

Abstract
In concert with the stress-induced activation of human heat shock factor 1 (HSF1), the factor becomes inducibly phosphorylated and accumulates into nuclear granules. To date, these processes are not fully understood. Here, we show that although stress caused by the proteasome inhibitors MG132 and clasto-lactacystine β-lactone induces the expression of Hsp70, the formation of HSF1 granules is affected differently in comparison to heat shock. Furthermore, proteasome inhibition increases serine phosphorylation on HSF1, but to a lesser extent than heat stress. Our results suggest that, depending on the type of stress stimulus, the multiple events associated with HSF1 activation might be affected differently.
INTRODUCTION
Activation of heat shock factor 1 (HSF1) is a multistep process, involving nuclear localization, trimerization, acquisition of DNA-binding activity, inducible phosphorylation, and obtaining of transcriptional activity. Coincident with the stress-induced activation, HSF1 relocalizes within the nucleus to form granules in human cells. Whereas the regulation of trimerization and acquisition of DNA-binding activity of HSF1 is well characterized, much less is known about the inducible phosphorylation and the formation of nuclear granules of HSF1 (for reviews, see Wu 1995; Morimoto 1998).
HSF1 is phosphorylated under non-stressful conditions (Larson et al 1988; Sorger and Pelham 1988; Sorger 1990; Sarge et al 1993; Cotto et al 1996; Fritsch and Wu 1999). Some of these constitutive phosphorylation sites have been mapped to serines 303 and 307, both of which are phosphorylated by glycogen synthase kinase 3β (GSK-3β) and the mitogen-activated protein kinase (MAPK), respectively (Chu et al 1996; Knauf et al 1996; Kline and Morimoto 1997). Recently, serine 363 was also reported to be phosphorylated under nonstressful conditions (Chu et al 1998). The role of the constitutive phosphorylation of HSF1 is to repress the transcriptional activity of HSF1 under nonstressful conditions and perhaps during recovery from stress. On stress-induced activation, HSF1 undergoes inducible phosphorylation, called hyperphosphorylation (Larson et al 1988; Sorger and Pelham 1988; Sorger 1990; Sarge et al 1993), that occurs at serine residues (Chu et al 1996; Cotto et al 1996; Kline and Morimoto 1997). However, the specific localization and number of inducible phosphorylation sites are unknown, as is the protein kinase(s)/phosphatase(s) responsible for HSF1 hyperphosphorylation. Several reports suggest that hyperphosphorylation of HSF1 is not required for DNA-binding activity but might be essential for transcriptional activation in mammalian cells (Sorger and Pelham 1988; Jurivich et al 1994, 1995; Lee et al 1995; Cotto et al 1996; Liu and Thiele 1996). Hence, regulation of HSF1 via phosphorylation appears to be very complex.
The appearance of discrete nuclear structures called HSF1 granules was first described in response to heat stress in HeLa cells (Sarge et al 1993). In addition to heat shock, stress caused by cadmium and azetidine has been shown to induce these brightly staining irregularly shaped HSF1 granules (Cotto et al 1997; Jolly et al 1999). The HSF1 granules have been found in fixed human primary and transformed cell lines as well as in living human cells (Cotto et al 1997; Jolly et al 1999). To date, no HSF1 granules have been detected in mouse or hamster cells (Sarge et al 1993; Mivechi et al 1994). The appearance of HSF1 granules correlates with the acquisition of DNA-binding activity, hyperphosphorylation, and transcriptional competence of HSF1 (Sarge et al 1993; Cotto et al 1997; Jolly et al 1999). During attenuation or recovery of the heat shock response, the HSF1 granules disappear and HSF1 redistributes diffusely in the cytoplasma and nucleus, resembling the localization found in unstressed cells (Sarge et al 1993; Cotto et al 1997; Jolly et al 1999). In addition, transient overexpression of GSK-3β facilitates the disappearance of HSF1 granules and reduces hsp70 transcription from a reporter construct after heat shock (He et al 1998). Another study, which shows that sodium salicylate does not induce HSF1 granules despite induction of HSF1 DNA-binding activity, supports the hypothesis that appearance of HSF1 granules is coupled to transcriptionally active HSF1 (Cotto et al 1997). However, HSF1 granules do not colocalize with active sites of hsp70 or hsp90 transcription (Jolly et al 1997), and HSF1 granules are also present in heat-shocked mitotic cells that are transcriptionally inactive (Martínez-Balbás et al 1995; Jolly et al 1999). The HSF1 granules do not coincide with other previously described nuclear compartments, such as nucleoli, coiled bodies, kinetochores, promyelocytic leukemia bodies, or the speckles enriched in splicling factors (Cotto et al 1997). Recently, heat stress was shown to cause recruitment of a novel heterogeneous nuclear ribonucleoprotein HAP to nuclear granules, which with delayed kinetics colocalizes with the HSF1 granules (Weighardt et al 1999). Since HAP does not appear to directly interact with HSF1, Weighardt and coworkers (1999) suggest the existence of an underlying nuclear structure that would serve as a recruiter for both proteins. In agreement, HSF1 granules have been shown to reform at the same nuclear positions on re-exposure to heat shock, and the number of HSF1 granules has been suggested to correlate with the ploidy of the cells, supporting the existence of chromosomal targets for HSF1 granules (Jolly et al 1997, 1999). To date, the functional relevance of HSF1 granules is an open question.
Elevated amounts of abnormal proteins have been suggested to trigger the activation of the stress response (Ananthan et al 1986; for review, see Morimoto 1998). Accumulation of misfolded proteins is caused by conditions such as heat shock or inhibition of proteasome-mediated protein degradation. Recently, selective inhibitors of the 26S proteasome have been identified, offering new tools for studying the involvement of the ubiquitin-proteasome pathway in various cellular processes. The natural products lactacystin and its derivative clasto-lactacystine β-lactone or the synthetic peptide aldehyde MG132 specifically inhibit the 26S proteasome, hence preventing protein degradation by the ubiquitin-proteasome pathway (for review, see Lee and Goldberg 1998a). These proteasome inhibitors have been shown to induce expression of heat shock proteins, for example, Hsp70 (Zhou et al 1996; Bush et al 1997; Lee and Goldberg 1998b; Meriin et al 1998). Of the multiple HSFs, HSF1, HSF2, and HSF3 have been reported to be affected by inhibitors of the proteasome (Zhou et al 1996; Kawazoe et al 1998; Mathew et al 1998; Kim et al 1999; Pirkkala et al 2000).
