is reasonable to hypothesize that modulation in the outcome of sepsis in heat shock–preconditioned rats may be related to the intensity of sepsis-induced apoptosis.
Therefore, this study was performed to determine how prior heat shock induction can influence the rate of apoptosis in animals that have experienced sepsis, with a discussion of the roles of Bcl-2 and Bax.
MATERIALS AND METHODS
Animals
Experiments were performed on adult male Sprague-Dawley rats (weighing from 270–350 g) obtained from the National Experimental Animal Center (Nan-Kang, Taipei, Taiwan). The experiments conducted in this study were approved by the Animal Committee of the Kaohsiung Medical College, and the authors adhered to the guidelines laid down by the National Institutes of Health for the use of experimental animals. The animals were divided into the following groups: sham operated group (no sepsis) (n = 6); sham heated early sepsis group (n = 6) of 9 h after CLP operation; sham heated late sepsis group (n = 6) of 18 h after CLP operation; and preheated late sepsis group with heat shock treatment 24 h before CLP operation (n = 6).
Induction of sepsis
Sepsis was induced by cecal ligation and puncture (CLP) as described previously (Yang et al 1998). Briefly, animals were deprived of food, but water was permitted for 6 h prior to the operation. Under light ether anesthesia, a laparotomy was performed through a midline abdominal incision. The cecum was pulled out and ligated just below the ileocecal valve. The ligated cecum was punctured twice at different sites with an 18-gauge needle, and the cecum was gently compressed until feces were extruded. The bowel was then returned to the abdomen, and the abdominal incision was closed in 2 layers. Control animals underwent a sham operation (a laparotomy was performed and the cecum manipulated but neither ligated nor perforated). All animals were given with 5 mL/100 g body weight of normal saline subcutaneously at the completion of surgery and also at 9 h postoperatively to provide replacement for the extracellular fluid sequestered during peritonitis. Animals were deprived of food but had free access to water after surgery. The thymus, spleen, Peyer's patch, lung, kidney, liver, and lymphocytes were collected and frozen as quickly as possible in liquid nitrogen and stored at −20°C.
Heat shock treatment
Rats of the preheated late sepsis group received heat shock by whole-body hyperthermia with an electric pad after pentobarbital anesthesia as described previously (Patriarca and Maresca 1990; Yang et al 1996). When the rectal temperature reached 41°C, it was maintained between 41°C and 42°C for 15 min. After the heating pad was removed, the rectal temperature was kept at 37°C until consciousness was completely regained. Attention was paid to ensure that the rat's airway was free of obstruction. The heated rats were put back in their cages to recover for 24 h. The rats of the sham heated control group were also anesthetized, but no heating was applied.
DNA extraction and qualitative analysis of DNA fragmentation by gel electrophoresis
Tissue preparation and DNA extraction were performed as described by Kaufman et al (1995). In brief, tissues of target organs were ground in a clean mortar in liquid nitrogen. The powder was submerged in a 2-mL microtube containing 8 to 10 volumes of lysis buffer (100 mM NaCl; 10 mM Tris-Cl, pH 8.0; 25 mM EDTA, pH 8.0; 0.5% sodium dodecyl suflate [SDS]; RNase at a concentration of 1 mg/mL/100 mg tissue) and then homogenized by a plastic homogenizer. The microtube was incubated at 37°C for 30 min. After proteinase K at a final concentration of 100 μL/mL was added, the homogenate was incubated overnight at 50°C. DNA was then extracted with equal volume of phenol/chloroform/isoamyl alcohol. After centrifugation for 10 min at 12 000 × g, the aqueous layer was decanted and a half volume of 7.5 M ammonium acetate and 2 volumes of absolute alcohol were added. The precipitated DNA was collected by centrifugation, rinsed with 70% ethanol, and then dissolved in TE buffer (10 mM Tris-HCl, pH 8.0; 1 mM EDTA). The concentration of DNA of each sample was estimated by measuring the optical density at 260 nm. Specimens containing 8 μg of DNA were applied to a 2% agarose gel and subjected to electrophoresis for 1 h at 100 V. The gel was stained with ethidium bromide and photographed under UV illumination.
