

Abstract
The nonclassical histocompatibility class I gene HLA-G has a tissue-restricted expression. To explore mechanisms involved in HLA-G transcriptional regulation, we have investigated the effect of stress, including heat shock and arsenite treatment, on HLA-G expression in tumor cell lines. We show that stress induces an increase of the level of the different HLA-G alternative transcripts without affecting other MHC class I HLA-A, -B, -E, and -F transcripts. A heat shock element (HSE) that binds to heat shock factor 1 (HSF1) on stress conditions was further identified within the HLA-G promoter. Considering the ability of HLA-G to modulate the function of immunocompetent cells, we hypothesize a new feature of HLA-G as a signal regulating the immune response to stress.
INTRODUCTION
During evolution, adaptive mechanisms, such as the heat shock response, which enables cells and tissues to detect and to respond to various environmental stress, have been conserved. This response is characterized by the rapid stress-induced synthesis of heat shock proteins (HSPs) that allows cells to restore protein homeostasis and to be protected against molecular damage (Morimoto and Santoro 1998).
In higher eukaryotes, the cellular response to acute exposure to a range of physiological and environmental trauma, including heat shock, oxidants, heavy metals, ultraviolet radiation, cytokines, hormones, and amino acid analogues, is tightly controlled at the level of transcription by a family of heat shock factors (HSFs), HSF1 to HSF4 (Morimoto 1998). In humans, 3 HSFs have been characterized (Rabindran et al 1991; Schuetz et al 1991; Nakai et al 1997). HSF1 is activated in cells exposed to high temperatures and other environmental stress conditions, whereas HSF2 appears to function in cells involved in processes of differentiation and development (Leppä and Sistonen 1997) and accumulates on arrest of proteasome activity (Mathew et al 1998). HSF4 is not an activator of transcription and may rather act as a negative regulator of heat shock gene expression (Nakai et al 1997). Recently, alternative splicing events generating functionally distinct protein isoforms were reported for these 3 HSFs (Fiorenza et al 1995; Goodson and Sarge 1995; Goodson et al 1995; Tanabe et al 1999). On activation by various stimuli, HSF1 undergoes a multistep process of activation, including trimerization, translocation to the nucleus, binding to a highly conserved upstream response element (heat shock element [HSE]), and hyperphosphorylation on serine residues (Cotto et al 1996). The HSEs, defined as a minimum of 3 contiguous repeats of the pentanucleotide NGAAN arranged in alternating orientation, are located in the promoter region of genes encoding HSPs and molecular chaperones (Perisic et al 1989). Stress-induced binding of HSFs to their HSEs targets alters the assembly of transcription factor complexes to their promoter, thus activating transcription (Brown and Kingston 1997; Mason and Lis 1997; Sandaltzopoulos and Becker 1998).
Stress-induced HSPs are not only essential for regulating the state of intracellular folding, assembly, and translocation of proteins, but are also potent immunogens that modulate immune responses. It is now widely recognized that stress exerts both stimulating and suppressive effects on immune responses, and, apart from their stressed inducible expression, altered expression of HSPs has also been observed in association with a diverse array of diseases (Morimoto and Santoro 1998). Immunity to HSPs is thus currently considered as a part of normal immunoregulatory T-cell responses with disease controlling potential (van Eden et al 1998) and has recently led to the use of HSPs in cancer immunotherapy protocols (Ménoret and Chandawarkar 1998; Srivastava et al 1998; Schild et al 1999). Other genes involved in immune responses, the major histocompatibility complex class I-chain related genes (MIC) A and MICB, have also been reported to be up-regulated by heat shock in Hela cells and quiescent intestinal epithelial cells (Groh et al 1996, 1998). In order to further analyze the influence of stress on the regulation of genes that modulate the immune response, we investigated the effect of elevated temperature and arsenite treatment on the expression of the nonclassical class I human major histocompatibility complex gene HLA-G.
In contrast with highly polymorphic classical HLA-A, HLA-B, and HLA-C class I genes expressed at the surface of most nucleated cells of the organism, the HLA-G gene exhibits low polymorphism and is characterized by a tissue-restricted pattern of protein expression, essentially limited to extravillous cytotrophoblast cells in the placenta (Kovats et al 1990; Le Bouteiller and Blaschitz 1999), to endothelial cells of fetal vessels present in the chorionic villi (Blaschitz et al 1997), and to amnion cells and amniotic fluid (McMaster et al 1998) as well as to thymic medullary and subcapsular epithelial cells (Crisa et al 1997). Although the exon/intron structure of HLA-G is similar to that of classical class I, the cytoplasmic tail is reduced to only 6 amino acids because of a premature stop codon in exon 6 (Geraghty et al 1987). A specific feature of the HLA-G gene is its alternative splicing in at least 6 HLA-G mRNAs (Ishitani and Geraghty 1992; Fujii et al 1994; Kirszenbaum et al 1994): (1) in addition to the full-length membrane-bound isoform HLA-G1, transcripts lacking exon 3 (-G2), exon 3, and 4 (-G3), or exon 4 (-G4) retain the capacity to be translated into membrane-bound proteins lacking 1 or 2 extracellular domains; (2) a soluble form of HLA-G1, HLA-G5 (or -G1s), is encoded by a transcript including intron 4, where a premature stop codon prevents the translation of exon 5 and downstream sequences that encode the transmembrane and cytoplasmic domains; and (3) another transcript, HLA-G6 (or -G2s), lacking exon 3 and including intron 4, also retains the capacity to be translated as a soluble protein.
Like classical HLA class I molecules, HLA-G is able to associate with a wide array of nonamer peptides (Lee et al 1995; Diehl et al 1996) and to bind the CD8 T-cell coreceptor (Sanders et al 1991). HLA-G serves as a restriction molecule that elicits a specific murine cytotoxic T-cell response in HLA-G transgenic mice (Horuzsko et al 1997; Schmidt et al 1997), but its capacity to generate a T-cell receptor restricted response in humans is still uncertain. HLA-G also interacts with different killer-cell inhibitory receptors (KIRs) present on natural killer (NK) cells and on subpopulations of cytotoxic CD8+ α β and γδT lymphocytes, thus modulating their cytotoxic effector activity, and also on lymphocyte B, monocyte, and dendritic cells (Colonna et al 1997; Cantoni et al 1998; Colonna et al 1998; Allan et al 1999; Rajagopalan and Long 1999). Although the HLA-G function is not clearly defined, a specific role in the maternal-fetal immune relationship during pregnancy was first proposed by providing evidence that HLA-G protects cytotrophoblasts against cytolysis by maternal uterine NK cells (Rouas-Freiss et al 1997; Ponte et al 1999). HLA-G was also recently detected in human colorectal cancer and melanoma biopsies (Fukushima et al 1998; Paul et al 1998, 1999), suggesting that HLA-G expression on tumor cells may favor their escape from cytotoxic antitumor responses.
The mechanisms that control HLA-G cell-specific protein expression are still undefined and involve regulation at the transcriptional level. Various patterns of HLA-G transcription can be observed: (1) high levels of all HLA-G alternative transcripts are observed in trophoblasts and in the choriocarcinoma JEG-3 cell line; (2) in peripheral blood mononuclear cells (PBMCs), low or moderate amounts of HLA-G transcripts are initially detectable and can be induced after exposure to type I and type II interferons (IFNs) (Yang et al 1996) as well as IL-10 for PBMCs (Moreau et al 1999)or in vitro culture for keratinocytes (Ulbrecht et al 1994); (3) in a wide variety of cells from fetal and adult tissues, the levels of mRNAs are low or very low and merely reflect a basal level of transcription (Carosella et al 1996); and (4) some cell types, including CD34+, NK, and first-trimester human fetal liver cells, are totally devoid of HLA-G transcript (Teyssier et al 1995; Amiot et al 1996; Moreau et al 1998), suggesting that a stringent negative control of transcription may operate in these cell types.