In this study, we wanted to use proteasome inhibitors as tools to examine the less well characterized processes occurring on HSF1 activation, specifically, the appearance of nuclear HSF1 granules and the phosphorylation state of HSF1. Our results show that although treatment with MG132 and clasto-lactacystine β-lactone induces appearance of HSF1 granules, these granules differ from the heat-induced HSF1 granules in a cell-type–specific manner. Furthermore, our study reveals that MG132 increases serine phosphorylation of HSF1, although the inducible phosphorylation of HSF1 is not as prominent on MG132 treatment as on heat shock as measured by in vivo 32P-labeling of HSF1. Taken together, our results show that the steps involved in activation of HSF1 are affected differently, depending on the type of stress stimulus.
MATERIALS AND METHODS
Cell culture, treatments, and preparation of cell extracts
Human K562 erythroleukemia cells and HeLa cervix carcinoma cells were maintained in RPMI 1640 and Dulbecco's modified Eagle's medium, respectively, supplemented with 10% fetal calf serum (FCS) and antibiotics in a humidified 5% CO2 atmosphere at 37°C. Heat shock treatments were performed at 42°C in a constant-temperature water bath. Clasto-lactacystin β-lactone (Calbiochem) or MG132 (Peptide Institute, Inc.) was added to the cells in a final concentration of 5 and 10 μM, respectively, and cells were incubated at 37°C for the indicated time periods. Whole cell extracts were prepared as described previously (Mosser et al 1988).
Purification of recombinant human HSF1 and preparation of polyclonal rabbit anti-hHSF1 antiserum
The expression and purification of recombinant human HSF1 (rhHSF1) was done mainly as previously described (Soncin et al 1997). BL21(DE3) cells were transformed with the human HSF1 cDNA containing expression vector pET-71, and the expression of rhHSF1 was induced for 3 h with 1 mM isopropyl-β-thiogalactopyranoside. Cells were collected by centrifugation, resuspended in buffer A (100 mM K2HPO4, pH 7.4, 20 mM KCl, 2 mM DTT, 0.1 mM EDTA), and frozen. For purification, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 μg/mL pepstatin A, 1 μg/mL leupeptin, and 0.1 mg/mL hen egg lysozyme were added and incubated on ice for 30 min. The cell extract was briefly sonicated and centrifuged, and rhHSF1 was precipitated in the presence of 30% ammonium sulfate saturation. The recovered pellet was dissolved and dialyzed in buffer A prior to being subjected to heparin affinity chromatographic purification using a POROS 20 HE1 column and a BioCAD workstation (Perseptive Biosystems, Inc.). The rhHSF1 was eluted with a KCl gradient (0 to 1 M) at a flow rate of 5 mL/min, desalted by dialysis in buffer A, and concentrated using a Centricon Plus-20 filter device (Millipore). The recovered rhHSF1 was shown to be composed of a major fraction of about 65–70 kDa and some smaller products, which probably are due to degradation and incomplete synthesis of the protein, as analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting (data not shown). The SDS-PAGE purified 65–70 kDa rhHSF1 was used for immunization of the rabbits. The specificity of the raised polyclonal rabbit anti-hHSF1 antibody was verified by Western blotting using rhHSF1 as well as human cell lysates. The HSF1 reactivity of the antibody was identical to that obtained with a previously described anti-mouse HSF1 antiserum (Sarge et al 1993; a kind gift from R. Morimoto, Northwestern University). The anti-hHSF1 antibody is also suitable for immunofluorescence analysis as well as for immunoprecipitation assays (Figs 1 and 3A).
Fig 1.
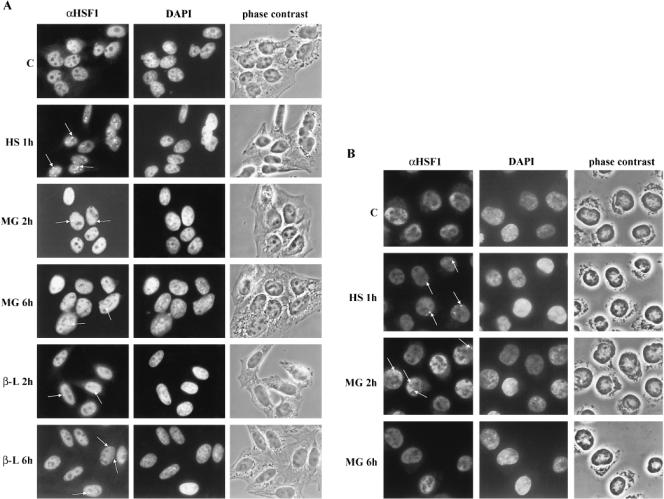
Formation of nuclear HSF1 granules is different in cells treated with proteasome inhibitors than in heat-shocked cells. (A) The localization of HSF1 in untreated (C), heat-shocked (HS; 42°C for 1 h), MG132-treated (MG; 10 μM for 2 and 6 h), and clasto-lactacystin β-lactone-treated (β-L; 5 μM for 2 and 6 h) HeLa cells was visualized using anti-hHSF1 antibodies (left panel). The middle and the right panels show the DAPI-stained DNA and the phase contrast image of the same fields as in the HSF1 panel. The arrows point to some of the granules. (B) Fluorescence micrographs of K562 cells treated as described in (A). Note that the spherical shape of the K562 suspension cells leads to detection of only a few granules in a focal level
Fig 3.
