

Abstract
Protein–protein interactions between human heat shock transcription factor 1 (hHSF1) and general transcription factors TFIIA-γ, TFIIB, TBP, TAFII32, and TAFII55 and positive coactivator PC4 were characterized in order to identify potential targets of contact in the transcriptional preinitiation complex. These contacts represent one of the final steps in the signal transfer of heat stress to the transcriptional apparatus. TATA-binding protein (TBP) and transcription factor IIB (TFIIB) were identified as major targets for HSF1 transcriptional activation domains AD1 and AD2 based on in vitro interaction assays. TBP showed affinity for AD2 and a fragment containing AD1, while the core domain of TFIIB interacted primarily with the AD1 fragment. Interactions were also detected between full-length HSF1 and the small subunit (γ) of TFIIA. PC4 interacted weakly with HSF2 and showed even less affinity for HSF1. Coimmunoprecipitation of transiently expressed TBP in HeLa cells demonstrated that HSF1 AD2 and AD1+AD2 are able to bind TBP in vivo. Assays based on transcriptional interference confirmed predictions that both TBP and TFIIB can interact with HSF1 activation domains in HeLa cells. The negative regulatory region (NR) of HSF1 did not interact with any general factors tested in vitro but did bind TFIID in nuclear extracts through contacts that probably involve TATA associated proteins (TAFs). These results suggest a model for transcriptional regulation by HSF1 that involves a shift between formation of dysfunctional TFIID complexes with the NR and transcriptionally competent complexes with the C-terminal activation domains.
INTRODUCTION
In metazoans the heat shock (HS) response entails a rapid and sustained activation of transcription on sudden exposure to high temperatures. A substantial body of evidence indicates that under non-HS conditions RNA polymerase II exists in a paused state at the 5′ end of HS genes in Drosophila and human cells (Rougvie and Lis 1988; Giardina et al 1992; Lee et al 1992; Giardina and Lis 1993; O'Brien and Lis 1993; Rasmussen and Lis 1993, 1995; Brown et al 1996; Li et al 1996; Brown and Kingston 1997; Weber et al 1997; Benjamin and Gilmour 1998). The paused polymerase initiates transcription prior to heat stress and synthesizes from 17 to 46 nucleotides of RNA before transcription is arrested (Rasmussen and Lis 1993). Within the Droshsp70 promoter, formation of the paused polymerase complex is dependent on the GAGA elements upstream of TATAA, DNA sequences near the transcription start site, and sequences at the pause site (Lee et al 1992). Consistent with the presence of a paused polymerase at the 5′ end of HS genes, chromatin reconstruction experiments indicate that the GAGA factor, TFIID, TFIIA, and RNA polymerase associate in vitro with the Droshsp70 promoter under non-HS conditions (Sandaltzopoulos and Becker 1998). Activated HSF binds to the promoter after HS and plays a role in releasing the paused RNA polymerase (Brown et al 1996; Sandaltzopoulos and Becker 1998). This early priming of the promoter facilitates a rapid response to stress exemplified by the Droshsp70 promoter, which is activated within 60 s of exposure to high temperatures (37°C) (O'Brien and Lis 1993). In subsequent firings, HSF is involved in sustaining a high rate of reinitiation of polymerase at the promoter (Sandaltzopoulos and Becker 1998). HSF1 has also been shown to be involved in the remodeling of chromatin downstream from the start site of transcription, a process that may facilitate elongation (Brown and Kingston 1997).
Human HSF1 is organized into functional domains that provide for DNA binding, trimerization, nuclear localization, transcriptional activation, and regulation of transcriptional activity. Regions involved in DNA binding (DBD), oligomerization (OD), and nuclear localization (NLS) are located in the N-terminal half of HSF1, whereas domains directly involved in transcriptional activation (AD1 and AD2) are present in the C-terminal portion of the protein (Wu 1995). Also present in the C-terminal region are the C-terminal hydrophobic repeat that regulates trimerization and a negative regulatory domain (NR) that plays a critical role in the on-off regulation of transcriptional activity for AD1 and AD2 (Green et al 1995; Shi et al 1995; Zuo et al 1995; Newton et al 1996). The core of the NR extends from amino acids (aa) 221 to 310 (Green et al 1995). DNA fragments containing this region are able to confer heat-inducible activation to the transcriptional activation domains of HSF1 and to the heterologous activation domain of VP16 (Newton et al 1996).
Presumably, interactions between HSF1 and general transcription factors are central to the activation of transcription, including the release of paused RNA polymerase and reinitiation at HS promoters. It is possible that these types of contacts are also involved in the on–off regulation of HSF1 transcriptional activation domains (AD1 and AD2) by the NR domain of HSF1. Drosophila HSF has been shown to interact directly with TBP in vitro in a manner that promotes cooperative binding of TBP and HSF to the hsp70 promoter (Mason and Lis 1997). The conserved C-terminal core of TBP was shown to bind to an N-terminal fragment of HSF containing the DNA-binding domain (DBD) and to a C-terminal fragment containing the transcriptional activation domain. Lis and colleagues proposed that the release of Drosophila RNA polymerase from the paused state by HSF may involve displacement of the H domain of the large subunit of RNA polymerase II from TBP (Usheva et al 1992; Lis and Wu 1993; Mason and Lis 1997).
In the present study we continue to explore the possible relationship between HSF interactions with general transcription factors and transcriptional activation by investigating interactions between hHSF1 and a series of general transcription factors, including TBP, TFIIA-γ, TAFII32, TAFII55, and PC4. Interactions between these factors and HSF1 were initially characterized by in vitro assays. For TBP and TFIIB, several additional approaches were employed to evaluate the potential for in vivo interactions with HSF1. Our findings suggest that major contacts occur in HeLa cells between HSF1 transcriptional activation domains and 2 general transcription factors, TBP and TFIIB. In addition, the negative regulatory domain of HSF1 has the potential to bind non-TBP components of TFIID (presumably TAFs) under both basal and HS conditions. We propose that interactions between TBP and the regulatory and transcriptional activation domains of HSF1 determine the capacity of HSF1 to activate transcription by forming transcriptionally active and nonactive complexes.