Transcriptional regulation of MHC classical class I genes is mediated by conserved regulatory elements clustered in the proximal promoter region. These conserved upstream DNA sequences include the enhancer A element, the interferon-stimulated response element (ISRE), site α and enhancer B elements, that are either deleted or altered in the HLA-G promoter (Le Bouteiller 1994; Gobin et al 1998). To date, experiments indicate that none of these regulatory elements or intronic sequences could account for tissue-specific expression of HLA-G observed in trophoblast-derived cell lines (Gobin et al 1998; Lefebvre et al 1999). Finally, although several agents, including IFN-γ alone (Yang et al 1996; Chu et al 1999) or in combination with IL-2 or GM-CSF (Amiot et al 1998) and IL-10 (Moreau et al 1999), have been shown to stimulate HLA-G expression in vitro in myelomonocytic cells, the mechanisms by which they act are still generally unknown.
In an attempt to identify the regulatory mechanisms that participate in physiological and tumor-specific activation of HLA-G transcription, we have studied the effects of various stress conditions (heat shock, arsenite) in a human melanoma cell line previously characterized as lacking HLA-G transcripts (Paul et al 1998). We examined the possibility that HLA-G may also represent a target gene induced by stress treatment. We have shown that various forms of stress induce HLA-G transcripts levels as well as in vitro binding of HSF1 to an HSE found within the HLA-G gene promoter. We thus propose that in addition to its known immunomodulary capacity, HLA-G might be considered as a stress-inducible gene.
MATERIALS AND METHODS
Cell culture and stress treatment
Human melanoma cell line M8 (HLA-A1, -A2, -B12, and B40/male), kindly provided by J.G. Guillet (INSERM U445, Paris, France), was maintained in RPMI 1640 medium with l-glutamine supplemented with 10% heat-inactivated fetal calf serum, 10 μg/mL gentamicin and 1X anti-PPLO (Life Technologies). The human HLA-G positive JEG-3 choriocarcinoma cell line (American Type Culture Collection) was cultured in DMEM with glutamax-I, sodium pyruvate, glucose, and pyridoxine supplemented with 10% heat-inactivated fetal calf serum, gentamicin, and anti-PPLO. The human glioblastoma cell line T98G (European Collection of Cell Cultures) was cultured in EMEM (EBSS) supplemented with 10% heat-inactivated fetal calf serum, 2 mM l-glutamine, 1% nonessential amino acids, 1 mM sodium pyruvate, and antibiotics. The cells were cultured in a 37°C, 5% CO2-humidified incubator. Cells were stressed by a heat shock treatment at 42°C for 2 h or by addition of an arsenite solution (Fluka) at a final concentration of 100 μM for 2 h. The cells were allowed to recover at 37°C for various times following stress treatment. Actinomycin D (Sigma) was added to the cell culture medium at 10 μg/mL just prior to heat shock treatment (42°C, 2 h) and was removed when cells returned to nonstressful conditions. Human peripheral blood was obtained from healthy volunteer donors. Mononuclear cells were isolated by Ficoll-Hypaque (1077) density centrifugation, and RNA was extracted immediately.
Reverse transcriptase-–polymerase chain reaction analysis
Total mRNA was extracted by using the RNA NOW reagent (Biogentex) according to the manufacturer's recommendations. The quality of RNA was checked by electrophoresis in a 1.5% agarose-denaturing gel. cDNAs were prepared from 2.5 μg of total RNA by using oligo-(dT)12–18 primer (Pharmacia Biotech) and Moloney murine leukemia virus reverse transcriptase (Life Technologies). Polymerase chain reaction (PCR) amplifications (DNA thermal cycler, Cetus, Perkin Elmer) were performed using exon 2–specific primer G.257 (5′-GGAAGAGGAGACACGGAACA-3′) (Kirszenbaum et al 1994) and 3′-untranslated region-specific primer GA.3U (5′-GGCTGGTCTCTGCACAAAGAGA-3′) (Onno et al 1994) or G.1004R (5′-CCTTTTCAATCTGAGCTCTTCTTT-3′), exon 3–specific primer G.526 (5′-CCAATGTGGCTGAACAAAGG-3′) (Kirszenbaum et al 1994) and intron 4–specific primer G.i4b (5′-AAAGGAGGTGAAGGTGAGGG-3′) (Moreau et al 1995a), or exon 2–/exon 4–specific primerG.-3 (5′-ACCAGAGCGAGGCCAACCCC-3′) and 3′-untranslated region-specific primer G.1216 (5′-GACGGAGACATCCCAGCCCC-3′) (Moreau et al 1995a), respectively, allowing the detection of all alternatively spliced HLA-G mRNAs, specifically HLA-G5 mRNA or HLA-G2 and HLA-G6 mRNA. Specific amplification of HLA-E was performed using exon 2–specific primer E.251 (5′-ACACGGAGCGCCAGGGACAC-3′) and 3′-untranslated region-specific primer E.1272 (5′-GTGTGAGGAAGGGGGTCATG-3′). Levels of HLA-F transcripts were evaluated by hybridization with an HLA-F–specific probe of cDNA products amplified using exon 2–specific primer Pan ClI (F) (5′-TCCCACTCCATGAGGTATTTC-3′) and exon 4–specific primer PanClI (R) (5′-TCCAGAAGGCACCACCACAG-3′) allowing the detection of all nonclassical HLA class I mRNAs (Houlihan et al 1992). PCR conditions used are as follows: 94°C for 1 min, 65°C (G.257/GA.3U, G.-3/G.1216 and Pan ClI [F]/Pan ClI [R]) or 61°C (G.257/G.1004R, G.526/G.i4b, and E.251/E.1272) for 1 min 30 s, 72°C for 2 min (35 cycles); the last extension step at 72°C was prolonged to 7 min. Coamplification of β-actin cDNA was carried out in each experiment with β-actin amplimer sets (Clontech) for 16 cycles to evaluate comparative amounts of cDNAs in samples. Absence of contaminant DNA was controlled by concomitant amplification of the PCR mixture without a template (H2O). PCR products were analyzed by electrophoresis in 1.5% agarose gel and stained with ethidium bromide. The specificity of PCR products was confirmed by alkaline blotting of the fragments in 0.4 M NaOH onto nylon membranes (Hybond N+, Amersham). After a 2-h prehybridization in 5X SSPE, 5X Denhardt's solution, 0.5% SDS, 100 mg of sonicated salmon sperm DNA/mL, at 60°C (G.257/GA.3U and G.257/G.1004R) or 55°C (G.526/G.i4b, G.-3/G.1216 and E.251/E.1272) or 65°C (Pan ClI (F)/Pan ClI (R)), hybridization was performed with 32P-labeled oligonucleotide probe exon 2–specific G.R (5′-GGTCTGCAGGTTCATTCTGTC-3′) (Houlihan et al 1992) or intron-4–specific G.I4F (5′-GAGGCATCATGTCTGTTAGG-3′) (Moreau et al 1995b) or exon 3-specific E.R (5′-ATCATTTGACTTTTGCTCGGA-3′) or exon 2–specific F.R (5′-GGCGTACCCTGTGGTCCACTC (Houlihan et al 1992), respectively, using [γ-32P]dATP (Amersham) and a 5′-end labeling kit (Appligene). After 2 washes for 15 min at room temperature and 2 washes for 15 min between 50° and 62°C in the presence of 2X SSC and 0.1% SDS, the blots were exposed to Biomax films (Kodak) with amplifying screens at −80°C. The blots were stripped in boiled 0.5% SDS solution and hybridized with a β-actin probe (5′-ATCATGTTTGAGACCTTCAACACCCCAGCC-3′).
Cloning and sequencing of HLA-G6
cDNAs of M8 cells treated for 2 h with arsenite and allowed to recover for 45 min were amplified by 2 rounds of PCR using specific primer -22ATG XbaI located in the 5′-untranslated region (5′-GCTCTAGAGCTCCCCAGACGCCAAGGATG-3′) and 3G.U KpnI located in the 3′-untranslated region (5′-GGGGTACCGGCTGGTCTCTGCACAAAGA-3′). After purification on agarose gel, a PCR product was cloned into the pcDNA3.1/hygro(−) eukaryotic expression vector (Invitrogen). Sequencing was performed with an automated DNA sequencer 377 XL using the PRISM AmpliTaq Big Dye Terminator diChloroRhodamine kit (Perkin Elmer). Sequences were compared with the GenBank database using the BLAST algorithm.