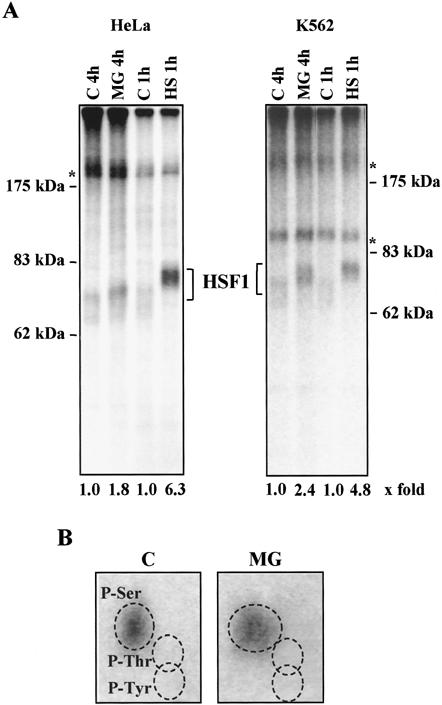
The phosphorylation state of HSF1 is increased by MG132, but to a lesser extent than by heat shock. HeLa and K562 cells were in vivo labeled with [32P]orthophosphate for 3 h before they were subjected to heat shock (HS; 42°C for 1 h) or MG132 treatment (MG; 10 μM for 4 h). HSF1 was immunoprecipitated with anti-hHSF1 antibodies, and the samples were resolved on an 8% SDS-PAGE. (A) Autoradiograph of HSF1. The control samples (C 1h, C 4h) were in vivo 32P-labeled for the same time period as the heat-shocked and the MG132-treated samples, respectively. The 32P-labeled HSF1 was quantified using a PhosphorImager. The values for the fold induction of HSF1 phosphorylation are shown relative to the control, which was assigned a fold induction value of 1. The experiments were repeated twice with identical results. The asterisk indicates unknown phosphoprotein. (B) Phosphoamino acid analysis of HSF1 immunoprecipitated from untreated (C) or MG132-treated (MG, 10 μM for 4 h) HeLa cells. The relative positions of phosphoserine, phosphothreonine, and phosphotyrosine are as indicated
Indirect immunofluorescence
For immunofluorescence analysis, HeLa cells growing on coverslips were washed with phosphate-buffered saline (PBS) and fixed for 10 min with 3% paraformaldehyde in PBS at room temperature. K562 suspension cells washed with PBS were plated onto the coverslips using a Cytospin 3 (Shandon) for 3 min at 300 rpm before fixation. After 2 rinses with PBS, the cells were permeabilized with 0.1% Triton X-100 in PBS. Then the cells were incubated for 30 min with a blocking solution containing 2% bovine serum albumin (BSA) in PBS before the rabbit anti-hHSF1 antiserum (1:500 dilution) was added for 1.5 h. After washes with PBS, the primary antibody was detected using FITC-conjugated goat anti-rabbit antibodies (1:5000 dilution for 1 h, Alexa™ 488, Molecular Probes). The DNA was stained with DAPI (0.3 μg/mL, 4′6-diamidino-2-phenylindol, Sigma) for 1 min before mounting with Mowiol 4-88 (Aldrich) containing 2.5% DABCO (1,4-diazobicyklo-[2.2.2]-octan, Aldrich). The cells were analyzed using a Leica TCS40 confocal microscope or a Leica DMR fluorescence microscope equipped with a digital Hamamatsu ORCA CCD camera. Images were further processed using Adobe Photoshop and PowerPoint software and printed with a Tekronix dye sub printer. In the experiments where formation of HSF1 granules was examined, 200–250 cells were analyzed per time point.
Gel mobility shift assay
Whole cell extracts (15 μg) were incubated with a 32P-labeled oligonucleotide, representing the proximal heat shock element (HSE) of the human hsp70 promoter. The protein-DNA complexes were analyzed on a native 4% polyacrylamide gel as described previously (Mosser et al 1988). The synthetic oligonucleotide was 32P-labeled with T4 polynucleotide kinase (Promega). For antibody supershift assay, rabbit polyclonal antibodies raised against human HSF1 were added to whole cell extracts and incubated at room temperature for 15 min prior to the gel mobility shift assay.
Western blot analysis
Whole cell extracts (10 μg) were subjected to an 8% SDS-PAGE and transferred to nitrocellulose membrane (Schleicher & Schuell) by using a semidry transfer apparatus (Bio-Rad). The equal loading of proteins was checked using Ponceau S staining (Sigma). The membrane was blocked for 1 h in 3% nonfat dry milk in PBS and then incubated for 1 h with a 1:5000 dilution of rabbit polyclonal anti-human HSF1 (anti-hHSF1) antibodies in 0.5% BSA-PBS containing 0.3% Tween 20. Mouse monoclonal antibodies were used to detect Hsp70 (4g4, Affinity Bioreagents, Inc., 1:20 000 dilution) and rat monoclonal antibodies to detect Hsc70 (StressGen, 1:5000). After washes with PBS-0.3% Tween 20, the membrane was incubated for 1 h with horseradish peroxidase-conjugated anti-rabbit antibodies (Promega, 1:20 000 dilution), anti-mouse, and anti-rat antibodies (Amersham Life Science, 1:50 000 dilution). The detection was performed using an enhanced chemiluminescence method (ECL, Amersham Life Science).
Northern blot analysis
Total RNA was isolated using RNAzol™B (Tel-Test Inc.). RNA (10 μg) was separated on a 1% agarose-formaldehyde gel, transferred to a nylon membrane (Hybond-N, Amersham Life Science), and hybridized at 65°C with [α-32P]dCTP-labeled probes for human hsp70 (pH 2.3; Wu et al 1985) and rat glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Fort et al 1985). The plasmid DNA was 32P-labeled using a nick translation kit (Promega).
Transcriptional nuclear run-on analysis
In vitro run-on transcription reactions were performed with equal number of isolated HeLa nuclei in the presence of 100 μCi of [α-32P]dUTP (3000 Ci/mmol, Amersham Pharmacia) as previously described (Banerji et al 1984; Holmberg et al 1997). The radiolabeled mRNA was hybridized to nitrocellulose-immobilized plasmids for human hsp70 (pH 2.3; Wu et al 1985), human β-actin (pHFβA-1; Gunning et al 1983), and a Bluescript vector (Stratagene). The hybridizations were carried out in 50% formamide − 6 × SCC (1 × SCC is 0.15 M sodium chloride and 0.017 M sodium citrate) − 10 X Denhardt's solution (1 × Denhardt's solution is 0.02% Ficoll, 0.02% polyvinylpyrrolidone, and 0.02% bovine serum albumin) − 0.2% SDS at 42°C for 72 h. The filters were washed with high stringency conditions (0.2 × SSC − 0.2% SDS at 65°C) and visualized by autoradiography.