Histopathological evaluation of apoptosis by Tdt-mediated dUTP nick end-labeling (TUNEL) method
The samples were paraffin embedded, cut into 4-μm-thick slices, and stained by the fluorescence TUNEL method. The TUNEL assay was performed using the In Situ Cell Death Detection Kit (Boehringer Mannheim GmbH, Germany). In brief, the samples were dewaxed, rehydrated, and then incubated with proteinase K (20 μg/mL in 10 mM Tris/HCl, pH 8.0) at 37°C for 30 min. After they were washed twice with PBS, 50 μL TUNEL reaction mixture were added to the samples. Slides were incubated in a humidified chamber at 37°C for 60 min in the dark and then rinsed three times in PBS. The fluorescent TUNEL-labeled slides were photographed using a fluorescence microscope.
Western blot and immunochemical analysis
Total cell extraction was prepared and estimated quantitatively using an assay kit (Bio-Rad Co., Hercules, USA) and spectrophotometer (model U-2000, Hitachi Co., Tokyo, Japan) at 595 nm. Equal amounts (20ug) of protein extract were loaded and separated by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to a membrane by semidry transblotter. Hsp72 and β-tubulin were detected simultaneously. Monoclonal antibodies specific for Hsp72 (Boehringer Mannheim) and for β-tubulin (Boehringer Mannheim) were utilized as the primary antibody, while anti-mouse immunoglobulin G conjugated with peroxidase (Boehringer Mannheim) was used as the secondary antibody. The expression of Bcl-2 and Bax were analyzed on 16% SDS-polyacrylamide gels. Polyclonal antibodies of Bcl-2 (Transduction Laboratories, USA) and Bax (Santa Cruz Biotechnology Inc., USA) were applied as the primary antibody, while anti-mouse and anti-rabbit immunoglobulin G conjugated with peroxidase (Boehringer Mannheim) were used as the secondary antibody, respectively. The blots were developed with the BM Chemiluminescence Western blotting kit (Boehringer Mannheim) and autoradiographed. The results were quantified by a densitometer and analysis software (Bio-1D V.97 software, Vilber Lourmat, France).
RESULTS
Detection of DNA ladder in different tissues from rats of sham heated-late sepsis
Results of DNA fragmentation assays were shown in Figure 1. In sham heated late stage of sepsis, DNA ladder formation could be detected in the kidney, lung, spleen, Peyer's patch, and most apparently in the thymus. No DNA ladder was visualized in the liver or lymphocytes by this method.
Fig 1.
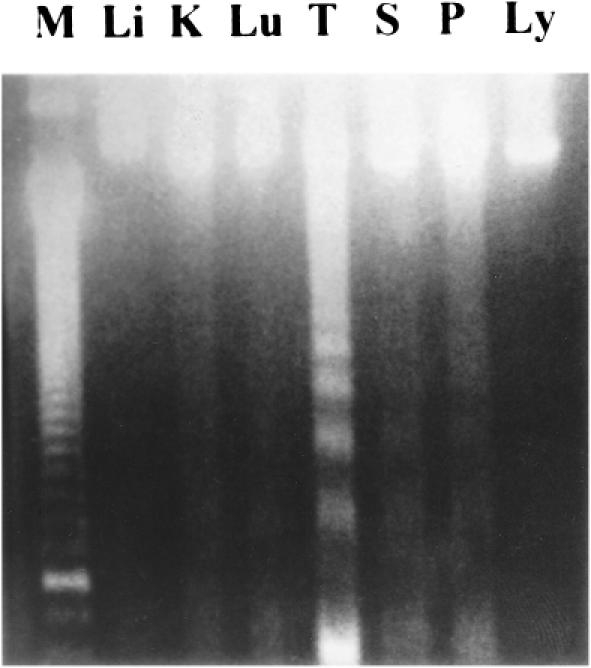
Analysis of DNA fragmentation in various organs of late sepsis by agarose gel electrophoresis. Organs were obtained from rats 18 h after cecal ligation and puncture (CLP) operation. DNA was prepared by chloroform/phenol method. Equal amount (8 μg) of DNA was applied to each lane. Li: liver, K: kidney, Lu: lung, T: thymus, S: spleen, P: Peyer's patch, Ly: lymphocytes, M: DNA ladder marker
Protective effect of heat shock pretreatment on apoptosis in thymus of late sepsis
The thymus was selected as the target organ for its highest expression of apoptosis in late sepsis. As shown in Figure 2, no DNA fragmentation was detected in the sham operated group (lane S) by this method. Following the CLP-operation, the ladder developed in both sham heated early sepsis (lane E) and sham heated late sepsis (lane L), and the ladder in late sepsis was more distinct than that in early stage, suggesting a qualitatively time-dependent effect. As predicted, the DNA ladder of the thymus in the preheated late sepsis group (lane HL) was less apparent compared with that of the sham heated group.