Northern blot analysis
Ten micrograms of total cellular RNAs were separated on a 1% formaldehyde agarose gel and transferred to a Nytran-Plus nylon membrane (Schleicher and Schuell) with 2X SSC buffer. RNAs were immobilized by UV cross-linking. After a 3 h prehybridization in 0.5 M Na2HPO4, pH 7.2, 7% SDS, 1% BSA, 1 mM EDTA at 65°C, hybridization was carried out overnight with a 32P-labeled DNA probe obtained by random priming (Megaprime DNA labeling systems, Amersham) with [α-32P]dCTP (Amersham). The filters were sequentially hybridized with the following probes: the HLA-A–specific probe is a 1.2-kb PvuII-XbaI fragment derived from the 3′ region of the HLA-A3 gene (Chimini et al 1988), and the HLA-B probe is a 0.55-kb PvuII-PvuII fragment from the 3′ region of the HLA-B7 gene (Coppin et al 1985). Washing was performed 3 times at 65°C in 40 mM Na2HPO4, pH 7.2, 5% SDS, 0.5% BSA, 1 mM EDTA. Membranes were exposed to Biomax films (Kodak) with amplifying screens at −80°C for 3 d. Between 2 hybridizations, blots were stripped in boiled 0.1% SDS solution. GAPDH probe was used as a control to quantify RNA on Northern blots.
Nuclear extracts
Cells (0.5 to 1.106) were washed twice with TBS (20 mM Tris-HCl pH 7.6, 137 mM NaCl). Pellets were gently resuspended and kept on ice 10 min with 800 μL of cold solution A (10 mM HEPES pH 7.8, 10 mM KCl, 2 mM MgCl2, 0.1 mM EDTA) supplemented just before use with 1 mM DTT, 0.1 mM phenylmethyl sulfonyl fluoride (PMSF), 4 μg/mL leupeptin. Cells were lysed by addition of 10% Nonidet P-40 (50 μL) and centrifuged for 2 min at 800× g and 4°C. Pellets were resuspended and kept for 20 min on ice with 400 μL of cold solution B (50 mM HEPES pH 7.8, 300 mM NaCl, 0.1 mM EDTA, 10% glycerol) supplemented just prior to use with 1 mM DTT, 0.1 mM PMSF, 4 μg/mL leupeptin. Nuclear ghosts were centrifuged 3 min at 800× g and 4°C. Supernatants were centrifuged again 5 min at 9000× g and frozen with 30% glycerol at −80°C.
Electrophoretic mobility shift assay
Binding reactions were performed by incubating 2 μL of nuclear extracts with approximately 50 000 cpm (0.6 ng) of 32P-labeled probe using double-stranded oligonucleotide probes described in Figure 3 and a 100-fold excess of cold oligonucleotide competitor (COI) in the presence of 2 μg of poly(dI-dC)-poly(dI-dC) (Pharmacia) in a final volume of 10 μL containing 20 mM HEPES pH 7.8, 40 mM KCl, 1 mM MgCl2, 0.1 mM EDTA, 0.5 mM DTT, 0.4% Ficoll. After 20 min of incubation at room temperature, the binding reactions were loaded onto a 4% polyacrylamide gel (29:1 acrylamide-bis ratio) in 44.6 mM Tris, 44.5 mM boric acid, and 1 mM EDTA. Gels were run at room temperature for 1 h 40 min at 200 V, fixed with 30% methanol and 10% acetic acid, transferred to Whatman 3MM papers, and autoradiographed. The 32P-labeled probes were prepared by 5′-end labeling 1 strand with polynucleotide T4 kinase (Appligene) using standard procedures. The radiolabeled strand was annealed to the complementary strand, and the unincorporated nucleotides were then removed by Sephadex G-25 (Pharmacia) chromatography. For experiments involving addition of antibodies to nuclear extracts prior to gel shift analysis, 1 μL of immune serum was added to 2 μL of extract and incubated at room temperature for 15 min before the addition of the binding solution. The rabbit polyclonal antibody against HSF1 (α-HSF1) was kindly provided by L. Sistonen (Turku Centre for Biotechnology, Turku, Finland), and a rabbit polyclonal serum (CAS) was used as a nonspecific control antiserum in supershift experiments.
Fig 3.

Sequences of the oligonucleotides used as probe and/or cold competitor in EMSA experiments. Sequences used are HSEHSP derived from the human hsp70 proximal promoter (position -119 to -87 upstream of the transcription start site), the wild-type HSEG (position -487 to -466 upstream of the HLA-G translation initiation codon) and mutant MUTHSEG, and an oligonucleotide corresponding to positions -1285 to -1254 of HLA-G promoter IR used as an irrelevant competitor. The residues altered in MUTHSEG are indicated in bold type. NGAAN sequences, comprising an array of inverted units characteristic of HSEs, are underlined. Palindromes formed by alignment of HSEs lying face to face on the DNA helix are framed
RESULTS
Heat shock or arsenite exposure increase amounts of HLA-G transcripts
In order to define a model in which a modulation of the HLA-G transcription could be observed, the effects of thermal and chemical stresses on levels of HLA-G transcripts were investigated by RT-PCR in the M8 human melanoma cell line. The cells were subjected to a mild heat shock (42°C, 2 h) or to a severe chemical stress (100 μM As2O3, 2 h), and RNAs were extracted at various times after a recovery period at 37°C. Results of an RT-PCR analysis using pan-HLA-G PCR primers (G.257/GA.3U) amplifying all known HLA-G isoforms and revealed by Southern blot with an exon 2–specific probe (G.R) are shown in Figure 1A. As previously described, we did not detect any HLA-G transcript in untreated M8 melanoma cells (Paul et al 1998), while high levels of HLA-G transcripts were detected in the JEG-3 positive control cell line. Heat shock (Fig 1A, left panel) or arsenite (Fig 1A, right panel) treatment of M8 cells resulted in a similar pattern of induction of the levels of HLA-G alternative transcripts. One hour post stress treatment, we observed a marked and selective induction of a signal corresponding to a transcript whose length lies between HLA-G1 and HLA-G2. Since the use of pan-HLA-G primers did not allow precise discrimination of HLA-G1 and HLA-G5 signals, both of which appear as a 1000-bp band, further identification of HLA-G transcripts encoding the HLA-G5 soluble isoform was conducted by hybridization with probes derived from intron 4 that specifically detect transcripts corresponding to HLA-G5 and -G6 isoforms (Fig 1B) and by HLA-G5–specific amplification PCR using specific primers G.526 and G.i4b (Fig 1C). The kinetics of HLA-G5 transcript activation, which encodes a full-length soluble HLA-G molecule, paralleled that observed using primers detecting all alternatively spliced HLA-G transcripts with a maximum activation rate between 3 and 6 h after recovery from stress treatment. We confirmed the specific kinetics of induction of the HLA-G6 transcript post stress treatment by (1) specific hybridization of the same membranes used for Figure 1A using an intron 4–specific probe (I4F), allowing the detection of both HLA-G5 and -G6 transcripts corresponding to soluble protein isoforms (Fig 1B), and (2) specific amplification of HLA-G2 and -G6 transcripts and selective detection of G6 cDNAs on Southern blots using an intron 4–specific probe (Fig 1D). Further cloning of an RT-PCR product purified from M8 cells treated for 2 h with 100 μM arsenite and allowed to recover for 45 min identifies HLA-G6 as the first transcript induced in this early phase of stress recovery. Three hours after recovery, all the known HLA-G transcripts—that is, HLA-G1/G5, HLA-G2/G4 (indiscriminable by size of cDNA products using pan-HLA-G primers), HLA-G6, and HLA-G3—were detected in large amounts in stressed M8 cells and reached levels that are comparable to that observed in the JEG-3 choriocarcinoma cell line. High levels of alternatively spliced HLA-G transcripts were maintained up to 21 to 22 h after the return to nonstressful conditions and progressively declined thereafter. HLA-G transcripts were still detectable 3 d after stress treatment, suggesting either that a residual transcriptional activity persisted or that a gain in mRNAs stability allowed them to keep their integrity at least for 2 d.