In vivo 32P-labeling of cells, immunoprecipitation of HSF1, and phosphoamino acid analysis
Subconfluent HeLa cells were incubated in 7 mL of phosphate-free Minimal Essential Media Eagle (Sigma) supplemented with 10% dialyzed FCS and 0.3 mCi/mL of [32P]-orthophosphate (Amersham Pharmacia Biotech) for 3 h at 37°C before the treatments. K562 cells (7 × 106/7 mL) were maintained in RPMI 1640 as described previously, and 0.3 mCi/mL of [32P]-orthophosphate was added for 3 h prior to the treatments. The cells were further heat shocked at 42°C for 1 h or treated with 10 μM MG132 for 4 h at 37°C. Untreated cells were kept at 37°C for the same time periods. The cells were harvested, washed with ice-cold PBS, and lysed in 1 mL of ice-cold RIPA-buffer (10 mM Tris pH 7.4, 150 mM NaCl, 1% sodium deoxycholate, 1% Triton X-100, 1 mM PMSF, and 2 μg/μL of leupeptin and pepstatin A). The lysates were cleared by centrifugation and the supernatants incubated with 6 μL of polyclonal rabbit anti-hHSF1 serum for 1 h at 4°C under rotation. After addition of 30 μL of a 1:1 slurry of Protein A-Sepharose-PBS, the samples were incubated for another hour at 4°C under rotation. The beads were washed 6 times with RIPA-buffer containing 0.1% SDS, resuspended in 30 μL of Laemmli buffer, and boiled for 5 min. The proteins were separated on an 8% SDS-PAGE and visualized by autoradiography. Quantitation of phosphorylated HSF1 was performed using a FujiFilm Bas-1800 II PhosphorImager. The values for the fold induction of HSF1 phosphorylation were calculated relative to the control, which was assigned a fold induction value of 1. For phosphoamino acid analysis, the proteins separated on the SDS-PAGE were transferred to a polyvinylidene difluoride membrane (Hybond-P, Amersham Life Science) and visualized by autoradiography. The HSF1 band was cut out and subjected to acid hydrolysis (6 M HCl for 1 h at 110°C) and further 2-dimensional thin-layer electrophoresis (HTLE-7000, CBS Scientific) together with phosphoserine, phosphothreonine, and phosphotyrosine standards (van der Geer et al 1993). The first-dimension electrophoresis was carried out at pH 1.9 for 37 min using 1.3 kV, and the second-dimension electrophoresis was run at pH 3.5 for 25 min (1.3 kV). The phosphoamino acids were visualized with 0.25% ninhydrin staining and autoradiography (van der Geer et al 1993).
RESULTS
Proteasome inhibitors induce appearance of nuclear HSF1 granules
Appearance of nuclear HSF1 granules has been detected on exposure to various stresses, such as heat shock, cadmium, and the amino acid analogue azetidine (Cotto et al 1997; Jolly et al 1999). In this study, we wanted to examine whether stress induced by proteasome inhibition affects the localization of HSF1 in human cells. Therefore, we performed indirect immunofluorescence analysis of HSF1 in HeLa cells treated for 15 min to 9 h with the synthetic peptide aldehyde MG132. In comparison, the cells were subjected to heat stress at 42°C. Our anti-hHSF1 antibody revealed some cytoplasmic but predominant nuclear localization of HSF1 in untreated HeLa cells, which is in agreement with a previous study (Mercier et al 1999). Exposure of HeLa cells to heat shock resulted in 5–15 brightly staining nuclear HSF1 granules of various sizes in every cell (Fig 1A). In agreement with earlier results, the HSF1 granules disappeared on several hours of continuous heat treatment (data not shown; Cotto et al 1997). Contrary to heat shock, exposure to the proteasome inhibitor MG132 resulted in formation of nuclear HSF1 granules in some but not in all HeLa cells (Fig 1A). The granules were detectable after 2 h of treatment in 3% of the cells and were most prominent after 6 h (ie, in 15% of the cells). Clasto-lactacystin β-lactone, a more specific inhibitor of the 26S proteasome, also induced only a few HSF1 granules in some cells with the same kinetics as MG132 (Fig 1A). Sporadic HSF1 granules were still detected after 9 h of MG132 treatment, but the prolonged exposures to MG132 affected the morphology of the cells (data not shown). Compared to the heat-induced HSF1 granules (Fig 1A), the granules formed on proteasome inhibition differed in many respects in HeLa cells. For example, in heat-shocked cells, HSF1 granules appeared in every cell, whereas only 15% of the MG132- or clasto-lactacystin β-lactone-treated cells contained HSF1 granules. Furthermore, the HSF1 granules detected in HeLa cells treated with the proteasome inhibitors were more diffusely stained, smaller, and fewer per cell than those observed in heat-shocked cells (Fig 1A). Finally, the kinetics of HSF1 granule formation on stress induced by proteasome inhibitors was slower than on induction by heat shock.
To address the question whether the HSF1 granule formation by proteasome inhibitors occurred in other cells than HeLa cells, we exposed K562 cells to MG132 treatment, after which the localization of HSF1 was studied. As shown in Figure 1B, both heat shock and MG132 treatment induced formation of HSF1 granules with a similar staining pattern. Because of the spherical shape of the suspension K652 cells, only a few HSF1 granules could be detected at a given focal plane. The appearance of HSF1 granules in most K562 cells treated with heat shock or MG132 was confirmed by confocal microscopy, but the visualization of the HSF1 granules was not improved by merging of several focal layers (data not shown). Similar to the MG132-treated HeLa cells, no HSF1 granules were detected after 1 h of MG132 treatment in K562 cells (data not shown) but appeared 2 h after initiation of the MG132 treatment (Fig 1B). However, the granules disappeared by 6 h of MG132 treatment in K562 cells (Fig 1B). Taken together, the appearance of HSF1 granules on MG132 treatment was clearly different in HeLa and K562 cells.
Proteasome inhibitors induced HSF1 HSE-binding activity, retarded migration of HSF1 on SDS-PAGE, and expression of Hsp70
As the appearance of nuclear HSF1 granules has previously been shown to coincide with activation of HSF1, we next wanted to analyze the HSF HSE-binding activity in HeLa and K562 cells treated with MG132 or clasto-lactacystin β-lactone. Interestingly, the proteasome inhibitors, like heat shock, induced a strong HSF HSE-binding in both cell lines, as shown by gel mobility shift assay in Figure 2A. The MG132-induced HSF HSE-binding activity was readily detected by 1 h, markedly increased in intensity by 2 h, and was still maintained at 9 h. The presence of HSF1 in the HSF-HSE complex was confirmed by antibody supershift assay (Fig 2B).
Fig 2.