Fig 2.
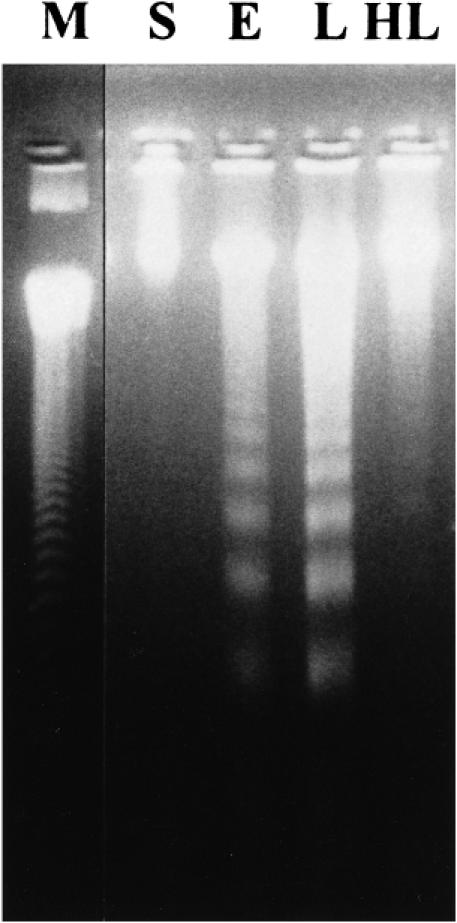
Effect of hyperthermia pretreatment on sepsis-inducedapoptosis in the thymus by DNA ladder detection. Hyperthermia pretreatment was performed 24 h before the cecal ligation and puncture (CLP) operation. Equal amounts (8 μg) of DNA were loaded in each lane. S: sham operation, E: sham heated early stage of CLP-induced sepsis, L: sham heated late stage of CLP-induced sepsis, HL: late stage of CLP-induced sepsis with hyperthermia pretreatment, M: DNA ladder marker
Histopathological study of apoptosis in the thymus by TUNEL assay
TUNEL assay was utilized to observe the severity and distribution of apoptotic cells in the thymus, and the results are shown in Figure 3. Apoptotic cells could be detected and visualized by fluorescent emission. By fluorescent microscopy, there were few apoptotic cells in the sham operated group (panel A). Under high magnification, apoptotic cells showed typical features, such as intensive staining, highly condensed chromatin, fragmented nuclei, and cellular fragmentation into apoptotic bodies (panel E). In the sham heated sepsis specimens, apoptotic cells were well stained and more distributed in the cortex than the medulla of the thymus. The number of apoptotic cells in the cortex increased after CLP-operation (panels B and C). As shown in panel D, sepsis-induced apoptosis was clearly inhibited by heat shock pretreatment.
Fig 3.