Fig 1.

Induced expression of all the known HLA-G transcripts in stress-treated cells. (A and B) Pan-HLA-G primers G.257 (exon 2) and GA.3U (3′-untranslated region) were used for PCR amplification of HLA-G transcripts corresponding to all the known HLA-G isoforms (HLA-G1: 874 bp; HLA-G2: 598 bp; HLA-G3: 325 bp; HLA-G4: 601 bp; HLA-G5: 996 bp; HLA-G6: 720 bp) and obtained from heat-shocked cells, 42°C, 2 h (left panels), or from arsenite-treated cells, 100 μM As2O3, 2 h (right panels). HLA-G–specific transcripts were revealed by hybridization with either the exon 2–specific G.R probe (A) or an intron 4–specific G.I4F probe (B). (C) Specific RT-PCR amplification of HLA-G5 transcripts (489 bp), which corresponds to a soluble isoform, was performed with primers G.526 (exon 3) and G.i4b (intron 4). cDNAs were revealed by hybridization with a G.I4F probe. (D) Specific RT-PCR amplification of HLA-G2 and -G6 transcripts was performed with primers G.-3 (exon 2/exon 4) and G.1216 (3′-untranslated region). HLA-G6 cDNAs (745 bp) were revealed by hybridization with a G.I4F probe. (E) RT-PCR analysis of the effect of actinomycin D on heat shock–mediated HLA-G mRNA expression. M8 cells were untreated or heat shocked (42°C, 2 h) in the absence (−) or the presence (+) of actinomycin D and allowed to recover for various times. Pan-HLA-G primers G.257 (exon 2) and G.1004R (3′-untranslated region) were used for PCR amplification of HLA-G transcripts corresponding to all the known HLA-G isoforms (HLA-G1: 764 bp; HLA-G2: 488 bp; HLA-G3: 215 bp; HLA-G4: 491 bp; HLA-G5: 886 bp; HLA-G6: 610 bp). (F) Induced expression of HLA-G transcripts in a stress-treated glioblastoma cell line (T98G). RT-PCR analysis of HLA-G mRNAs obtained from untreated, heat-shocked cells (42°C, 2 h) or arsenite-treated (100 μM As2O3, 2 h) T98G cells and allowed to recover for 4 h. Pan-HLA-G primers G.257 (exon 2) and GA.3U (3′-untranslated region) were used for PCR amplification of HLA-G transcripts corresponding to all the known HLA-G isoforms. The JEG-3 choriocarcinoma cell line was used as a control for high HLA-G transcription. Absence of contaminant DNA was controlled by concomitant amplification of the PCR mixture without a template (H2O). Bands corresponding to HLA-G1, -G2, -G3, -G4, -G5, and -G6 are indicated by arrows. PCR products coamplified in the same reaction by β-actin primers were detected on the same membrane by a β-actin probe
To further determine whether the accumulation of HLA-G transcripts results from a stress-induced increase of the mRNA stability, M8 cells were incubated with or without the transcriptional inhibitor actinomycin D during a heat shock treatment. Actinomycin D completely blocked heat shock-mediated increases of HLA-G mRNA levels (Fig 1E), suggesting that stress induces HLA-G gene expression by activating at least its transcription.
High levels of induction of HLA-G transcripts were also observed after heat or arsenite treatment of the T98G glioblastoma cell line (Fig 1F), indicating that the activation of HLA-G expression by stress is not restricted to melanoma cell type.
Stress conditions affect HLA-G mRNA levels with a relative specificity with regard to other HLA class I transcripts
Northern blot analysis (Fig 2A) of RNAs extracted at various times after stress treatment of M8 cells demonstrated that HLA-A or HLA-B transcripts are detectable in M8 melanoma cells as well as in positive control PBMC and are not significantly affected by thermal or chemical stress. Likewise, RT-PCR analysis of levels of other nonclassical class I HLA-E (Fig 2B) and HLA-F (Fig 2C) transcripts on the same samples used to analyze HLA-G transcripts revealed that their amounts are not modified by stress conditions. These results indicate that among the class I genes tested so far, HLA-G is specifically transactivated by stress in M8 melanoma cells.
Fig 2.
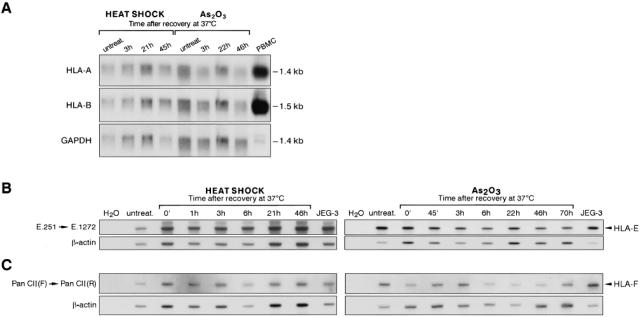
Stress does not modify levels of HLA-A, -B, -E, and -F class I transcripts. (A) Northern blot analysis of mRNAs isolated from heat-shocked (42°C, 2 h) and arsenite-treated (100 μM As2O3, 2 h) cells at various times after recovery at 37°C. The filters were hybridized with HLA-A, HLA-B locus-specific probes, or GAPDH probe used as a control to quantify RNA in each sample. RNAs extracted from PBMCs were used as a positive control for HLA class Ia expression. (B) primers E.251 (exon 2) and E.1272 (3′-untranslated region) were used for PCR amplification of HLA-E transcripts from heat-shocked cells, 42°C, 2 h (left panel), or from arsenite-treated cells, 100 μM As2O3, 2 h (right panel). HLA-E–specific transcripts were revealed by hybridization with the E.R-specific probe located in exon 3. (C) RT-PCR amplification of HLA-F transcripts was performed with primers Pan clI (F) located in exon2 and Pan clI (R) located in exon 4. HLA-F-specific transcripts were revealed by hybridization with the F.R-specific probe located in exon 2. The JEG-3 choriocarcinoma cell line was used as a control for high HLA-G transcription. Absence of contaminant DNA was controlled by concomitant amplification of the PCR mixture without a template (H2O). PCR products coamplified in the same reaction by β-actin primers were detected on the same membrane by a β-actin probe
Stress-induced HSF1 binding to HSEs in the promoter of HLA-G and hsp70 genes are detected in M8 melanoma cells
A computer search for heat shock consensus elements within the HLA-G promoter sequence identified a sequence positioned 480-bp upstream of the translation initiation codon of the HLA-G gene as a putative target for HSF binding (Fig 3). We thus tested the eventuality that the increased HLA-G mRNA levels described in stressed M8 cells could be induced through this putative HSE, designated HSEG. DNA-binding activity to the HSEG was evaluated by electrophoretic mobility shift assays (EMSAs) using nuclear extracts from heat shocked or arsenite treated M8 cells after various recovery times at 37°C. Specificity of binding was evaluated using wild-type or mutated HSEs derived from hsp70 and HLA-G gene promoters as cold competitor in a 100-fold excess relative to the labeled probe (Fig 3).