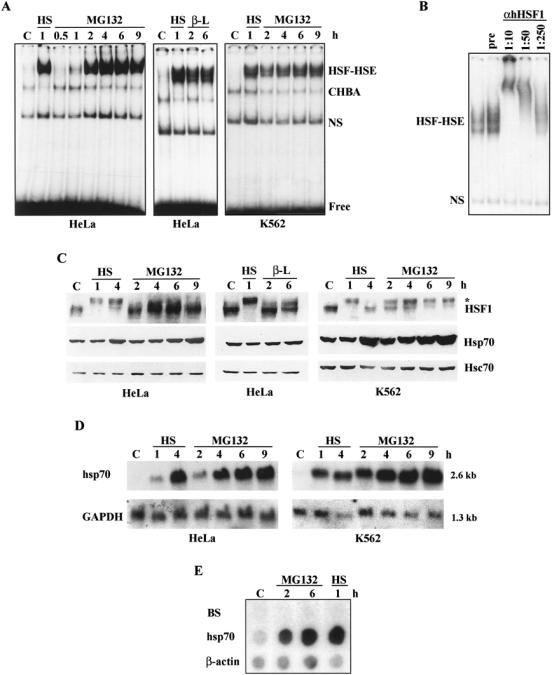
Proteasome inhibitors induce activation of HSF1 and Hsp70 expression. HeLa or K562 cells were exposed to heat shock (HS; 42°C), MG132 (MG; 10 μM), and clasto-lactacystin β-lactone (β-L; 5 μM) for the indicated time periods. Untreated cells are marked with C. (A) The HSF HSE-binding activity of the whole cell extracts was analyzed by gel mobility shift assay. CHBA denotes constitutive HSE-binding activity, NS denotes nonspecific DNA-binding activity, and free indicates unbound HSE oligonucleotide. (B) The existence of HSF1 in the HSF-HSE complex was identified using antibody supershift assay. Whole cell extracts from HeLa cells treated with MG132 for 3 h were incubated with indicated dilutions of anti-hHSF1 antibodies or with preimmune serum (pre, 1:10 dilution) prior to the gel mobility shift assay. (C) For protein analysis, whole cell extracts were examined by Western blotting using antibodies against HSF1, Hsp70, and Hsc70. The asterisk indicates hyperphosphorylated HSF1. (D) The steady-state levels of hsp70 mRNA were analyzed by Northern blotting using 32P-labeled probes for hsp70 and GAPDH. GAPDH was used as a control of equal loading. The size of the mRNA is indicated on the right. (E) The transcription rate of hsp70 was analyzed by nuclear run-on assay. Equal number of nuclei isolated from HeLa cells exposed to the indicated treatments were used for in vitro 32P-UTP labeling of transcripts, which were then hybridized to DNA corresponding to human hsp70, β-actin, and the Bluescript vector (BS). β-actin was used as an internal control
On activation by various stresses, HSF1 becomes inducibly phosphorylated. This hyperphosphorylated HSF1 can indirectly be detected by a slower migration on SDS-PAGE compared to HSF1 in unstressed samples (Sarge et al 1993; Cotto et al 1996). As shown by the Western blot analysis (Fig 2C), treatment of HeLa cells with MG132 or clasto-lactacystin β-lactone induced a slightly slower migrating form of HSF1 than in the untreated cells. However, this form of HSF1 did not migrate as slowly as the heat-shocked HSF1, indicating that the proteasome inhibitors might induce an intermediate phosphorylated state of HSF1 in HeLa cells. On the other hand, in K562 cells exposed to MG132, the migration of HSF1 on the SDS-PAGE resembled more that of HSF1 detected on heat shock (Fig 2C). At 2 to 4 h of MG132 treatment, there seemed to be 2 populations of HSF1 since 1 band migrated as HSF1 in heat-stressed K562 cells and the other band migrated as HSF1 in unstressed cells. Despite the differences in the migration of HSF1 on SDS-PAGE, MG132 treatment induced accumulation of Hsp70 to the same extent as heat shock in both cell types (Fig 2C). Treatment with clasto-lactacystin β-lactone for 6 h induced a slight increase in Hsp70 accumulation (Fig 2C), which was further enhanced by longer exposure to the proteasome inhibitor (data not shown). The proteasome inhibitors did not affect the accumulation of Hsc70, which was used as an internal control for equal loading of the samples (Fig 2C). To examine in more detail the effect of proteasome inhibition on the expression of Hsp70, the steady-state levels of hsp70 mRNA were analyzed by Northern blotting. Similarly to heat shock, treatment with MG132 strongly increased the steady-state levels of hsp70 mRNA (Fig 2D). Furthermore, our nuclear run-on analysis demonstrates that the rate of hsp70 transcription was markedly and rapidly elevated on MG132 treatment as well as heat shock in HeLa (Fig 2E), as has been shown in K562 cells (Pirkkala et al 2000). In agreement with the HSE-binding analysis (Fig 2A), the MG132-induced hsp70 transcription persisted for at least 6 h (Fig 2E).
The phosphorylation of HSF1 is increased by MG132, but not to the same extent as by heat shock
To directly study the phosphorylation of HSF1 during stress condition caused by proteasome inhibition, HeLa and K562 cells were in vivo labeled with [32P]-orthophosphate. The cells were prelabeled for 3 h before exposure to a 4-h MG132 treatment or a 1-h heat shock. Control cells were labeled for 7 and 4 h, respectively. After the treatment, HSF1 was immunoprecipitated and resolved on an 8% SDS-PAGE. As shown in the autoradiogram in Figure 3A, treatment of HeLa cells with MG132 induced a 1.8-fold increase in relative phosphorylation of HSF1 compared to a 6.3-fold increase on heat shock. In K562 cells, the relative phosphorylation of HSF1 was increased by 2.4- and 4.8-fold on treatment with MG132 or heat shock, respectively (Fig 3A). Hence, MG132 treatment enhanced the phosphorylation of HSF1 to a different extent in HeLa and K562 cells (ie, with 29% in HeLa cells and with 50% in K562 cells). In these calculations, the corresponding heat-induced increase in HSF1 phosphorylation was assigned to 100%. The migration patterns of 32P-labeled HSF1 on the autoradiograph corresponded to those observed by Western blotting (Figs 3A and 2C).
Phosphoamino acid analysis of the immunoprecipitated 32P-labeled HSF1 showed that MG132 treatment increased the phosphorylation of serine residues of HSF1 (Fig 3B). In agreement with earlier reports (Chu et al 1996; Cotto et al 1996; Kline et al 1997), heat shock increased HSF1 serine phosphorylation (data not shown).