MATERIALS AND METHODS
Expression vectors and clones
HSF1 and general transcription factors were translationally fused to glutathione-S-transferase (GST) and expressed in Escherichia coli using vector pGEX-SB, which was derived from pGEX-KG (Guan and Dixon 1991) by removing the EcoR I and Hind III sites and inserting a Bgl II site adjacent to the Sal I site. All HSF and ScTBP (yeast TBP; aa 1–82) translational fusions were inserted between Sal I (N-terminus) and Bgl II sites in pGEX-SB and expressed in E. coli strain BL21 by 0.1 mM IPTG induction for 3 h at room temperature. The bacterial pellets from 50 mL of culture (OD600 = 1–2) were suspended in 3 mL of buffer A100 and lysed by sonication on ice. The protein lysates were collected after a brief centrifugation (4°C) and stored at −20°C. GST fusion proteins were purified using glutathione Sepharose 4B beads (Pharmacia) and were stored in buffer A100 without BSA. Buffer A100 (buffer A with 100 mM NaCl) contained 1.25 mL 1-M HEPES pH 7.5, 0.25 mL 1-M MgCl2, 12.5 μL 0.4-M EDTA, 6 mL glycerol, 0.29 g NaCl, 2 μL β-mercaptoethanol, 50 μL NP-40, and 50 μL 4% BSA per 50 mL (0.004% final). Protein quantification was estimated by SDS-PAGE and Coomassie Brilliant Blue staining of protein immobilized on beads to ensure that assays contained approximately equal amounts of GST fusion proteins.
Sources for plasmids and vectors were as follows: GST-VP16 (aa 413–490), GST-VP16 (Δ456-FP442), and T7 epitope-tagged hTFIIB in pET5a (Novagen) were kindly provided by S.G.E. Roberts and M.R. Green (University of Massachusetts) (Roberts and Green 1994). A human TBP cDNA clone was provided by R.G. Roeder (The Rockefeller University) (Hoffman et al 1990) and was tagged by cloning into vector p24d-SB, which was derived from pET24d (Novagen) by blunt-ending the Bgl II site and inserting a linker with sites for Sal I positioned upstream and Bgl II downstream. Clones for PC4 (Ge and Roeder 1994), hTAFII32 (Hisatake et al 1995), hTAFII55 (Chiang and Roeder 1995), and TFIIA-γ (Ozer et al 1994) were obtained from the HeLa S3 Matchmaker cDNA library (Clontech) by PCR cloning using gene-specific primers.
Protein-binding assays
The binding assays (pull-down assays) were conducted in 100 μL of buffer A150, or A300, with approximately 5 μg of GST-fusion protein attached to Sepharose beads and 5 μL of T7-tagged protein (ligand) lysate. The binding incubation was 2 h at 4°C with gentle rocking. The beads were then extensively washed with buffer A containing NaCl and 0.004% BSA. Proteins were released from the beads by boiling after addition of 40 μL of SDS loading buffer and were resolved by 10% SDS-PAGE. Monoclonal antibodies against the T7 epitope (Novagen) and a chemiluminescense detection system (ECL; Amersham) were used to detect T7-tagged proteins by Western blot analysis.
HeLa cell transfection and heat shock treatment
HeLa cells were grown at 37°C in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal calf serum and transfected by the calcium phosphate precipitation method (Ausubel et al 1991). Transfections contained 0.5 μg of Gal4-DBD fusion constructs in vector pCI-neo (Promega), 0.5 μg of human growth hormone reporter (pXGH5; Nichols Institute Diagnostics) for monitoring transfection efficiency, and 2 μg of pG5Luc luciferase reporter DNA (Patel et al 1995). Forty-four hours after transfection, cells were incubated for 2 h at either 42°C (heat shock) or 37°C (control) and allowed to recover for 2 h at 37°C before harvesting. Transcriptional efficiency was determined based on synthesis of human growth hormone estimated with the hGH-TGES 100T Kit according to the suppliers protocol (radioimmune assay; Nichols Institute Diagnostics). Luciferase activity was determined using a kit from Promega. All pCI-neo vectors and all derived vectors were modified by elimination of the original Bgl II site to be compatible with cloning strategies.
Transcriptional interference assay for analysis of in vivo interactions
A series of transcriptionally inactive mutants of human transcription factors TFIIA-γ, TBP, TFIIB, TAF32, and PC4 were cloned into the Sal I and Bgl II sites of the mammalian protein expression vector pT7-NLS. pT7-NLS was derived from pCI-neo by addition of a T7-tag for the detection of expressed proteins and by insertion a 13 amino acid nuclear localization signal (NLS) from human DNA ligase I (Montecucco et al 1995) for transporting expressed proteins into the nucleus. HeLa cells were cotransfected with 4 plasmids: 3 μg of the transcriptionally inactive mutants in the pT7-NLS vector, 2 μg of reporter plasmid pG5luc containing GAL4-binding sites upstream of luciferase gene, 0.5 μg of the effector construct containing GAL4-DBD/AD1+2 fusion in the pCI-neo vector, and 0.5 μg of pXGH5 (growth hormone reporter). The final amount of DNA for each transfection was equalized by substituting empty vector pCI-neo DNA where appropriate. All transfections were conducted in triplicate, and activities of the luciferase reporter gene were normalized for transfection efficiency against units of human growth hormone.
Coimmunoprecipitation
A second method used to detect in vivo associations between the activation domains of hHSF1 and general transcription factors was based on their coimmunoprecipitation. The vector pNG-SB was derived from pCI-neo by the addition of aa 1–97 of the GAL4 DBD (Xho I/Sal I) and modification of the polylinker to include a unique Bgl II site for construction of C-terminal fusions (Sal I/Bgl II). The vector pNT7-SB was constructed in a similar manner with the T7 epitope substituted for the GAL4 DBD. C-terminal fragments of HSF1 containing AD2, mutated AD2, and AD1+2 were cloned into pNG-SB as GAL4 DBD fusions, and TBP was cloned into pNT7-SB. Vectors containing HSF1 activation domains and TBP were cotransfected into HeLa cells. Forty-eight hours after transfection, cells were harvested and lysed by 3 rounds of freezing and thawing in buffer A500 plus a cocktail of proteinase inhibitors (0.1 mM PMSF, 2 μg/mL leupeptin, 3 μg/mL pepstatin A, 40 μg/mL antipain). The lysates were cleared by centrifugation for 5 min at 4°C and then diluted with buffer A0 to a final salt concentration of 150 mM (buffer A150). GAL4-DBD fusions were coimmunoprecipitated from cellular lysates with anti-GAL4-DBD monoclonal antibody (mAb) (Santa Cruz Biotechnology), which was covalently cross-linked to protein A-Sepharose beads using DMP (dimethylpimelimidate) (Harlow and Lane 1988). The beads were washed twice with buffer A150 and boiled for 5 min in SDS loading buffer. Released proteins were resolved by 10% SDS-PAGE and blotted to Immobilon PVDF membrane (Millipore). The membrane was then probed with mAb against the T7-epitope to detect the T7-tagged TBP.