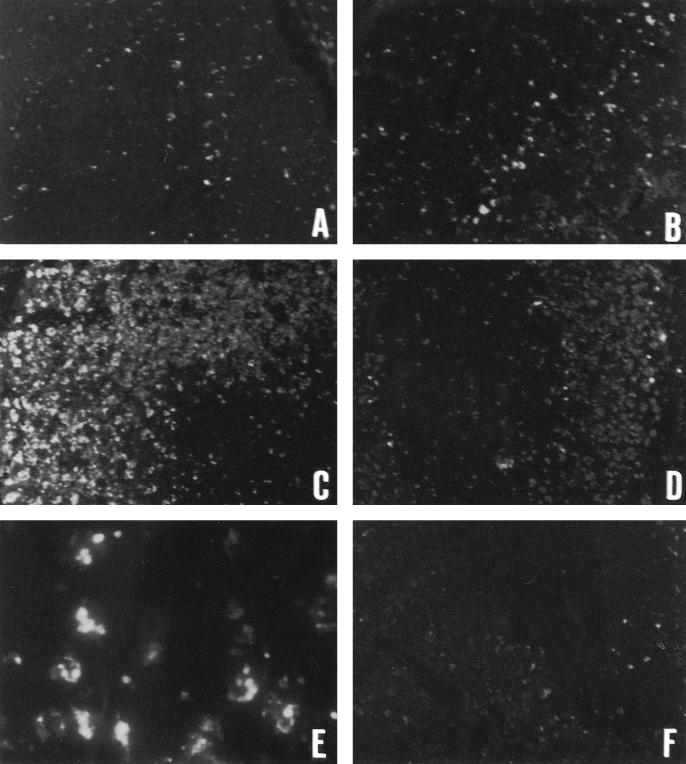
Immunohistochemical detection of apoptotic cells in the thymus during cecal ligation and puncture (CLP)-induced sepsis by TUNEL assay. Apoptotic cells were visualized by fluorescence stain and appeared as bright green spots under fluoromicroscopy. Panels A, B, C, and D indicate the specimens obtained from rats of sham operation, sham heated early stage, sham heated late stage of CLP-induced sepsis, and late stage of CLP-induced sepsis with hyperthermia pretreatment (100×). Panel E indicates high-power magnification of apoptotic cells from sham heated late sepsis (1000×), while panel F is the negative control
Detection of Hsp72 in the thymus
In this study, tubulin was detected simultaneously, acting as an internal standard. As shown in Figure 4, Hsp72 could be detected in the sham operated group. There was, however, no excessive expression after CLP-operation, neither in the sham heated early nor in the sham heated late stage of sepsis. The specimens of the preheated group showed a significant induction of Hsp72 synthesis, which was 3 times more than that of the nonheated group, indicating the presence of a heat shock response.
Fig 4.
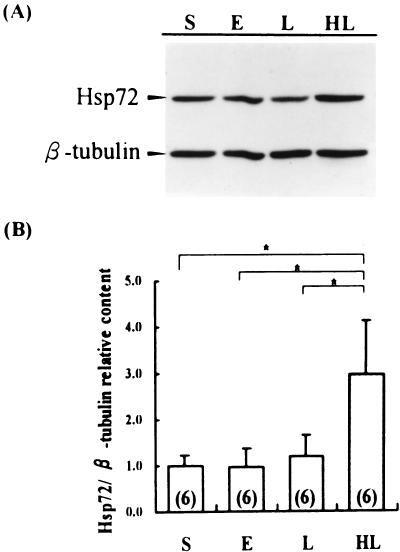
Detection of Hsp72 in the thymus by Western blotting and immunochemical study. Equal amount of cellular extract was loaded to each lane. Bands of Hsp72 and β-tubulin, acting as the internal standard, were quantified using a densitometer. Samples from preheated rats were statistically compared with those of nonheated rats. (A) Immunochemical study. (B) Statistical analysis of relative content of Hsp72 (ratio of ODHsp72 to tubulin). S: sham operation, E: sham heated early stage of CLP-induced sepsis, L: sham heated late stage of CLP-induced sepsis, HL: late stage of CLP-induced sepsis with hyperthermia pretreatment. Number of rats is shown in parentheses in each column. * = p < 0.05
Detection of Bcl-2 and Bax in the thymus of sepsis
The expression of Bcl-2 in the thymus of sham heated late sepsis decreased with a significant difference in the sham operated group (P < 0.05). There was, however, no significant change in the Bcl-2 expression during late sepsis between the sham heated and the preheated group, indicating that sepsis induced suppression of Bcl-2, which was not influenced by heat shock preconditioning. In the Bax expression, despite no significant change in the thymus of the sham operated, the sham heated early and the sham heated late sepsis, it decreased significantly in preheated late sepsis (P < 0.05). As a result, the ratio of Bcl-2 to Bax apparently increased in the preheated group of late sepsis (Fig 5).