DISCUSSION
Our results reveal that the appearance of nuclear HSF1 granules and phosphorylation of HSF1 are induced differently by proteasome inhibitors and by heat stress in the human HeLa and K562 cell lines. The current study provides new evidences for the intricate regulation of HSF1 activation on stress, showing that the multiple events that occur concomitantly with HSF1 activation might be affected differently, depending on the stress stimulus. Only 15% of the HeLa cells treated with the proteasome inhibitors MG132 and clasto-lactacystin β-lactone contained HSF1 granules, whereas these structures were found in all cells exposed to heat shock. In addition, the overall size of the HSF1 granules induced by proteasome inhibitors was smaller compared to the heat-induced granules, which have been measured to vary between 0.3 and 3 μm (Jolly et al 1999). It is possible that the number of HSF1 granule-positive cells and the sizes of the granules reflect the severity of stress since heat shock at 43°–45°C has been reported to more rapidly increase the portion of cells containing HSF1 granules and the size of the granules as compared to a moderate heat stress at 42°C (Sarge et al 1993; Jolly et al 1997). It is plausible that the severity of stress induced by proteasome inhibitors could be recognized in a cell-type–specific fashion. In K562 cells, the response to proteasome inhibition, as measured by formation of HSF1 granules, was very similar to the heat-induced response, whereas the granule formation on exposure to proteasome inhibitors was more modest in HeLa cells. Nevertheless, treatment with MG132 induced the heat shock response, resulting in increased Hsp70 expression in both cell types.
Although the function of HSF1 granules is not known, they appear in concert with the DNA-binding, hyperphosphorylated, and transcriptionally active state of HSF1 (Sarge et al 1993; Cotto et al 1997; Jolly et al 1997, 1999). Moreover, the number of HSF1 granules has been suggested to correlate with the chromosomal ploidy (Cotto et al 1997; Jolly et al 1997). For example, HeLa cells have been demonstrated to contain about 6–10 heat-induced HSF1 granules, with a substantial cell-to-cell variation (Cotto et al 1997; Jolly et al 1999). Interestingly, we found that the number of HSF1 granules in HeLa cells treated with the proteasome inhibitors MG132 and clasto-lactacystin β-lactone was not the same as in the heat-shocked cells, suggesting that the formation of HSF1 granules does not strictly correspond to the ploidy. In addition to heat shock, exposure to other stressors, such as cadmium or the amino acid analogue azetidine, have been reported to induce the formation of HSF1 granules (Cotto et al 1997; Jolly et al 1999), but with a slower kinetics than by heat stress (Jolly et al 1999). This is consistent with the delayed activation of HSF1 induced by cadmium and azetidine (Mosser et al 1988; Williams and Morimoto 1990). Similarly, delayed kinetics of HSF1 granule formation was detected on proteasome inhibition. It is worth noticing that the life cycle of the HSF1 granules did not correlate with the DNA-binding activity of HSF1 in cells treated with proteasome inhibitors. These results suggest that attenuation of HSF1 DNA-binding activity might not be linked to the disappearance of HSF1 granules. Alternatively, HSF1 might not remain bound to HSE in vivo, although a sustained DNA-binding activity of HSF1 can be detected in vitro in response to stress induced by proteasome inhibitors.
In the case of stress caused by heat shock or cadmium treatment, the HSF1 granules have been shown to coincide with hyperphosphorylation of HSF1 (Sarge et al 1993; Cotto et al 1997; Jolly et al 1999). Treatment with azetidine has also been shown to induce HSF1 granules (Cotto et al 1997), but it was originally thought that this treatment does not affect the phosphorylation of HSF1 (Sarge et al 1993). Recently, Jolly and coworkers (1999) presented that azetidine treatment induces both HSF1 granules and hyperphosphorylation of HSF1. Proteasome inhibitors, such as the synthetic peptide aldehydes ALLN and MG132 as well as the natural product lactacystin, have been shown to affect the phosphorylation of HSF1, as analyzed by migration in the Western blots (Zhou et al 1996; Mathew et al 1998; Kim et al 1999; Pirkkala et al 2000). In the present study, we demonstrate that the migration of HSF1 on SDS-PAGE was not identically influenced in cells treated with MG132 or clasto-lactacystin β-lactone as in heat-shocked cells. The effect of MG132 treatment on HSF1 phosphorylation was verified by in vivo 32P-labeling of HSF1. Indeed, treatment with the proteasome inhibitor MG132 increased the phosphorylation state of HSF1. This is in agreement with a previous study showing that phosphorylation of the chicken HSF1 and HSF3 is increased by MG132 (Kawazoe et al 1998). Similar to the heat-induced hyperphosphorylation of HSF1, we found that MG132 treatment increased the serine phosphorylation of HSF1, but the increase in the level of HSF1 phosphorylation on MG132 treatment was less prominent than on heat shock. In this respect, the response was not identical in both cell lines. Combining the results from Western blotting and in vivo 32P-labeling analysis, our study indicates that in HeLa cells only an intermediate phosphorylated state of HSF1 was induced, whereas hyperphosphorylated HSF1 was obtained in K562 cells on prolonged treatment with proteasome inhibitors.
The inducible phosphorylation of HSF1 has been linked to the transcriptionally competent HSF1 (Sorger and Pelham 1988; Jurivich et al 1994, 1995; Lee et al 1995; Cotto et al 1996; Liu and Thiele 1996; Holmberg et al 1997, 1998). Here, we show that although the degree of inducible phosphorylation of HSF1 was less prominent in cells treated with proteasome inhibitors than in heat-shocked cells, both treatments equally enhanced the steady-state levels of hsp70 mRNA and the accumulation of Hsp70. The increase in hsp70 mRNA is due to transcriptional induction of the hsp70 gene since our data demonstrate that treatment with MG132 induces hsp70 transcription (Fig 2E; Pirkkala et al 2000). This is in agreement with a study by Zhou and coworkers (1996), showing an increase in hsp70 transcription on treatment with ALLN, another potent proteasome inhibitor.
Taken together, our results provide evidence that the appearance of nuclear HSF1 granules more closely correlates with the phosphorylation state of HSF1 than with its HSE-binding activity. There is a possibility that granule formation is required in some of the molecular mechanisms underlying the inducible phosphorylation and transcriptional activation of HSF1 in human cells. However, to be able to dissect and characterize the differences in HSF1 phosphorylation on stress, induced by heat or proteasome inhibition, further studies on the identification of new phosphorylation sites of HSF1 will be needed. To date, only 3 constitutive phosphorylation sites of HSF1 have been reported (Chu et al 1996; Knauf et al 1996; Kline and Morimoto 1997; Chu et al 1998), but work is in progress to identify the other constitutively phosphorylated sites and, more important, the inducible sites that are critical for the transactivating capacity of HSF1.