Pull-down assay to detect binding to TBP transiently expressed in HeLa cells
Human TBP and its deletions were cloned into Sal I and Bgl II sites of vectors pNT7-SB (without added NLS for wild-type TBP) or pT7-NLS (with added NLS for deletions of TBP) and transiently expressed (48 h) in HeLa cells. Cells were lysed in buffer A500 plus proteinase inhibitors. The lysates were collected after a 10-min centrifugation (4°C) and incubated with rocking for 2 h at 4°C in buffer A150 with 5μg of GST-AD1 or GST-AD1+2 fusion proteins immobilized on Sepharose-4B beads. The beads were washed 3 times with buffer A150 and boiled for 5 min in SDS loading buffer. Released proteins were analyzed by Western blotting as described.
TFIID Complex pull-down assays using GST-fusions of HSF1
For detection of interactions of HSF1 AD2 and AD1+2 with the TFIID complex, binding reactions consisted of 180 μL of buffer A100 plus 0.1% NP-40, 50 μL of the HeLa nuclear extract, and 20 μL of immobilized GST-AD2 or GST-AD1+2. The reaction mixtures were adjusted to 100 mM, 150 mM, or 250 mM by adding 3 M KCl and then incubated for 2 h at 4°C with gentle rocking. The beads were washed twice with buffer A150 and bound proteins eluted by buffer A500 and visualized by Western blotting. Endogenous TBP in the TFIID complex was detected by anti-hTBP mAb from Promega. For detection of interactions between the negative regulatory region (NR) and the TFIID complex, 500 μL of buffer A100 and 500 μl HeLa nuclear extract were mixed and passed through a GST-NR affinity column (150 μL of GST-NR) by gravity at 4°C. The beads were washed with 1.5 mL of buffer A100 and bound proteins eluted with buffer A500. Visualization was performed by Western blotting using anti-hTBP antibody.
In order to identify potential targets of HSF contact involved in the activation of transcription, interaction studies were conducted in vitro using immobilized glutathione S-transferase (GST) fusion constructs of hHSF1 and various proteins present in the preinitiation complex (PIC). Potential target proteins included in this study were 2 general transcription factors (TBP and TFIIB), TAFs 32 and 55, and the positive coactivator PC4. The GST-HSF fusion proteins were bound to glutathione-Sepharose beads forming an affinity matrix that was incubated with extracts containing recombinant peptides derived from general transcription factors expressed in E. coli or HeLa cells. The binding reactions were conducted at near physiological salt concentrations and pH to approximate conditions in vivo. Ligand proteins and their respective deletion constructs were T7-epitope-tagged to facilitate detection of bound proteins by Western blotting. Since 13% of the residues in HSF1 activation domain 2 (AD2) are acidic (Green et al 1995), binding assays using GST fusions with the acidic activator VP16 (GST-VP16) were conducted for comparison. Negative controls for no or little binding were conducted using GST, the VP16 (Δ456-FP442) mutant that does not bind TFIIB in vitro (Roberts et al 1993), and an N-terminal truncation mutant of yeast TBP, scTBP (aa 1–82). The nonmutated VP16 activation domain (aa 413–490) was also used as a positive control for binding where appropriate. The binding reactions were initially optimized to determine suitable levels of input for free ligand protein (data not shown). Subsequent experiments were then conducted with appropriate positive and negative controls to provide context for assessing the strength of interactions. The choice of factor proteins selected for testing was determined by availability and by previously demonstrated ability to interact with the acidic activator VP16. A limited number of interactions were tested using GST fusion constructs of hHSF2.
A similar pattern of interaction with HSF1 was observed for TFIIB (Fig 1C). In addition to interacting with full-length HSF1, TFIIB also showed affinity for HSF1 constructs containing hydrophobic region B (HR-B) of the OD (constructs 5 and 6) and with a fragment containing AD1 present in construct 7. As with TBP, no interactions were seen between TFIIB and the negative control region (NR) demonstrated with HSF1 constructs 10 through 13. TFIIB differed from TBP by showing a strong preference for the AD1 fragment, whereas TBP interacted strongly with both AD1 and AD2. Although interactions with HSF2 were not mapped in detail, it does have potential for TFIIB interaction as demonstrated in Figure 1C, lane H2.
Fig 1.
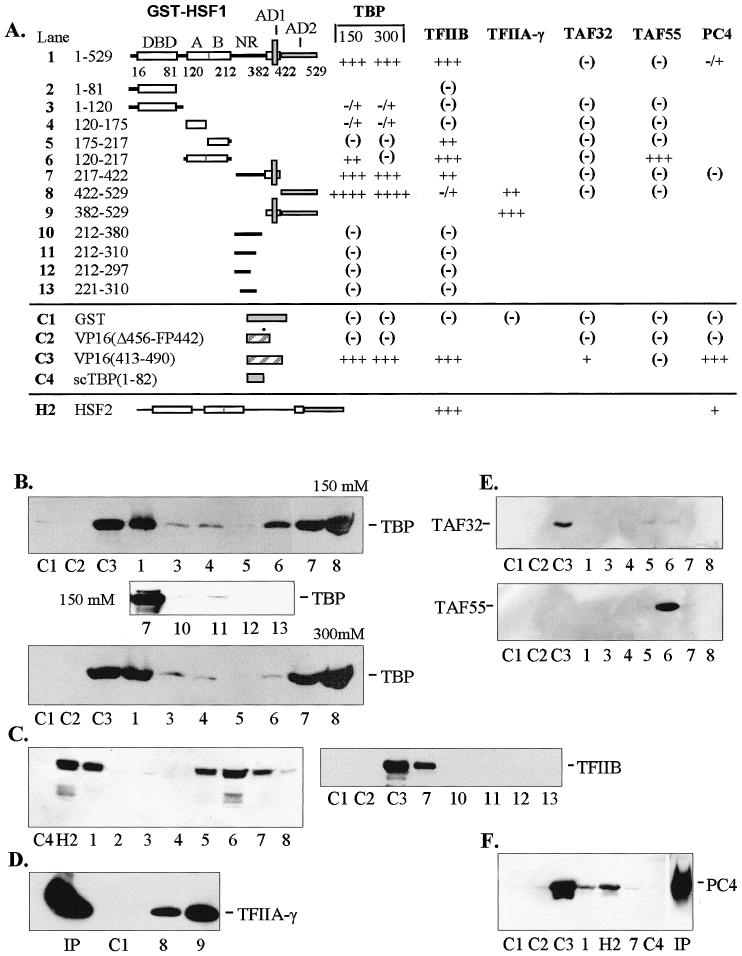