In a first experiment, an oligonucleotide, corresponding to the 2 overlapping HSEs located between positions -119 and -87 in the human hsp70 promoter and called HSEHSP, was used as a probe to assay binding of factors found in M8 nuclear extracts. A specific HSE-binding activity was induced immediately on a heat shock or an arsenite treatment and disappeared 6 h after recovery at 37°C (Fig 4A, upper panel; data not shown for samples corresponding to the recovery from the arsenite treatment). Formation of the stress-induced DNA complex was specifically competed by a 100-fold excess of unlabeled HSEHSP but not by an irrelevant competitor (IR), demonstrating the sequence specificity of the DNA-protein interactions. Moreover, an unlabeled excess of HSEG but not mutated MUTHSEG oligonucleotides could compete for the formation of this complex. This indicates that the same protein complex, which is able to interact with HSEHSP, could recognize an HLA-G promoter-derived sequence and that this interaction occurs through an HSE motif present in HSEG and altered by a 3-bp mutation in MUTHSEG (Fig 3). By using HSEG as the radiolabeled probe (Fig 4A, lower panel), we confirmed that a specific and transient DNA-binding activity that involves an HSE motif, found in the HLA-G promoter, is induced in nuclear extracts from heat shock or arsenite treated M8 cells. Kinetics of activation of heat shock or arsenite induced DNA-binding activity to both HSEHSP and HSEG elements and exhibit similar patterns using EMSA analysis (Fig 4A; data not shown).
Fig 4.
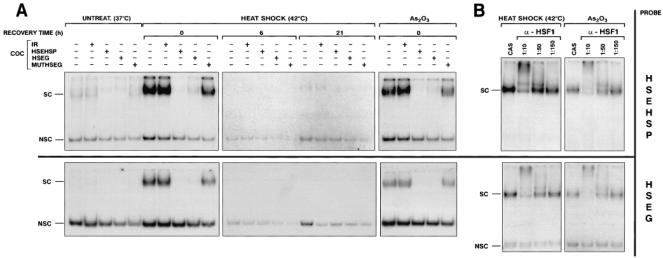
Heat shock induces binding of HSF1 to HLA-G–and hsp70-derived HSEs. (A) EMSA analysis of the kinetic of stress-induced HSE-binding activities using 32P-labeled HSEHSP (upper panel) or HSEG probes (lower panel) in nuclear extracts prepared from either untreated or heat-shocked M8 cells (42°C, 2h) at various recovery time at 37°C or arsenite-treated cells (100 μM As2O3, 2 h) in absence or presence of a 100-fold excess of cold oligonucleotide competitors (COC). (B) nuclear extracts, prepared from stressed cells immediately after the end of stress treatment, were incubated either alone or in presence of various dilutions of anti-HSF1 (α-HSF1) serum or in presence a nonspecific rabbit polyclonal control antiserum (CAS) for 15 min at room temperature before addition of the binding solution. SC and NSC denote specific and nonspecific DNA-protein complexes, respectively
In order to further characterize the presence of HSF1 in protein complexes that interact with HSEG and HSEHSP, EMSA, were performed by preincubating the nuclear extracts from heat-shocked and arsenite-treated M8 cells with a rabbit polyclonal serum that specifically recognizes HSF1 (α-HSF1). When we used HSEHSP as a probe (Fig 4B, upper panel), we observed a supershift of specific thermo-induced HSE complexes with α-HSF1, which decreased with dilution of the serum. Conversely, we did not observe any supershift with a control rabbit polyclonal serum (CAS), suggesting that HSF1 was present in the specific stress-induced HSE complex. An identical pattern was observed when HSEG was used as a probe (Fig 4B, lower panel). We thus conclude that HSF1, present in nuclear extracts prepared from heat-shocked or arsenite-treated M8 cells, can bind in vitro on an HLA-G promoter-derived sequence containing an HSE.
DISCUSSION
The differential constitutive and inducible levels of HLA-G transcripts found in various cell types suggest that this gene is subjected to complex regulation, involving transcriptional control mechanisms that influence the tissue-restricted pattern of expression of HLA-G proteins. Sequence comparison of HLA-G and classical HLA-class I gene promoters reveals that regulatory sequences that have been implicated in the control of MHC class I transcriptional activation, mainly the enhancer A palindrome, the interferon-stimulated response element (ISRE), the site α, and the enhancer B are disrupted within the HLA-G promoter, which may in part explain why low levels of HLA-G transcript are detected in most cell types (Boucraut et al 1993; Gobin et al 1998). A 250-bp positive regulatory region located 1.1 kb upstream of the HLA-G translation start site has been shown to target activation of transgene transcription in mice placenta (Schmidt et al 1993). Moreover, differential profiles of DNA-binding complexes interacting with this regulatory region were detected in HLA-G–positive or HLA-G–negative cells, suggesting that regulatory mechanisms are involved in repressing HLA-G in some cell types (Moreau et al 1998; Lefebvre et al 1999). We thus cannot exclude that tissue-specific activation of HLA-G gene occurs through release of inhibitory mechanisms that repress promoter activity in most cell types. Recently, it was proposed that an interferon-γ–activated site (GAS), located 740 bp upstream of the translation start site of HLA-G, could control induction of steady-state levels of HLA-G mRNA by IFNs (Yang et al 1996; Chu et al 1999). Despite these preliminary approaches, the factors and the sequences that determine the tissue or tumor-specific activation of HLA-G gene expression are unknown.
HLA-G gene expression may be activated by various factors, such as growth factors, cytokines, or hormones whose presence is associated with a particular physiological state such as pregnancy or that are found in the microenvironment of HLA-G–positive tissues such as thymus. Specific detection of high levels of HLA-G transcripts in melanoma biopsies also suggests that tumor microenvironment may favor activation of HLA-G transcript levels during malignant transformation (Paul et al 1999). Heat shock treatment modulates expression of proteins that will allow cytoprotection, and stress-induced proteins have also been implicated in balancing immune responses during the course of various diseases (Morimoto and Santoro 1998). This prompted us to evaluate the effect of stress on expression of the HLA-G gene, which encodes an antigen sharing many features that fit in the regulatory scheme of stress-induced immune regulation.
Our results demonstrate that heat shock or arsenite treatment induce an increase in the levels of HLA-G transcript both in human melanoma and in glioblastoma cell lines, initially characterized as negative for HLA-G gene expression. Levels of all known alternatively spliced HLA-G transcripts are highly and transiently up-regulated between 3 h and 24 h after the cells have recovered from the stress treatment. We report differential kinetics in the induction of HLA-G alternative transcripts since the transcript encoding the soluble isoform HLA-G6 is induced prior to the other HLA-G transcripts, during the early recovery time after stress. Moreover, the lack of accumulation of HLA-G transcripts in the presence of actinomycin D during the stress period supports a transcriptional mechanism for control of HLA-G gene expression.
Unusual expression and localization of HSPs have been reported in human tumor cells, particularly in melanoma cells (Ferrarini et al 1992; Dressel et al 1998), probably conferring an adaptive advantage to these cells in resisting stress conditions. Severe heat shock treatment (45°C for mammalian cells) is known to transiently inhibit pre-mRNA splicing, and it was suggested that HSPs themselves help to protect the splicing machinery during the stress conditions (Yost et al 1990). Treating M8 cells with a severe heat shock induced only a partial inhibition of HLA-G mRNA processing (data not shown), probably reflecting the constitutive protection of the splicing machinery by HSPs. Alteration of HLA-G alternative splicing has already been shown in melanoma biopsies. Differential levels of alternative transcripts encoding a soluble protein (HLA-G5) and the other HLA-G transcripts have been described (Paul et al 1999), suggesting that splicing of HLA-G pre-mRNA may be selectively regulated in melanoma cells. Altogether, these results suggest that control of alternative splicing after stress treatment in tumor cells may represent a way to refine selective expression of a subset of isoforms whose specific functions remain to be defined.