Fig 5.
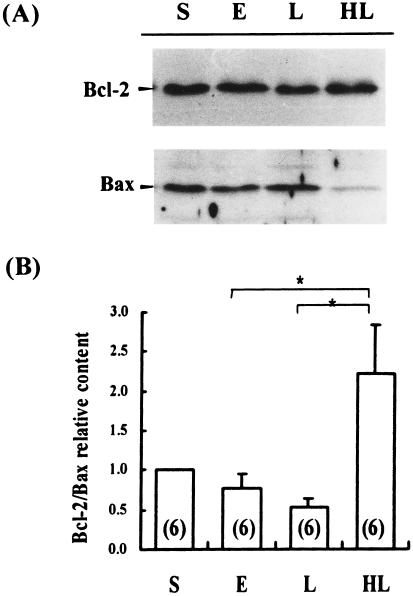
Detection of Bcl-2 and Bax in the thymus by Western blotting and immunochemical study. Equal amounts of cellular extract were loaded in each lane. Immunochemical detection of Bcl-2 and Bax was carried out simultaneously on the same nitrocellulose membrane, and the bands were quantified by a densitometer. The ratio was calculated and analyzed statistically. (A) Immunochemical study. (B) Statistical analysis of Bcl-2:Bax ratio. S: sham operation, E: sham heated early stage of CLP-induced sepsis, L: sham heated late stage of CLP-induced sepsis, HL: late stage of CLP-induced sepsis with hyperthermia pretreatment. Number of rats is shown in parentheses in each column. * = p < 0.05
DISCUSSION
Apoptosis is an active gene-directed cellular self-destruction that may occur under physiologic and pathologic conditions, with distinct morphologic and biochemical changes. Disorders in the regulation of apoptosis seem to have been vital determinants in the pathogenesis of diseases, such as cancer, AIDS, or neuron degenerative disorders (Hale et al 1996). Apoptosis is also an important cause of cell death in the immune system (eg, in the thymus, spleen, and lymph nodes, during and after viral and bacterial infection or lipopolysaccharide injection; Hiramatsu et al 1997). Despite limited research, it has been speculated that apoptosis may contribute to organ failure (Ayala et al 1995; Hiramatsu et al 1997).
In this study, we chose the CLP-induced sepsis animal model because of its exhibition of a biphasic dynamic change in the disease process, which is similar to the clinical findings in septic patients (Hwang et al 1994). The DNA electrophoresis in agarose gel showed that the DNA ladder was detected most clearly in the thymus obtained from CLP-operated rats. Characteristic findings were also observed in the spleen, lung, kidney, and Peyers' patch but not in the liver or lymphocytes. These results were in agreement with a previous study of Hotchkiss et al (1997). It was shown that the thymus is one of the most sensitive organs for CLP-induced apoptosis. Results were confirmed by the TUNEL method in the present study. Furthermore, the severity of apoptosis represented a progressive response in the course of sepsis.
Apoptosis has been reported to be a self-protective phenomenon of organisms in the face of complex pathophysiological conditions during sepsis, allowing the body to down-regulate the intense inflammatory response caused by necrosis (Cobb et al 1996). However, most studies have suggested that extensive apoptosis does harm to general physiological functions of tissues or organs. Ayala et al (1996) reported that apoptosis in mucosal B cells (eg, Peyer's patch) induces the breakdown of gut barrier integrity during sepsis, leading to a marked influx of enteric antigens. In addition, LPS induces a disseminated form of endothelial apoptosis that may be necessary for the development of endotoxic shock (Haimovitz-Friedman et al 1997). Apoptosis also takes place in the bone marrow, leading to a failure in the development of host immunity by immaturation of T cell and/or B cell during sepsis (Hotchkiss et al 1997). Furthermore, apoptosis has been reported to contribute to the progression of multiple organ failure during sepsis (Haimovitz-Friedman et al 1997; Hotchkiss et al 1997; Hiramatsu et al 1997). It is obvious that nonphysiological apoptosis is harmful to cells or organisms.