In vitro interactions between general transcription factors and human HSF1. (A) Diagrammatic summary of interactions between immobilized GST-HSF1 and TBP, TFIIB, TFIIA-γ, TAFII32, TAFII55, and PC4. The number of pluses (+) corresponds to the intensity of Western blot bands (panels B–F). Blanks indicate that the interaction was not tested. Positive and negative controls are designated C1 through C4 and represent constructs containing GST alone, VP16 mutant (Δ456-FP442), nonmutated VP16 (aa 413–490), and the N-terminus of yeast TBP (scTBP, aa 1–82), respectively. Construct numbers refer to lanes on Western blots shown in panels B through F. All transcription factors were expressed in E. coli and contained the T7 epitope at the N-terminus. All binding reactions were conducted with equal amounts of crude E. coli extracts containing full-length recombinant factors at 150 mM KCl unless otherwise indicated. (B) Interactions with human TBP at 150 and 300 mM KCl. With full-length GST-HSF1, approximately 80% on input TBP was bound under these conditions. (C) Interactions with TFIIB. Under these conditions, approximately 50% of input TFIIB was bound. (D) Interactions with TFIIA-γ. (E) Interactions with TAFII32 (top) and TAFII55 (bottom). (F) Interactions with positive coactivator PC4. Human HSF2 (H2) was tested with TFIIB and PC4. Input (Ip) lanes contained 10% of total present in binding assays. DBD, DNA-binding domain; A and B, hydrophobic regions A and B of the oligomerization domain (HR-A and HR-B); NR, negative domain; AD1 and AD2, activation domains 1 and 2
Other limited studies were conducted with TFIIA-γ and positive coactivator PC4. Binding studies conducted with full-length TFIIA-γ indicated moderate affinity for AD2 of HSF1 (Fig 1D, construct 8). The more intense band seen with construct 9 suggests that TFIIA-γ also interacts with the fragment containing AD1 since this construct contains both activation domains. PC4 interacted with full-length HSF2 weakly (Fig 1F, lane H2) and even more weakly with HSF1 (Fig 1F, lane 1).
Two components of TFIID, TAFII32 and TAFII55, were tested for interactions with the full series of HSF deletions. TAFII32 is known to interact with VP16 acidic activator (Klemm et al 1995), whereas TAFII55 does not bind VP16 in vitro (Chiang and Roeder 1995). HSF1 affinities clearly differed from those of VP16 in showing no binding to TAFII32 (Fig 1E, lane C3). Another difference between VP16 and HSF1 was evident by the interaction between TAFII55 and the OD (Fig 1E, TAFII55 blot, construct 6). However, the TAFII55 binding to the OD is difficult to interpret since no binding was observed using full-length HSF1 (Fig 1E, TAFII55 blot, construct 1).
Core domains of the TBP and TFIIB interact with HSF1
The site of interaction of TBP with HSF1 was mapped in vitro at a salt concentration of 150 mM KCl. Binding reactions were conducted 2 ways: with HSF1 activation domains AD1 plus AD2 (Fig 1A, construct 9) or with TBP deletion mutants immobilized on beads. Interactions required the presence of TBP conserved core repeat 1 (R1; aa 156–221) as indicated by the binding of TBP construct 2 and the lack of binding by TBP constructs 4 and 5 (Fig 2B,C). Interactions with construct 5 were weak and variable (compare panels B and D with C). The reciprocal experiment using GST-AD1+2 as the matrix-bound protein and T7-tagged deletion fragments of TBP (expressed in HeLa cells) as the ligand gave identical results (Fig 2, panel D). From these experiments it is unclear whether core repeat 1 alone binds HSF1 or whether binding requires TBP repeats 1 and 2 together since isolated core repeats may not assume the proper structure.
Fig 2.
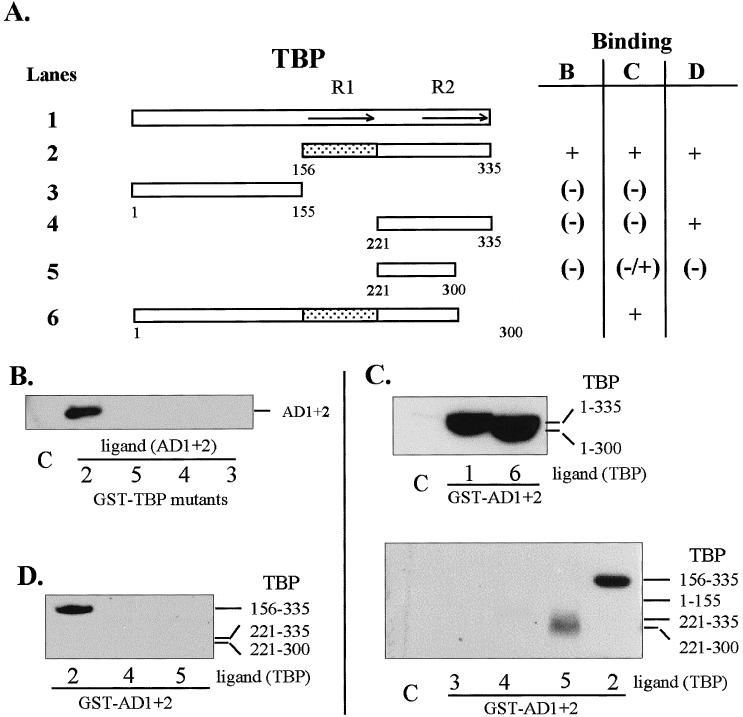
Regions of TBP that interact in vitro with HSF1 AD1+2. (A) Diagrammatic summary of interactions between TBP and AD1+AD2. Binding reactions contained GST-fusion proteins (TBP or HSF1 AD1+2) immobilized on glutathione-Sepharose beads and equal amounts of the ligand proteins present in crude extracts from E. coli or in a nuclear extract from transformed HeLa cells. The ligand proteins in the extracts were tagged with the T7 epitope at the N-terminus. The shaded region corresponds to TBP sequences interacting with HSF1. R1 and R2 indicate repeated sequences of the conserved core. (B) Western blot of AD1+2 in E. coli extract bound to immobilized GST-TBP fusion proteins corresponding to the TBP deletions depicted in panel A. (C) Western blot of TBP deletions bound to GST-AD1+2. TBP deletions from E. coli lysate were used in binding reactions with immobilized GST-AD1+2 fusion protein. (D) Western blot of TBP deletions bound to GST-AD1+2. TBP deletions from transformed HeLa cell lysates were used to interact with GST-AD1+2 fusion protein and were detected with anti-T7 tag antibody. The bars indicate the expected positions of bands representing deleted TBP proteins. Lane numbers correspond to TBP constructs in panel A. Lane C in all panels represents the negative control showing the amount of nonspecific interaction between full-length TBP (ligand) and immobilized GST
Similar experiments were conducted using various deletion constructs of T7-tagged TFIIB as shown in Figure 3. In these assays GST-AD1+2 was immobilized on beads and incubated with recombinant TFIIB from E. coli lysates in binding reactions containing either 150 or 250 mM KCl. Full-length TFIIB, as well as constructs containing TFIIB core repeat 1 (R1), showed strong affinity for AD1+2 as indicated with constructs 1, 2, 5, and 6. An exception was seen with construct 4, which contained the isolated R1 from aa 109 to 207 and showed much weaker binding. The fact that other TFIIB constructs with deletion boundaries at residue 109 (constructs 5 and 6) or at residue 207 (construct 2) showed stronger binding to AD1+2 than isolated R1 (construct 4) may indicate that regions of repeat 2 also contribute either directly or indirectly to interactions with AD1+2.