Considering the tissue-restricted localization and the immunotolerant function assigned to date to the HLA-G gene, we were interested in analyzing the effect of stress conditions on several highly homologous members of the HLA class I gene family. Our results show that stress specifically induces the HLA-G gene expression since none of the HLA-A, -B, -E, or -F class I genes transcripts appeared to be affected by stress treatment. One of the hallmarks of the MHC is its high concentration of genes with immunological functions, and recently, a group of genes contiguous to the class I region, involved in stress, inflammation, or infection, was designated as the class IV region (Gruen and Weissman 1997). This region includes hsp70 genes, the TNF family cluster, and MIC genes. Highly divergent human class I–related genes, MICA and MICB, have also been shown to function as stress inducible genes and are thought to play an essential role in immune surveillance of stressed or infected intestinal epithelial cells (Groh et al 1996, 1998). These results strengthen the notion that despite overall conservation of HLA class I coding sequences, specific regulatory mechanisms have been selected to target selective stress-induced activation of a subset of nonclassical class I genes that may allow fine-tuning of recognition of the stressed cells. In humans, exposure to environmental trauma such as heat shock or arsenite treatment, as used in this study, is directly or indirectly sensed by HSF1. Immediately after stress stimulus, pre-existing HSF1 trimerize, translocate to the nucleus, bind to contiguous alternative repeats of the pentanucleotide NGAAN (HSE), and are hyperphosphorylated. Through HSEs, present in all heat shock genes, the HSF family regulates induction of heat shock gene expression at the transcriptional level in response to a plethora of stress signals and also to nonstress conditions, including cell growth, development, and pathophysiological states. The presence of HSEs in genes of the MHC class IV region, which do not belong to the HSPs family, suggest that regulatory mechanisms have been conserved through evolution to control selective expression of immune response genes encoded in the MHC in response to stress. MICA and MICB genes have been shown to bear an HSE in their promoter (Groh et al 1996). An HSE consisting of 2 inverted repeats of the NGAAN motif-oriented face to face on the DNA helix was also identified within the 5′ promoter region of the HLA-G gene, 480 bp upstream of the translation start site. EMSA analysis with nuclear extracts prepared from heat-shocked and arsenite-treated M8 cells confirmed that the HSE derived from the HLA-G gene promoter was transiently recognized by HSF1, as were the hsp70 promoter-derived HSEs.
Multhoff and co-workers have reported that cell surface expression of hsp70 is selectively inducible by nonlethal heat on human tumor cell lines, resulting in an increased sensitivity to lysis mediated by natural killer cells (Multhoff et al 1997; Botzler et al 1999). On the other hand, it has recently been demonstrated that HLA-G protein could down-modulate cytotoxic activity of NK and T cells, both directly through interaction with killer cell immunoglobulin-like receptors (KIRs) or indirectly by providing a leader peptide to allowing another nonclassical class antigen HLA-E to be expressed at the cell surface and inhibit NK cell activity through interaction with CD94/NKG2A receptors (Lanier 1999). The presence of HLA-G receptors at the surface of myelomonocytic cells and B lymphocytes also suggests that HLA-G may have a immunomodulatory role on the function of these cells. In this context, stress-induced HLA-G expression on tumor cells or viral infected cells, resulting from a high temperature and a plethora of factors released in their microenvironment, could provide these cells with an additional mechanism to escape immunosurveillance.
In summary, we report here a new feature of HLA-G as a stress-inducible gene, in addition to being involved in down-modulation of the aggressiveness of the human immune system and in the establishment of tumor escape mechanisms.
Acknowledgments
We thank F. Adrian-Cabestre, S. Lefebvre, and P. Moreau for their daily support and helpful comments and M. O'Brien for rereading the manuscript. We thank J-Y. Thuret for critically reviewing the manuscript. We thank L. Sistonen (Turku Centre for Biotechnology, Turku, Finland) for kindly providing HSF1 antibody. We express our gratitude to C. Gazin (INSERM U462, Paris) for his constant support throughout this study. We also thank C. Massard (Centre d'Etude du Polymorphisme Humain, Paris) for sequencing. This work was supported by grants from the French Commissariat à l'Energie Atomique, the Association pour la Recherche sur le Cancer and the Etablissement Français des Greffes. E.I. is a recipient of a grant from the Ministère de l'Education Nationale de la Recherche et de la Technologie.
REFERENCES
- Allan DS, Colonna M, Lanier LL, Churakova TD, Abrams JS, Ellis SA, McMichael AJ, Braud VM. Tetrameric complexes of human histocompatibility leukocyte antigen (HLA-G) bind to peripheral blood myelomonocytic cells. J Exp Med. 1999;189:1149–1156. doi: 10.1084/jem.189.7.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiot L, Onno M, Drénou B, Monvoisin C, Fauchet R. HLA-G class I gene expression in normal and malignant hematopoietic cells. Hum Immunol. 1998;59:524–528. doi: 10.1016/s0198-8859(98)00041-x. [DOI] [PubMed] [Google Scholar]
- Amiot L, Onno M, Renard I, Drénou B, Guillaudeux T, Le Bouteiller P, Fauchet R. HLA-G transcription studies during the different stages of normal and malignant hematopoiesis. Tissue Antigens. 1996;47:408–413. doi: 10.1111/j.1399-0039.1996.tb02576.x. [DOI] [PubMed] [Google Scholar]
- Blaschitz A, Lenfant F, Mallet V, Hartmann M, Bensussan A, Geraghty DE, Le Bouteiller P, Dohr G. Endothelial cells in chorionic fetal vessels of first trimester placenta express HLA-G. Eur J Immunol. 1997;27:3380–3388. doi: 10.1002/eji.1830271237. [DOI] [PubMed] [Google Scholar]
- Botzler C, Ellwart J, Günther W, Eissner G, Multhoff G. Synergistic effects of heat and ET-18-OCH3 on membrane expression of hsp70 and lysis of leukemic K562 cells. Exp Haematol. 1999;27:470–478. doi: 10.1016/s0301-472x(98)00055-1. [DOI] [PubMed] [Google Scholar]
- Boucraut J, Hawley S, Robertson K, Bernard D, Loke YW, Le Bouteiller P. Differential nuclear expression of enhancer A DNA-binding proteins in human first trimester trophoblast cells. J Immunol. 1993;150:3882–3894. [PubMed] [Google Scholar]
- Brown SA, Kingston RE. Disruption of downstream chromatin directed by a transcriptional activator. Genes Dev. 1997;11:3116–3121. doi: 10.1101/gad.11.23.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantoni C, Verdiani S, Falco M, et al. p49, a putative HLA class I-specific inhibitory NK receptor belonging to the immunoglobulin superfamily. Eur J Immunol. 1998;28:1980–1990. doi: 10.1002/(SICI)1521-4141(199806)28:06<1980::AID-IMMU1980>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Carosella ED, Dausset J, Kirszenbaum M. HLA-G revisited. Immunol Today. 1996;17:407–409. doi: 10.1016/0167-5699(96)30055-8. [DOI] [PubMed] [Google Scholar]