Acknowledgments
We thank Stuart Calderwood for kindly providing the pET-71 plasmid. We are grateful to John Eriksson and Päivi Nykänen for critical comments on the manuscript. This work was supported by the Academy of Finland (L.S., M.K.), the Sigrid Jusélius Foundation, the Finnish Cancer Organizations (L.S.), and INCO Copernicus (A.M.). C.I.H. was supported by the Turku Graduate School of Biomedical Sciences and the Victoria Foundation.
REFERENCES
- Ananthan J, Goldberg AL, Voellmy R. Abnormal proteins serve as eukaryotic stress signals and trigger the activation of heat shock genes. Science. 1986;232:522–524. doi: 10.1126/science.3083508. [DOI] [PubMed] [Google Scholar]
- Banerji SS, Theodorakis NG, Morimoto RI. Heat shock-induced translational control of HSP70 and globin synthesis in chicken reticulocytes. Mol Cell Biol. 1984;4:2437–2448. doi: 10.1128/mcb.4.11.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush KT, Goldberg AL, Nigam SK. Proteasome inhibition leads to a heat-shock response, induction of endoplasmic reticulum chaperones, and thermotolerance. J Biol Chem. 1997;272:9086–9092. doi: 10.1074/jbc.272.14.9086. [DOI] [PubMed] [Google Scholar]
- Chu B, Soncin F, Price BD, Stevenson MA, Calderwood SK. Sequential phosphorylation by mitogen-activated protein kinase and glycogen synthase kinase 3 represses transcriptional activation by heat shock factor 1. J Biol Chem. 1996;271:30847–30857. doi: 10.1074/jbc.271.48.30847. [DOI] [PubMed] [Google Scholar]
- Chu B, Zhong R, Soncin F, Stevenson MA, Calderwood SK. Transcriptional activity of heat shock factor 1 at 37°C is repressed through phosphorylation on two distinct serine residues by glycogen synthase kinase 3α and protein kinases Cα and Cζ. J Biol Chem. 1998;273:18640–18646. doi: 10.1074/jbc.273.29.18640. [DOI] [PubMed] [Google Scholar]
- Cotto JJ, Fox SG, Morimoto RI. HSF1 granules: a novel stress-induced nuclear compartment of human cells. J Cell Sci. 1997;110:2925–2934. doi: 10.1242/jcs.110.23.2925. [DOI] [PubMed] [Google Scholar]
- Cotto JJ, Kline M, Morimoto RI. Activation of heat shock factor 1 DNA binding precedes stress-induced serine phosphorylation. J Biol Chem. 1996;271:3355–3358. doi: 10.1074/jbc.271.7.3355. [DOI] [PubMed] [Google Scholar]
- Fort P, Marty L, Piechaczyk M, El Sabrouty S, Dani C, Jeanteur P, Blanchard JM. Various rat adult tissues express only one major mRNA species from the glyceraldehyde-3-phosphate-dehydrogenase multigenic family. Nucleic Acids Res. 1985;13:1431–1442. doi: 10.1093/nar/13.5.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch M, Wu C. Phosphorylation of Drosophila heat shock transcription factor. Cell Stress Chaperones. 1999;4:102–117. doi: 10.1379/1466-1268(1999)004<0102:podhst>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunning P, Ponte P, Okayama H, Engel J, Blau H, Kedes L. Isolation and characterization of full-length cDNA clones from human α-, β-, and γ-actin mRNAs: skeletal but not cytoplasmic actins have an amino-terminal cysteine that is subsequently removed. Mol Cell Biol. 1983;3:787–795. doi: 10.1128/mcb.3.5.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Meng Y-H, Mivechi NF. Glycogen synthase kinase 3β and extracellular signal-regulated kinase inactivate heat shock transcription factor 1 by facilitating the disappearance of transcriptionally active granules after heat shock. Mol Cell Biol. 1998;18:6624–6633. doi: 10.1128/mcb.18.11.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg CI, Leppä S, Eriksson JE, Sistonen L. The phorbol ester 12-Otetradecanoylphorbol 13-acetate enhances the heat-induced stress response. J Biol Chem. 1997;272:6792–6798. doi: 10.1074/jbc.272.10.6792. [DOI] [PubMed] [Google Scholar]
- Holmberg CI, Roos PMK, Lord JM, Eriksson JE, Sistonen L. Conventional and novel PKC isoenzymes modify the heat induced stress response but are not activated by heat shock. J Cell Sci. 1998;111:3357–3365. doi: 10.1242/jcs.111.22.3357. [DOI] [PubMed] [Google Scholar]
- Jolly C, Morimoto RI, Robert-Nicoud M, Vourc'h C. HSF1 transcription factor concentrates in nuclear foci during heat shock: relationship with transcription sites. J Cell Sci. 1997;110:2935–2941. doi: 10.1242/jcs.110.23.2935. [DOI] [PubMed] [Google Scholar]
- Jolly C, Usson Y, Morimoto RI. Rapid and reversible relocalization of heat shock factor 1 within seconds to nuclear stress granules. Proc Natl Acad Sci USA. 1999;96:6769–6774. doi: 10.1073/pnas.96.12.6769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurivich DA, Pachetti C, Qiu L, Welk JF. Salicylate triggers heat shock factor differently than heat. J Biol Chem. 1995;270:24489–24495. doi: 10.1074/jbc.270.41.24489. [DOI] [PubMed] [Google Scholar]
- Jurivich DA, Sistonen L, Sarge KD, Morimoto RI. Arachidonate is a potent modulator of human heat shock gene transcription. Proc Natl Acad Sci USA. 1994;91:2280–2284. doi: 10.1073/pnas.91.6.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawazoe Y, Nakai A, Tanabe M, Nagata K. Proteasome inhibition leads to the activation of all members of the heat-shock-factor family. Eur J Biochem. 1998;255:356–362. doi: 10.1046/j.1432-1327.1998.2550356.x. [DOI] [PubMed] [Google Scholar]
- Kim D, Kim S-H, Li GC. Proteasome inhibitors MG132 and lactacystin hyperphosphorylate HSF1 and induce hsp70 and hsp27 expression. Biochem Biophys Res Commun. 1999;254:264–268. doi: 10.1006/bbrc.1998.9840. [DOI] [PubMed] [Google Scholar]