Fig 3.
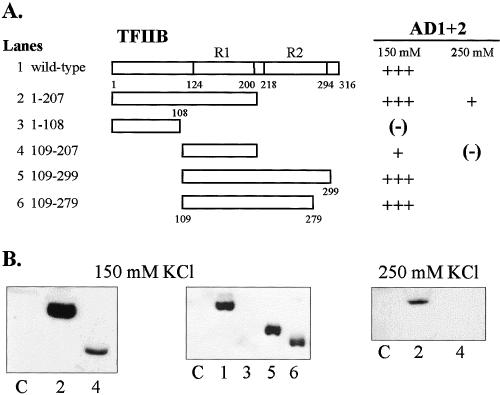
Regions of TFIIB that interact in vitro with HSF1 AD1+2. All proteins were expressed in E. coli, and approximately equal amounts of each TFIIB construct were added to each binding reaction. GST-AD1+2 fusion proteins were immobilized on glutathione-Sepharose beads, and wild-type and truncated TFIIB proteins (ligand) were T7 epitope-tagged. (A) Summary of interactions with schematics representing various truncations of TFIIB. Repeat domains (R1 and R2) of the conserved core are indicated. (B) Western blots demonstrating interactions between TFIIB constructs 1 through 6 at 150 mM or 250 mM KCl. Lane numbers correspond to TFIIB truncations depicted in panel A. Lane C represents the negative control showing nonspecific interactions between full-length TBP (ligand) and immobilized GST
HSF1 activation domains bind endogenous TBP and TFIID from HeLa nuclear extracts
Since human TBP is associated with TAFs in the TFIID complex in vivo, it is important to determine whether the activation domains of HSF1 are capable of interacting with TBP as a component of the native TFIID complex. In Figure 4A, immobilized GST-AD2 was incubated with whole-cell HeLa extracts from cells expressing T7-tagged TBP. GST-AD2 was able to bind the transiently expressed TBP with high efficiency as seen by comparison with the input amount (Ip). In a second experiment, immobilized GST-fusions with either AD2 or AD1+2 were used in binding assays with HeLa nuclear extract prepared from nontransformed cells. The bands seen in Figure 4B indicate that endogenous TBP was able to interact with GST-AD1+2 in salt concentrations up to 250 mM KCl. This result does not differentiate between the possibilities that HSF1 interactions may occur with TBP directly and/or with TAFIIs associated with TBP. The much weaker binding to AD2 in Figure 4B compared to panel 4A may be due to the overexpression of the T7-tagged TBP and the possibility of enhanced binding to AD1+2 due to the synergism of binding TBP by the 2 activation domains. Bands for AD2 binding in Figure 4B were visible on longer exposures (not shown).
Fig 4.
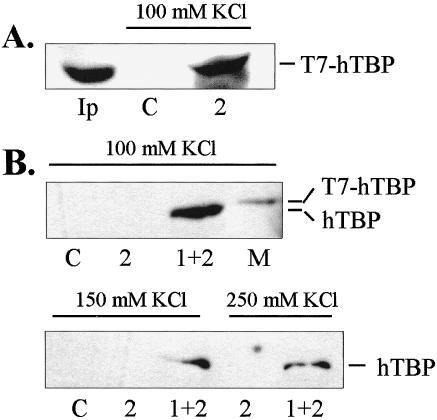
Native TFIID complex in HeLa whole-cell extracts binds immobilized HSF1 C-terminal activation domains in vitro. (A) Western blot of transiently expressed TBP (T7-tagged) from whole-cell extracts bound to GST-AD2. (B) Western blot of endogenous TBP from HeLa whole-cell extracts bound to GST-AD2 and GST-AD1+2. Fifty microliters of whole-cell extract and 20 μL of GST-AD1+2 were used for the binding assays at the salt concentrations indicated. TBP was visualized by anti-T7 epitope monoclonal antibody or anti-hTBP. Lanes: C = GST negative control; Ip = input amount; 2 = AD2; 1+2 = AD1+2; M = marker consisting of T7-tagged TBP expressed in E. coli.
Coimmunoprecipitation suggests in vivo interaction between TFIID and HSF1 transcriptional activation domains
As a further test of the potential for interaction between the transcriptional activation domains of HSF1 and TBP, coimmunoprecipitation experiments were conducted using proteins expressed in HeLa cells. Activation domain constructs were fused to the GAL4 DBD to ensure nuclear transport and to provide an epitope for immunoprecipitation. Western blot analysis of proteins precipitated by the GAL4 DBD antibody demonstrated the presence of TBP (T7-tagged) in complexes containing GAL4-AD2 and GAL4-AD1+2 (Fig 5, lanes 4 and 5, lower band). A clear increase in binding was evident with the GST-AD1+2 construct compared to GST-AD2 alone. Significantly, the AD2 mutant (MA-1) was unable to bind TFIID as evidenced by the absence of TBP in lane 3 (Fig 5). This lack of interaction correlates with the drastically reduced transcriptional activity of the MA-1 mutant (aa substitutions: D477V, D482A, L515P, and P522K) (Yuan et al 1997), suggesting that AD2-TBP interactions in vivo may be required for transcriptional activity of HSF1.