- Chimini G, Pontarotti P, Nguyen C, Toubert A, Boretto J, Jordan BR. The chromosome region containing the highly polymorphic HLA class I genes displays limited large scale variability in the human population. EMBO J. 1988;7:395–400. doi: 10.1002/j.1460-2075.1988.tb02826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu W, Yang Y, Geraghty DE, Hunt JS. Interferons enhance HLA-G mRNA and protein in transfected mouse fibroblasts. J Reprod Immunol. 1999;42:1–15. doi: 10.1016/s0165-0378(98)00077-1. [DOI] [PubMed] [Google Scholar]
- Colonna M, Navarro F, Bellon T, et al. A common inhibitory receptor for major histocompatibility complex class I molecules on human lymphoid and myelomonocytic cells. J Exp Med. 1997;186:1809–1818. doi: 10.1084/jem.186.11.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M, Samaridis J, Cella M, et al. Human myelomonocytic cells express an inhibitory receptor for classical and nonclassical MHC class I molecules. J Immunol. 1998;160:3096–3100. [PubMed] [Google Scholar]
- Coppin HL, Denny DW, Weissman SM, McDewitt HD. HLA-B locus polymorphism: studies with a specific hybridization probe. Proc Natl Acad Sci USA. 1985;82:8614–8618. doi: 10.1073/pnas.82.24.8614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotto JJ, Kline M, Morimoto RI. Activation of heat shock factor 1 DNA binding precedes stress-induced serine phosphorylation. J Biol Chem. 1996;271:3355–3358. doi: 10.1074/jbc.271.7.3355. [DOI] [PubMed] [Google Scholar]
- Crisa L, McMaster MT, Ishii JK, Fisher SJ, Salomon DR. Identification of thymic epithelial cell subset sharing expression of the class Ib HLA-G molecule with fetal trophoblasts. J Exp Med. 1997;186:289–298. doi: 10.1084/jem.186.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl M, Munz C, Keilholz W, Stevanovic S, Holmes N, Loke YW, Rammensee HG. Nonclassical HLA-G molecules are classical peptide presenters. Curr Biol. 1996;6:305–314. doi: 10.1016/s0960-9822(02)00481-5. [DOI] [PubMed] [Google Scholar]
- Dressel R, Johnson JP, Günther E. Heterogeneous patterns of constitutive and heat shock induced expression of HLA-linked HSP70-1 and HSP70-2 heat shock genes in human melanoma cell lines. Melanoma Res. 1998;8:482–492. doi: 10.1097/00008390-199812000-00002. [DOI] [PubMed] [Google Scholar]
- Ferrarini M, Heltai S, Zocchi MR, Rugarli C. Unusual expression and localization of heat-shock proteins in human tumor cells. Int J Cancer. 1992;51:613–619. doi: 10.1002/ijc.2910510418. [DOI] [PubMed] [Google Scholar]
- Fiorenza MT, Farkas T, Dissing M, Kolding D, Zimarino V. Complex expression of murine heat shock transcription factors. Nucleic Acids Res. 1995;23:467–474. doi: 10.1093/nar/23.3.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii T, Ishitani A, Geraghty DE. A soluble form of the HLA-G antigen is encoded by a messenger ribonucleic acid containing intron 4. J Immunol. 1994;153:5516–5524. [PubMed] [Google Scholar]
- Fukushima Y, Oshika Y, Nakamura M, et al. Increased expression of human histocompatibility leukocyte antigen-G in colorectal cancer cells. Int J Mol Med. 1998;2:349–351. doi: 10.3892/ijmm.2.3.349. [DOI] [PubMed] [Google Scholar]
- Geraghty DE, Koller BH, Orr HT. A human major histocompatibility complex class I gene that encodes a protein with a shortened cytoplasmic segment. Proc Natl Acad Sci USA. 1987;84:9145–9149. doi: 10.1073/pnas.84.24.9145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobin SJP, Keijsers V, Van Zutphen M, Van den Elsen PJ. The role of enhancer A in the locus-specific transactivation of classical and nonclassical HLA class I genes by nuclear factor κB. J Immunol. 1998;161:2276–2283. [PubMed] [Google Scholar]
- Goodson ML, Park-Sarge O-K, Sarge KD. Tissue-dependent expression of heat shock factor 2 isoforms with distinct transcriptional activities. Mol Cell Biol. 1995;15:5288–5293. doi: 10.1128/mcb.15.10.5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodson ML, Sarge KD. Regulated expression of heat shock factor 1 isoforms with distinct leucine zipper arrays via tissue-dependent alternative splicings. Biochem Biophys Res Commun. 1995;211:943–949. doi: 10.1006/bbrc.1995.1903. [DOI] [PubMed] [Google Scholar]
- Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc Natl Acad Sci USA. 1996;93:12445–12450. doi: 10.1073/pnas.93.22.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh V, Steinle A, Bauer S, Spies T. Recognition of stress-induced MHC molecules by intestinal epithelial γδT cells. Science. 1998;279:1737–1740. doi: 10.1126/science.279.5357.1737. [DOI] [PubMed] [Google Scholar]
- Gruen JR, Weissman SM. Evolving views of the major histocompatibility complex. Blood. 1997;90:4252–4265. [PubMed] [Google Scholar]
- Horuzsko A, Antoniou J, Tomlinson P, Portik-Dobos V, Mellor AL. HLA-G functions as a restriction element and a transplantation antigen in mice. Int Immunol. 1997;9:645–653. doi: 10.1093/intimm/9.5.645. [DOI] [PubMed] [Google Scholar]
- Houlihan JM, Biro PA, Fergar-Payne A, Simpson KL, Holmes CH. Evidence for the expression of non-HLA-A,-B,C class I genes in the human fetal liver. J Immunol. 1992;149:668–675. [PubMed] [Google Scholar]
- Ishitani A, Geraghty DE. Alternative splicing of HLA-G transcripts yields proteins with primary structures ressembling both class I and class II antigens. Proc Natl Acad Sci USA. 1992;89:3947–3951. doi: 10.1073/pnas.89.9.3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirszenbaum M, Moreau P, Gluckman E, Dausset J, Carosella ED. An alternatively spliced form of HLA-G mRNA in human trophoblasts and evidence for the presence of HLA-G transcript in adult lymphocytes. Proc Natl Acad Sci USA. 1994;91:4209–4213. doi: 10.1073/pnas.91.10.4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovats S, Main EK, Librach C, Stubblebine M, Fisher SJ, DeMars R. A class I antigen, HLA-G, expressed in human trophoblasts. Science. 1990;248:220–223. doi: 10.1126/science.2326636. [DOI] [PubMed] [Google Scholar]
- Lanier LL. Natural killer cells fertile with receptors for HLA-G? Proc Natl Acad Sci USA. 1999;96:5343–5345. doi: 10.1073/pnas.96.10.5343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bouteiller P. HLA class I chromosomal region, genes, and products: facts and questions. Crit Rev Immunol. 1994;14:89–129. doi: 10.1615/critrevimmunol.v14.i2.10. [DOI] [PubMed] [Google Scholar]
- Le Bouteiller P, Blaschitz A. The functionality of HLA-G is emerging. Immunol Rev. 1999;167:233–244. doi: 10.1111/j.1600-065x.1999.tb01396.x. [DOI] [PubMed] [Google Scholar]
- Lee N, Malacko AR, Ishitani A, Chen M-C, Bajorath J, Marquardt H, Geraghty DE. The membrane-bound and soluble forms of HLA-G bind identical sets of endogenous peptides but differ with respect to TAP association. Immunity. 1995;3:591–600. doi: 10.1016/1074-7613(95)90130-2. [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Moreau P, Guiard V, et al. Molecular mechanisms controlling constitutive and IFN-γ-inducible HLA-G expression in various cell types. J Reprod Immunol. 1999;43:213–224. doi: 10.1016/s0165-0378(99)00035-2. [DOI] [PubMed] [Google Scholar]
- Leppä S, Sistonen L. Heat shock response—pathophysiological implications. Ann Med. 1997;29:73–78. doi: 10.3109/07853899708998745. [DOI] [PubMed] [Google Scholar]
- Mason PB, Lis JT. Cooperative and competitive protein interaction at the hsp70 promoter. J Biol Chem. 1997;26:33227–33233. doi: 10.1074/jbc.272.52.33227. [DOI] [PubMed] [Google Scholar]
- Mathew A, Mathur SK, Morimoto RI. Heat shock response and protein degradation: regulation of HSF2 by the ubiquitin-proteasome pathway. Mol Cell Biol. 1998;18:5091–5098. doi: 10.1128/mcb.18.9.5091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMaster M, Zhou Y, Shorter S, Kapasi K, Geraghty D, Lim K-H, Fisher S. HLA-G isoforms produced by placental cytotrophoblasts and found in amniotic fluid are due to unusual glycosylation. J Immunol. 1998;160:5922–5928. [PubMed] [Google Scholar]