- Kline MP, Morimoto RI. Repression of the heat shock factor 1 transcriptional activation domain is modulated by constitutive phosphorylation. Mol Cell Biol. 1997;17:2107–2115. doi: 10.1128/mcb.17.4.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauf U, Newton EM, Kyriakis J, Kingston RE. Repression of human heat shock factor 1 activity at control temperature by phosphorylation. Genes Dev. 1996;10:2782–2793. doi: 10.1101/gad.10.21.2782. [DOI] [PubMed] [Google Scholar]
- Larson JS, Schuetz TJ, Kingston RE. Activation in vitro of sequence-specific DNA binding by a human regulatory factor. Nature. 1988;335:372–375. doi: 10.1038/335372a0. [DOI] [PubMed] [Google Scholar]
- Lee BS, Chen J, Angelidis C, Jurivich DA, Morimoto RI. Pharmacological modulation of heat shock factor 1 by antiinflammatory drugs results in protection against stress-induced cellular damage. Proc Natl Acad Sci USA. 1995;92:7207–7211. doi: 10.1073/pnas.92.16.7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 1998a;8:397–403. doi: 10.1016/s0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- Lee DH, Goldberg AL. Proteasome inhibitors cause induction of heat shock proteins and trehalose, which together confer thermotolerance in Saccharomyces cerevisiae. Mol Cell Biol. 1998b;18:30–38. doi: 10.1128/mcb.18.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X-D, Thiele DJ. Oxidative stress induces heat shock factor phosphorylation and HSF-dependent activation of yeast metallothionein gene transcription. Genes Dev. 1996;10:592–603. doi: 10.1101/gad.10.5.592. [DOI] [PubMed] [Google Scholar]
- Mathew A, Mathur SK, Morimoto RI. Heat shock response and protein degradation: regulation of HSF2 by the ubiquitin-proteasome pathway. Mol Cell Biol. 1998;18:5091–5098. doi: 10.1128/mcb.18.9.5091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Balbás MA, Dey A, Rabindran SK, Ozato K, Wu C. Displacement of sequence-specific transcription factors from mitotic chromatin. Cell. 1995;83:29–38. doi: 10.1016/0092-8674(95)90231-7. [DOI] [PubMed] [Google Scholar]
- Mercier PA, Winegarden NA, Westwood JT. Human heat shock factor 1 is predominantly a nuclear protein before and after heat stress. J Cell Sci. 1999;112:2765–2774. doi: 10.1242/jcs.112.16.2765. [DOI] [PubMed] [Google Scholar]
- Meriin AB, Gabai VL, Yaglom J, Shifrin VI, Sherman MY. Proteasome inhibitors activate stress kinases and induce Hsp72: diverse effects on apoptosis. J Biol Chem. 1998;273:6373–6379. doi: 10.1074/jbc.273.11.6373. [DOI] [PubMed] [Google Scholar]
- Mivechi NF, Murai T, Hahn GM. Inhibitors of tyrosine and Ser/Thr phosphatases modulate the heat shock response. J Cell Biochem. 1994;54:186–197. doi: 10.1002/jcb.240540207. [DOI] [PubMed] [Google Scholar]
- Morimoto RI. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998;12:3788–3796. doi: 10.1101/gad.12.24.3788. [DOI] [PubMed] [Google Scholar]
- Mosser DD, Theodorakis NG, Morimoto RI. Coordinate changes in heat shock element-binding activity and HSP70 gene transcription rates in human cells. Mol Cell Biol. 1988;8:4736–4744. doi: 10.1128/mcb.8.11.4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirkkala L, Alastalo T-P, Zuo X, Benjamin IJ, Sistonen L. Disruption of heat shock factor 1 reveals an essential role in the ubiquitin proteolytic pathway. Mol Cell Biol. 2000;20:2670–2675. doi: 10.1128/mcb.20.8.2670-2675.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarge KD, Murphy SP, Morimoto RI. Activation of heat shock gene transcription by HSF1 involves oligomerization, acquisition of DNA binding activity, and nuclear localization and can occur in the absence of stress. Mol Cell Biol. 1993;13:1392–1407. doi: 10.1128/mcb.13.3.1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soncin F, Prevelige R, Calderwood SK. Expression and purification of human heat shock factor 1. Prot Exp Purific. 1997;9:27–32. doi: 10.1006/prep.1996.0672. [DOI] [PubMed] [Google Scholar]
- Sorger PK. Yeast heat shock factor contains separable transient and sustained response transcriptional activators. Cell. 1990;62:793–805. doi: 10.1016/0092-8674(90)90123-v. [DOI] [PubMed] [Google Scholar]
- Sorger PK, Pelham HRB. Yeast heat shock factor is an essential DNA-binding protein that exhibits temperature-dependent phosphorylation. Cell. 1988;54:855–864. doi: 10.1016/s0092-8674(88)91219-6. [DOI] [PubMed] [Google Scholar]
- van der Geer P, Luo K, Sefton BM, and Hunter T. 1993 Phosphopeptide mapping and phosphoamino acid analysis on cellulose thin-layer plates. In: Protein Phosphorylation: A Practical Approach, ed Hardie DG. IRL Press, Oxford University Press, New York, 31–59. [Google Scholar]
- Weighardt F, Cobianchi F, Cartegni L, Chiodi I, Villa A, Riva S, Biamonti G. A novel hnRNP protein (HAP/SAF-B) enters a subset of hnRNP complexes and relocates in nuclear granules in response to heat shock. J Cell Sci. 1999;112:1465–1476. doi: 10.1242/jcs.112.10.1465. [DOI] [PubMed] [Google Scholar]
- Williams GT, Morimoto RI. Maximal stress-induced transcription from the human Hsp70 promoter requires interactions with the basal promoter elements independent of rotational alignment. Mol Cell Biol. 1990;10:3125–3136. doi: 10.1128/mcb.10.6.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. The heat shock factors: structure and regulation. Annu Rev Cell Dev Biol. 1995;11:441–469. doi: 10.1146/annurev.cb.11.110195.002301. [DOI] [PubMed] [Google Scholar]
- Wu B, Hunt C, Morimoto RI. Structure and expression of the human gene encoding major heat shock protein HSP70. Mol Cell Biol. 1985;5:330–341. doi: 10.1128/mcb.5.2.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Wu X, Ginsberg HN. Evidence that a rapidly turning over protein, normally degraded by the proteasomes, regulates hsp72 gene transcription in HepG2 cells. J Biol Chem. 1996;9:24769–24775. doi: 10.1074/jbc.271.40.24769. [DOI] [PubMed] [Google Scholar]