Fig 5.
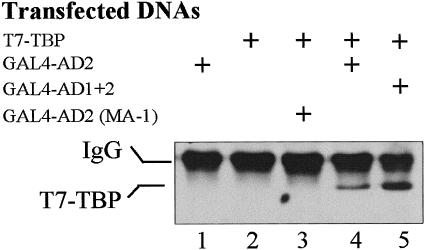
Interactions between TBP and HSF1 demonstrated by coimmunoprecipitation. Chimeric proteins GAL4-AD2, GAL4-AD1+2, or GAL4-AD2 (MA-1) were coimmunoprecipitated with T7-tagged TBP coexpressed in HeLa cells. Plus sign (+) represents the presence of each plasmid in the transfection mixture. The AD2 (MA-1) construct contains a mutated AD2 that is severely impaired in transcriptional activity when assayed in yeast cells (Yuan et al 1997). Protein complexes were first immunoprecipitated using anti-GAL4 DBD antibody covalently cross-linked to protein A-Sepharose beads. The presence of T7-tagged TBP in the complexes was then detected by probing the Western blot with anti-T7 tag antibody
Negative region of HSF1 binds TFIID from HeLa extracts
The GST pull-down assay was also used to detect possible interactions between native TFIID and the negative regulation domain (aa 221–310) of HSF1 (Fig 6). Nuclear extracts prepared from heat-stressed and non–heat-stressed HeLa cells were incubated with either GST-NR or GST in a binding reaction containing 100 mM KCl. Bound proteins were blotted and probed with anti-TBP antibody. The presence of TBP was evident in the pull-down assay using GST-NR, with the specificity of the interaction demonstrated by the lack of binding to GST alone. It is also significant that similar amounts of TBP were bound from heat-stressed and non–heat-stressed nuclear extracts. Since the NR showed no direct affinity for TBP in vitro (Fig 1B, lanes 10–13), TBP is assumed to interact indirectly through tight association with other proteins that contact the NR. Likely candidates for these bridging proteins include 1 or more of the TAFs.
Fig 6.

Negative region of HSF1 binds TFIID in HeLa nuclear extracts. Nuclear extracts (500 μL) prepared from heat-stressed (HS+) and non–heat stressed (−) cells were used in binding reactions with immobilized GST and GST-negative region fusion proteins (GST-NR) in 100 mM KCl. The GST-NR construct contained the region from aa 212 to 310 of hHSF1. TBP was visualized by probing the blotted proteins with anti-TBP antibody
Functional test for involvement of TBP in the transcriptional activation pathway of HSF1
An assay based on transcriptional interference (Colgan et al 1993) (Fig 7) served as a functional demonstration of protein-protein interactions in vivo between AD1+2 of HSF1 and TBP. Activity was monitored in transient assays using a reporter gene driven by GAL4-binding sites in the promoter (pG5luc). In these experiments, transcriptional activity of the GAL4-AD1+2 activator protein was inhibited by coexpression of a TBP mutant protein. To ensure that the activities obtained from the coexpression of the GAL4-AD1+2 activator and various TBP mutants were activator dependent, basal activities of the TBP wild-type and mutant constructs alone (no GAL4-AD1+2 effector) were measured and found to be less than 5% of activated transcription obtained with the GAL4-AD1+2 construct (Fig 7C, lane C). Titration of the GAL4-AD1+2 vector indicated that DNA levels between 0.2 and 2 μg showed no significant self-inhibition of activity (not shown). When 0.3 μg of the GAL4-AD1+2 plasmid was cotransfected with 3 μg of TBP mutant DNA encoding from aa 156 to 335 (Fig 7A, construct 2), a clear inhibition of activity was observed. This reduction in activity is consistent with the formation of a dysfunctional complex between the mutant TBP and the C-terminal activation domains of HSF1. A comparison of activities between TBP construct 2 and construct 4 indicated that the interaction requires core repeat 1 from aa 156 to 220, consistent with the in vitro studies (Fig 2). Alternatively, TBP may make contact with coactivators that are required in HSF1 activation of transcription. All TBP constructs showed similar protein expression levels as indicated by Western blot analysis (Fig 7B). The lack of inhibition by the TBP1–300 (construct 6) was surprising and seems to indicate that this C-terminal truncation of TBP is functional under these conditions; alternatively, the global structure of the mutant TBP may be perturbed in a manner that prevents HSF1 AD1+2 interaction in vivo.
Fig 7.
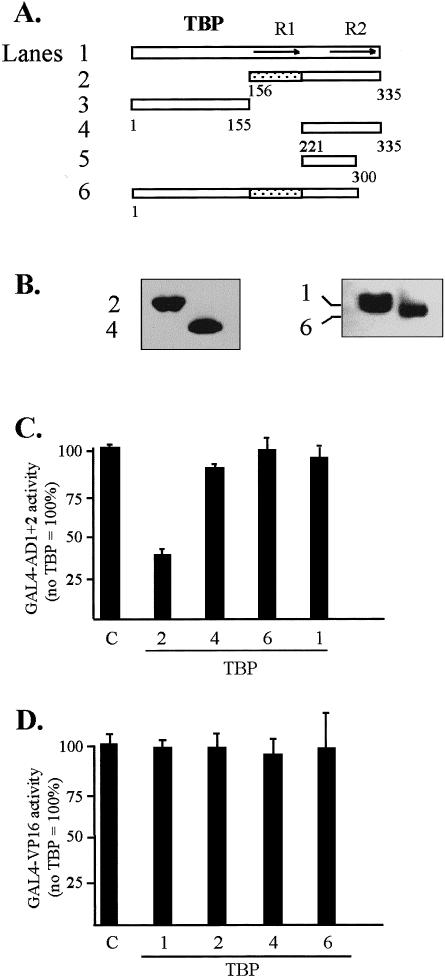
In vivo interactions between AD1+2 and TBP analyzed by the transcriptional interference assays. (A) Schematic shows full-length and truncated T7-tagged TBP proteins coexpressed with GAL4-AD1+2. (B) Western blot of T7-tagged TBP mutants transiently expressed in HeLa cells and detected with anti-T7-tag antibody. Numbers indicate constructs depicted in panel A. (C) Transcriptional interference assay demonstrates the effect of coexpressed TBP mutants on the transcriptional activity of AD1+2. One effector plasmid contained HSF1 activation domains AD1+2 fused to the GAL4 DBD in vector pNG-SB. The other effector consisted of the indicated TBP deletions with the T7-tag and nuclear localization signal cloned into vector pT7-NLS. The reporter was the luciferase gene with 5 