From both the results of agarose gel electrophoresis and those of the TUNEL method, it is clear that heat shock pretreatment attenuated the severity of apoptosis during late sepsis, when the Hsps were also overexpressed. Heat shock treatment may induce various metabolic or biochemical alterations in the body, but Hsp synthesis seems to be the major one. The relationships between Hsps and apoptosis have been well described in vitro. The induction of thermotolerance protects cells from heat and TNF alpha- and ceramide-induced apoptosis in mouse embryo fibroblasts, and Hsps have been categorized as a class of antiapoptotic proteins (Buzzard et al 1998). Enhancement to Hsp70 synthesis is correlated with resistance of DNA fragment development and apoptosis induction (Mosser and Martin 1992; Gordon et al 1997). On the contrary, quercetin inhibited the synthesis of Hsp70 and resulted in apoptosis. Furthermore, when tumor cells were first exposed to heat shock, no apoptosis was induced by quercetin (Wei 1994). In the present study, there is no evidence to show the direct correlation between Hsps and the attenuation of apoptosis. There is, however, no doubt that the heat shock response was successfully induced by the heat shock treatment as evidenced by Hsp72 synthesis. Our in vivo study confirmed the event that heat shock pretreatment inhibited the sepsis-induced apoptosis and that this may have contributed to the decrease in mortality revealed in previous studies.
Hsps have been known to influence general cellular metabolism through various mechanisms. Hsps functioning as molecular chaperones have been shown to help facilitate different stages of protein metabolism, such as transportation of premature protein, folding of native conformations of nascent polypeptides, and refolding of denature proteins (Gething and Sambrook 1992). Hsp assists in the transfer of newly synthesized protein into mitochondria, helping to maintain overall mitochondrial integrity (Stuart et al 1994). In terms of energy metabolic homeostasis, Hsps have been demonstrated to participate in ion compartmentalization, including a decrease in Ca2+ efflux from the endoplasmic reticulum, a crucial step of apoptosis (Kroemer et al 1998). In addition, Hsp may be involved in preserving cellular ATP levels (Wong et al 1997), producing ATP (Polla et al 1996) and preventing NAD+ and ATP consumption (Bellmann et al 1995). The regulation of energy metabolism and ion compartmentalization by Hsps may, in part, influence the progression of sepsis induced-apoptosis.
In terms of regulation of apoptosis, many factors have been reported, including Bcl-2 subfamily and Bax subfamily (Hale et al 1996). Some of Bcl-2 family members, such Bcl-2 and Bcl-χL, function as cell-death suppressors. Bcl-2 has been reported to participate in the antiapoptotsis effect by several pathways: prevention of activation of caspase cascade (Shimizu et al 1996), blockage of mitochondrial membrane potential loss (Shimizu et al 1996), inhibition in the release of cytochrome c from mitochondria (Kluck et al 1997; Yang et al 1997), or regulation of endoplasmic reticulum-associated Ca++ fluxes (Distelhorst et al 1996). When Bax was overexpressed relative to Bcl-2, the Bax-Bax heterodimer increased, and apoptotic death was accelerated in response to the death signal. In contrast, when Bcl-2 was overexpressed, it heterodimerized with Bax and homodimerized with itself, and death was repressed. The ratio of Bcl-2 to Bax, instead of each to itself, has been considered more important in determining the susceptibility to apoptosis (Oltvai et al 1993; Chao and Korsmeyer 1998; Reed 1998; Wang and Reed 1998). In this study, we found that Bax expression was down-regulated, even though the Bcl-2 did not change significantly. Accordingly, the Bcl-2:Bax ratio appeared to increase in the preheated late sepsis group. We suggest that heat shock pretreatment inhibits Bax expression, resulting in an increase in the ratio of Bcl-2 to Bax, and contributes to an inhibition of apoptosis induction at the terminal state of sepsis.