- Ménoret A, Chandawarkar R. Heat-shock protein-based anticancer immunotherapy: an idea whose time has come. Semin Oncol. 1998;25:654–660. [PubMed] [Google Scholar]
- Moreau P, Adrian-Cabestre F, Menier C, Guiard V, Gourand L, Dausset J, Carosella ED, Paul P. IL-10 selectively induces HLA-G expression in human trophoblasts and monocytes. Int Immunol. 1999;11:803–811. doi: 10.1093/intimm/11.5.803. [DOI] [PubMed] [Google Scholar]
- Moreau P, Carosella ED, Gluckman E, Gourand L, Prost S, Dausset S, Kirszenbaum M. Transcrits différentiels du gène du CMH de classe I non classique HLA-G dans le trophoblaste de 1ertrimestre de gestation et le placenta à terme. C R Acad Sci Paris. 1995a;318:837–842. [PubMed] [Google Scholar]
- Moreau P, Carosella E, Teyssier M, Prost S, Gluckman E, Dausset J, Kirszenbaum M. Soluble HLA-G molecule an alternatively spliced HLA-G mRNA form candidate to encode it in peripheral blood mononuclear cells and human trophoblasts. Hum Immunol. 1995b;43:231–236. doi: 10.1016/0198-8859(95)00009-s. [DOI] [PubMed] [Google Scholar]
- Moreau P, Lefebvre S, Gourand L, Dausset J, Carosella ED, Paul P. Specific binding of nuclear factors to the HLA-G gene promoter correlates with a lack of HLA-G transcripts in first trimester human fetal liver. Hum Immunol. 1998;59:751–757. doi: 10.1016/s0198-8859(98)00081-0. [DOI] [PubMed] [Google Scholar]
- Morimoto RI. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998;12:3788–3796. doi: 10.1101/gad.12.24.3788. [DOI] [PubMed] [Google Scholar]
- Morimoto RI, Santoro MG. Stress-inducible responses and heat shock proteins: new pharmacologic targets for cytoprotection. Nature Biotech. 1998;16:833–838. doi: 10.1038/nbt0998-833. [DOI] [PubMed] [Google Scholar]
- Multhoff G, Botzler L, Jennen L, Schmidt J, Ellwart J, Issels R. Heat shock protein 72 on tumor cells. J Immunol. 1997;158:4341–4350. [PubMed] [Google Scholar]
- Nakai A, Tanabe M, Kawazoe Y, Inazawa J, Morimoto RI, Nagata K. HSF 4, a new member of the human heat shock factor family which lacks properties of a transcriptional activator. Mol Cell Biol. 1997;17:469–481. doi: 10.1128/mcb.17.1.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onno M, Guillaudeux T, Amiot L, et al. The HLA-G gene is expressed at a low mRNA level in different human cells and tissues. Human Immunol. 1994;41:79–86. doi: 10.1016/0198-8859(94)90089-2. [DOI] [PubMed] [Google Scholar]
- Paul P, Adrian-Cabestre F, Le Gal FA, et al. Heterogeneity of HLA-G gene transcription and protein expression in malignant melanoma biopsies. Cancer Res. 1999;59:1954–1960. [PubMed] [Google Scholar]
- Paul P, Rouas-Freiss N, Khalil-Daher I, et al. HLA-G expression in melanoma: a way for tumor cells to escape from immunosurveillance. Proc Natl Acad Sci USA. 1998;95:4510–4515. doi: 10.1073/pnas.95.8.4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perisic O, Xiao H, Lis JT. Stable binding of Drosophila heat shock factor to head-to-head and tail-to-tail repeats of a conserved 5 bp recognition unit. Cell. 1989;59:797–806. doi: 10.1016/0092-8674(89)90603-x. [DOI] [PubMed] [Google Scholar]
- Ponte M, Cantoni C, Biassoni R, et al. Inhibitory receptors sensing HLA-G1 molecules in pregnancy: decidua-associated natural killer cells express LIR-1 and CD94/NKG2A and acquire p49, an HLA-G1-specific receptor. Proc Natl Acad Sci USA. 1999;96:5674–5679. doi: 10.1073/pnas.96.10.5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabindran SK, Giorgi G, Clos J, Wu C. Molecular cloning and expression of a human heat shock factor, HSF1. Proc Natl Acad Sci USA. 1991;88:6906–6910. doi: 10.1073/pnas.88.16.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan S, Long EO. A human histocompatibility leukocyte antigen (HLA)-G-specific receptor expressed on all natural killer cells. J Exp Med. 1999;189:1093–1100. doi: 10.1084/jem.189.7.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouas-Freiss N, Marchal-Bras Gonτalves R, Menier C, Dausset J, Carosella ED. Direct evidence to support the role of HLA-G in protecting the fetus from maternal uterine natural killer cytolysis. Proc Natl Acad Sci USA. 1997;94:11520–11525. doi: 10.1073/pnas.94.21.11520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandaltzopoulos R, Becker PB. Heat shock factor increases the reinitiation rate from potentiated chromatin templates. Mol Cell Biol. 1998;18:361–367. doi: 10.1128/mcb.18.1.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SK, Giblin PA, Kavathas P. Cell-cell adhesion mediated by CD8 and human histocompatibility leukocyte antigen G, a nonclassical major histocompatibility complex class 1 molecule on cytotrophoblasts. J Exp Med. 1991;174:737–740. doi: 10.1084/jem.174.3.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild H, Arnold-Schild D, Lammert E, Rammensee H-G. Stress proteins and immunity mediated by cytotoxic T lymphocytes. Curr Opin Immunol. 1999;11:109–113. doi: 10.1016/s0952-7915(99)80019-3. [DOI] [PubMed] [Google Scholar]
- Schmidt CM, Ehlenfeldt RG, Athanasiou MC, Duvick LA, Heinrichs H, David CS, Orr HT. Extraembryonic expression of the human MHC class I gene HLA-G in transgenic mice. J Immunol. 1993;5:2633–2645. [PubMed] [Google Scholar]
- Schmidt CM, Garrett E, Orr HT. Cytotoxic T lymphocyte recognition of HLA-G in mice. Human Immunol. 1997;55:127–139. doi: 10.1016/s0198-8859(97)00097-9. [DOI] [PubMed] [Google Scholar]
- Schuetz TJ, Gallo GJ, Sheldon L, Tempst P, Kingston RE. Isolation of a cDNA for HSF2: evidence for two heat shock factor genes in humans. Proc Natl Acad Sci USA. 1991;88:6911–6915. doi: 10.1073/pnas.88.16.6911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava PK, Ménoret A, Basu S, Binder RJ, McQuade KL. Heat shock proteins come of age: primitive functions acquire new roles in an adaptative world. Immunity. 1998;8:657–665. doi: 10.1016/s1074-7613(00)80570-1. [DOI] [PubMed] [Google Scholar]
- Tanabe M, Sasai N, Nagata K, Liu X-D, Liu PCC, Thiele DJ, Nakai A. The mammalian HSF4 gene generates both an activator and a repressor of heat shock genes by alternative splicing. J Biol Chem. 1999;274:27845–27856. doi: 10.1074/jbc.274.39.27845. [DOI] [PubMed] [Google Scholar]
- Teyssier M, Bensussan A, Kirszenbaum M, Moreau P, Gluckman E, Dausset J, Carosella E. Natural Killer cells are the unique lymphocyte cell subset which do not express HLA-G. Nat Immun. 1995;14:262–270. [PubMed] [Google Scholar]
- Ulbrecht M, Rehberger B, Strobel I, Messer G, Kind P, Degitz K, Bieber T, Weiss EH. HLA-G: expression in human keratinocytes in vitro and in human skin in vivo. Eur J Immunol. 1994;24:176–180. doi: 10.1002/eji.1830240127. [DOI] [PubMed] [Google Scholar]
- van Eden W, Van der Zee R, Paul AGA, Prakken BJ, Wendling U, Anderton SM, Wauben MHM. Do heat shock proteins control the balance of T-cell regulation in inflammatory diseases? Immunol Today. 1998;19:303–307. doi: 10.1016/s0167-5699(98)01283-3. [DOI] [PubMed] [Google Scholar]
- Yang Y, Chu W, Geraghty DE, Hunt JS. Expression of HLA-G in human mononuclear phagocytes and selective induction by IFN-γ. J Immunol. 1996;156:4224–4231. [PubMed] [Google Scholar]
- Yost HJ, Petersen RB, Lindquist S. RNA metabolism: strategies for regulation in the heat shock response. Trend Genet. 1990;6:223–227. doi: 10.1016/0168-9525(90)90183-7. [DOI] [PubMed] [Google Scholar]