GAL4 DNA-binding sites upstream of the TATA-box (pG5luc). Lane marked “C” contained pCI-neo as an empty-vector control with only polylinker downstream of the CMV promoter. The following DNAs were cotransfected into HeLa cells: 0.3 μg of GAL4-AD1+2, 3 μg of the TBP mutant, 2 μg of pG5luc, and 0.5 μg of the growth hormone reporter (pXGH5) as an internal standard. For all assays, luciferase activities of triplicate transfections were normalized against production of human growth hormone to establish relative activities. Activity of pCI-neo alone was assigned a value of 100%. (D) Transcriptional interference assay demonstrating little effect of TBP mutants on the transcriptional activity of VP16. VP16 activity was monitored using a mammalian 2-hybrid system to reconstitute VP16 activity by coexpression of GAL4 DBD-P and N-VP16 vectors (Patel et al 1995). Coexpression of TBP mutants and the empty-vector control were as described in panel C. The ratio of VP16 effector to TBP mutant DNAs was 1:10 in transfections. Activity of the VP16 effector alone was assigned a value of 100%. Activity of GAL4-HSFAD1+2 was 0.39 times the activity of the 2-hybrid GAL4-VP16
In order to determine whether the inhibition observed with TBP construct 2 was specific to the HSF1 activation domains, the acidic activator VP16 (aa 413–490) was coexpressed with TBP mutants. Using the same amounts of activator and TBP vector DNAs as in the GST-AD1+2 assay, no inhibition of activity was observed with any of the TBP mutants (Fig 7D). This result suggests that, unlike HSF1 activation domains, interactions between VP16 activation domains and the TBP mutants were not strong enough to inhibition transcription in vivo. Alternatively, the lack of inhibition using VP16 may result from a redundancy in pathways of activation for VP16 in vivo. We conclude that the inhibition in activity seen when GST-AD1+2 was coexpressed with the TBP aa 156 to 335 fragment (construct 2) was not due to a global reduction in transcription but was the result of specific interference with transcriptional activation by HSF1. It is noteworthy that, in this case, strong in vitro interactions between VP16 and TBP (Stringer et al 1990; Kim et al 1994) were not reliable predictors of strong interactions in living cells.
Functional test for the involvement of TFIIB in the activation pathway
Coexpression of the TFIIB(1–207) mutant protein with GAL4-AD1+2 in HeLa cells resulted in approximately 70% inhibition of activity suggesting that a dysfunctional complex was formed between the 2 proteins in vivo. It has been shown that interaction between the acidic activator VP16 and helix E1 of TFIIB core repeat 1 is disrupted by R185E and R193E point mutations (Roberts et al 1993). When these 2 mutations were introduced into the TFIIB(1–207) mutant protein, no relief of GST-AD1+2 inhibition was observed, suggesting that AD1+2 may not interact with TFIIB in the same manner as VP16 (Fig 8, construct 3).
Fig 8.
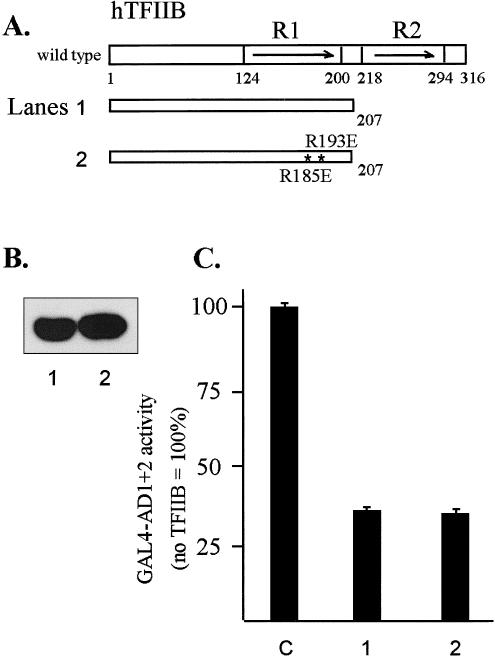
In vivo interactions between AD1+2 and TFIIB analyzed by transcriptional interference assays. (A) Diagram of TFIIB and mutant proteins. The asterisks indicate 2 point mutations in E1 helix of hTFIIB: R185 to E185 and R193 to E193. (B) Western blot of T7-tagged TFIIB mutants transiently expressed in HeLa cells and detected with anti-T7 tag antibody. (C) Inhibition of GAL4-AD1+2 activity by the wild-type and mutated TFIIB1-207 deletion. In transformations, the first effector was the GAL4-AD1+2 fusion in vector pNG-SB, and the second effector was the indicated TFIIB deletion fragment with the T7-epitope tag and nuclear localization signal in vector pT7-NLS. The luciferase reporter plasmid was pG5luc, and DNA from pCI-neo served as the empty-vector control (lane C). Cotransfected DNAs were as follows: 0.3 μg of GAL4-AD1+2, 3 μg of TFIIB mutant, 2 μg of luciferase reporter (pG5luc), and 0.5 μg of growth hormone reporter (pXGH5). Luciferase activity from triplicate assays was normalized using human growth hormone expression
No clear inhibition seen with PC4 mutants
Transcriptional interference assays conducted by coexpressing positive coactivator PC4 with GAL4-AD1+2 served as a negative control for the TBP and TFIIB studies since PC4 was expected to have only minimal involvement in the AD1+2-mediated activation based on the low-affinity interaction with HSF1 obtained in vitro (Fig 1). The results of the transcriptional interference assay (Fig 9A) were consistent with the in vitro observations, indicating that little or no interaction occurs between hHSF1 and PC4. A slight inhibition of AD1+2 activity was seen with the PC4(21–127) mutant, which was not observed with PC4(41–127). The slight degree of inhibition exhibited by PC4(21–127) seems consistent with the weak indication of interaction with HSF1 and AD2 obtained from the in vitro pull-down assay (Fig 1F). Western blot analysis (Fig 9C) indicated that similar levels of the 2 PC4 mutant proteins were expressed in HeLa.
Fig 9.