In conclusion, heat shock pretreatment prevents rats from sepsis-induced apoptosis that may contribute to the better outcome of experimental sepsis. Although further investigation is needed, the increase in the ratio of Bcl-2 to Bax expression by heat shock response may in part explain the molecular mechanism of the effect.
Acknowledgments
The present study was supported by a grant from the National Science Council, Taiwan, ROC (NSC 88-2314-B-037-008).
REFERENCES
- Ayala A, Herdon CD, Lehman DL, Ayala CA, Chaudry IH. Differential induction of apoptosis in lymphoid tissues during sepsis: variation in onset, frequency, and the nature of the mediators. Blood. 1996;87:4261–4275. [PubMed] [Google Scholar]
- Ayala A, Herdon CD, Lehman DL, DeMaso CM, Ayala CA, Chaudry IH. The induction of accelerated thymic programmed cell death during polymicrobial sepsis: control by corticosteroids but not tumor necrosis factor. Shock. 1995;3:259–267. doi: 10.1097/00024382-199504000-00003. [DOI] [PubMed] [Google Scholar]
- Bellmann K, Wenz A, Radons J, Burkart V, Kleemann R, Kolb H. Heat shock induces resistance in rat pancreatic islet cells against nitric oxide, oxygen radicals and streptozotocin toxicity in vitro. J Clin Invest. 1995;95:2840–2845. doi: 10.1172/JCI117989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzzard KA, Giaccia AJ, Killender M, Anderson RL. Heat shock protein 72 modulates pathway of stress-induced apoptosis. J Biol Chem. 1998;273:17147–17153. doi: 10.1074/jbc.273.27.17147. [DOI] [PubMed] [Google Scholar]
- Chao DT, Korsmeyer SJ. Bcl-2 family: regulators of cell death. Annu Rev Immunol. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- Cobb JP, Hotchkiss RS, Karl IE, Buchman TG. Mechanisms of cell injury and death. Br J Anaesth. 1996;77:3–10. doi: 10.1093/bja/77.1.3. [DOI] [PubMed] [Google Scholar]
- Distelhorst CW, Lam M, McCormick TS. Bcl-2 inhibit hydrogen peroxide-induced ER Ca2+ pool depletion. Oncogene. 1996;12:2051–2055. [PubMed] [Google Scholar]
- Gething MJ, Sambrook J. Protein folding in the cell. Nature. 1992;335:33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- Gordon SA, Hoffman RA, Simmons RL, Ford HR. Induction of heat shock protein 70 protects thymocytes against radiation-induced apoptosis. Arch Surg. 1997;132:1277–1282. doi: 10.1001/archsurg.1997.01430360023004. [DOI] [PubMed] [Google Scholar]
- Haimovitz-Friedman A, Cordon-Cardo C, Bayoumy S, et al. Lipoplysaccharide induces disseminated endothelial apoptosis requiring ceramide generation. J Exp Med. 1997;186:1831–1841. doi: 10.1084/jem.186.11.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale AJ, Smith CA, Sutherland LC, Stoneman VE, Longthorne VL, Culhane AC, Williams GT. Apoptosis: molecular regulation of cell death. Eur J Biochem. 1996;236:1–26. doi: 10.1111/j.1432-1033.1996.00001.x. [DOI] [PubMed] [Google Scholar]
- Hiramatsu M, Hotchkiss RS, Karl IE, Buckman TG. Cecal ligation and puncture (CLP) induced apoptosis in thymus spleen, lung and gut by endotoxin and TNF-independent pathway. Shock. 1997;7:247–253. doi: 10.1097/00024382-199704000-00002. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Swanson PE, Cobb JP, Jacobson A, Buchman TG, Karl IE. Apoptosis in lymphoid and parenchymal cells during sepsis: findings in normal and T- and B-cell deficient mice. Crit Care Med. 1997;25:1298–1307. doi: 10.1097/00003246-199708000-00015. [DOI] [PubMed] [Google Scholar]
- Hwang TL, Lau YT, Huang SF, Chen MF, Liu MS. Change of alpha 1-adrenergic receptor in human liver during intraabdominal sepsis. Hepatology. 1994;20:638–642. [PubMed] [Google Scholar]
- Kaufman PB, Wu W, Kim D, and Cseke LJ. 1995 Handbook of molecular and cellular methods in biology and medicine. CRC Press, London, 1–27. [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome C from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol. 1998;60:619–642. doi: 10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- Mosser DD, Martin LH. Induced thermotolerance to apoptosis in human T lymphocyte cell line. J Cell Physiol. 1992;151:561–570. doi: 10.1002/jcp.1041510316. [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman C, Korsmeyer SJ. Bcl-2 heterodimerizers in vivo with a conserved homology, Bax that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- Parrillo JE. Pathogenetic mechanisms of septic shock. N Engl J Med. 1993;328:1471–1477. doi: 10.1056/NEJM199305203282008. [DOI] [PubMed] [Google Scholar]
- Patriarca EJ, Maresca B. Acquired thermotolerance following heat shock protein synthesis prevents impairment of mitochondrial ATPase activity at elevated temperatures in Saccharomyces cerevisiae. Exp Cell Res. 1990;190:57–64. doi: 10.1016/0014-4827(90)90143-x. [DOI] [PubMed] [Google Scholar]
- Polla BS, Kantengwa S, Francois D, Slvioli S, Franceschi C, Marsac C, Cossarizza A. Mitochondria are selective targets for the protective effects of heat shock against oxidative injury. Proc Natl Acad Sci USA. 1996;93:6458–6463. doi: 10.1073/pnas.93.13.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JC. Bcl-2 family proteins. Oncogene. 1998;17:3225–3236. doi: 10.1038/sj.onc.1202591. [DOI] [PubMed] [Google Scholar]
- Ribeiro SP, Villar J, Downey GP, Edelson JD, Slutsky AS. Sodium arsenite induces heat shock protein-72 kilodalton expression in the lungs and protects rats against sepsis. Crit Care Med. 1994;22:922–929. doi: 10.1097/00003246-199406000-00008. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Eguchi Y, Kamiike W, Matsuda H, Tsujimoto Y. Bcl-2 expression prevents activation of the ICE protease cascade. Oncogene. 1996;12:2251–2257. [PubMed] [Google Scholar]
- Stuart RA, Cyr DM, Neupert W. Hsp70 in mitochondrial biogenesis: from chaperoning nascent polypetide chains to facilitation of protein degradation. Experientia. 1994;50:1002–1011. doi: 10.1007/BF01923454. [DOI] [PubMed] [Google Scholar]
- Villar J, Ribeiro SP, Mullen JB, Kuliszewski M, Post M, Slutsky AS. Induction of the heat shock response reduces mortality rate and organ damage in a sepsis-induced acute lung injury model. Crit Care Med. 1994;22:914–921. [PubMed] [Google Scholar]
- Wang HG, Reed JC. Mechanisms of Bcl-2 protein function. Histol Histopathol. 1998;13:521–530. doi: 10.14670/HH-13.521. [DOI] [PubMed] [Google Scholar]
- Wei YQ, Zhao X, Kariya Y, Fukata H, Teshigawara K, Uchida A. Induction of apoptosis by quercetin: involvement of heat shock protein. Cancer Res. 1994;54:4952–4957. [PubMed] [Google Scholar]
- Wong HR, Ryan M, Menendez IY, Denenberg A, Wispe JR. Heat shock protein induction protects human respiratory epithelium against nitric oxide-mediated cytotoxicity. Shock. 1997;8:213–218. doi: 10.1097/00024382-199709000-00010. [DOI] [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, et al. Prevention of apoptosis by Bcl-2: release of cytochrome C from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Yang RC, Wiang CI, Chen HW, Chou FP, Lue SI, Hwang KP. Heat shock treatment decreases the mortality of sepsis in rats. Kaohsiug J Med Sci. 1998;14:664–672. [PubMed] [Google Scholar]
- Yang RC, Yang SL, Chen HW, Lai SL, Chen SS, Ching CS. Previous heat shock treatment attenuates bicuculline-induced convulsion in rats. Exp Brain Res. 1996;108:18–22. doi: 10.1007/BF00242900. [DOI] [PubMed] [Google Scholar]