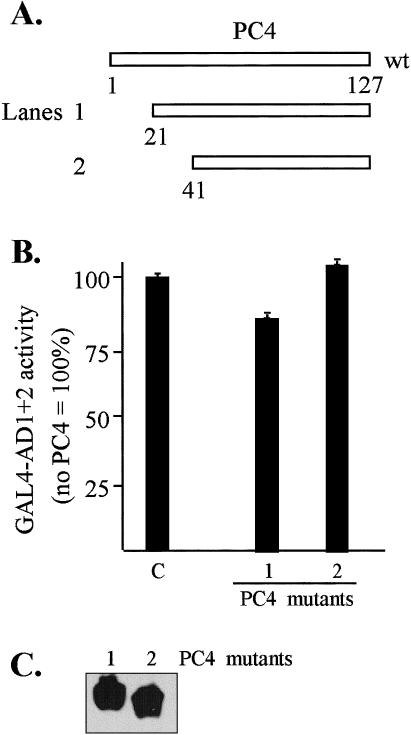
In vivo interactions between GAL4-AD1+2 and PC4 analyzed by transcriptional interference assays. (A) Schematic of PC4 constructs. (B) Test for inhibition of GAL4-AD1+2 activity by coexpression of PC4 mutant proteins. Effector plasmids consisted of the GAL4-AD1+2 fusion in vector pNG-SB and PC4 with the T7-tag and nuclear localization signal in vector pT7-NLS. The luciferase reporter plasmid was pG5luc, and DNA from pCI-neo served as the empty-vector control (lane C). Lane numbers correspond to construct designations. Cotransfected DNAs: 0.3 μg of GAL4-AD1+2, 3 μg of PC4 mutants, 2 μg of luciferase reporter (pG5luc), and 0.5 μg of growth hormone reporter (pXGH5). The luciferase activity of each transfection was normalized by human growth hormone expression. (C) Western blot of PC4 protein expression. The PC4 mutant proteins were transiently expressed in HeLa cells and detected with anti-T7 tag antibody
DISCUSSION
Using a combination of in vitro and in vivo approaches, we have identified a number of general transcription factors that show potential for interactions with HSF1. GST pull-down assays using recombinant proteins expressed in E. coli indicated strong interactions between HSF1 and TBP, TFIIA-γ, and TFIIB (Fig 1). Much weaker interactions were observed with PC4. No interactions were detected with TAFII32, and interactions with TAFII55 were confined to an isolated fragment of HSF1 containing HR-B of the oligomerization domain. Further indications of interaction were obtained using several methods that utilized proteins expressed in HeLa cells. GST pull-down assays demonstrated that a fragment containing AD1 and isolated AD2 could interact with complexes containing either transiently expressed or endogenous TBP from HeLa nuclear extracts. In addition, coimmunoprecipitation experiments using transiently expressed proteins in HeLa cells established a correlation between the transcriptional competence of AD2 and its ability to bind TBP (or a complex of proteins containing TBP). As an alternative approach, transcriptional interference assays provided a functional test for the ability of HSF1 activation domains to bind TBP, TFIIB, and PC4 in living cells. Of these, the strongest evidence for in vivo interaction was obtained for TBP and TFIIB.
There was a general correspondence between domains of HSF1 that bind to general transcription factors and those that have been shown previously to be involved in transcriptional activity. TFIIB exhibited a strong bias in binding to a fragment containing AD1 versus isolated AD2, whereas TBP, and possibly TFIIA-γ, showed strong interactions with both activation domains. Interestingly, human HSF1 showed more potential for targeting TFIIB than Drosophila HSF, which has only slight affinity for TFIIB in vitro (Mason and Lis 1997). Interactions between TBP and TFIIB with the OD of HSF1 were also evident using in vitro pull-down assays, although this region of HSFs has not been associated with transcriptional activity in previous studies. A weak interaction was also seen between TBP and the DBD of HSF1 at both moderate and high salt concentrations but was not investigated further. Surprisingly, the OD appeared to exhibit a higher affinity for TFIIB than to either of the 2 C-terminal activation domains. These observations regarding interactions with the OD raise the possibility that some contacts between TBP and TFIIB with HSF1 may not result in enhanced transcriptional activity or that some isolated interactions alone are insufficient to activate transcription.
Activation domains of HSF1 interact with a restricted set of target proteins compared to the archetypal acidic activator VP16. This result was not expected due to the similarity in activation potential of the respective activation domains regarding transcriptional initiation (including reinitiation) and elongation when assayed in vitro (Brown et al 1998). In our study, VP16 interacted with TBP, TFIIB, TAFII32, and PC4, whereas HSF1 showed no affinity for TAFII32 and only weak binding to PC4. Another difference between VP16 and HSF1 was seen in interactions with TFIIB, where point mutations in helix E1 of TFIIB known to disrupt VP16 binding had no effect in alleviating inhibition of HSF1 activation domains in transient assays (Fig 8). Although AD1 and AD2 show sequence similarity to the pattern of aromatic, hydrophobic, and charged residues typical of VP16, a relatively high proportion of prolines (18%) and serines (24%) are present in AD2.
From our protein interaction studies, it appears that HSF1 targets TBP, TFIIB, and TFIIA-γ in the transcriptional activation of HS genes; however, these experiments did not specifically address the issue of heat inducibility. Potentially, HSF1 activity may be controlled at multiple steps in its function, including oligomerization, nuclear localization, DNA binding, transcriptional activation, and the attenuation of activity after the heat shock response. At present, 2 domains of HSF1 appear to play major roles in the regulation of activation and deactivation. One of these domains is hydrophobic repeat C (HR-C, aa 384–409), which overlaps the N-terminal half of AD1 (aa 401–421). Deletion or mutation of HR-C results in constitutive nuclear localization and DNA-binding activity of human HSF1, Drosophila HSF (Rabindran et al 1993; Zuo et al 1994), and chicken HSF1 and HSF3 (Nakai and Morimoto 1993). Since some mutations within the OD also result in constitutive trimerization, it has been proposed that HR-C interacts with the hydrophobic repeats within the OD to maintain HSF1 in a folded monomeric form under non–heat shock conditions (Rabindran et al 1993; Zuo et al 1994; Wu 1995). Another important regulatory domain is the NR, which inhibits transcriptional activity of the C-terminal activation domains AD1 and AD2 (Green et al 1995; Shi et al 1995; Newton et al 1996). This conserved portion of this region extends from aa 221 to 310 and is present in HSF1 from humans, mice, and chickens. A fragment of HSF1 (aa 201–370) containing the NR is also capable of conferring heat-inducible regulation to the heterologous activation domain of VP16 (Newton et al 1996). This ability to regulate VP16 suggests that its mode of action does not rely on specific interactions with C-terminal activation domains and is independent of HR-C, which is absent in the heterologous activators. Little is known regarding how heat inducibility is conferred by the NR, but a single point mutation from lysine to alanine at residue 298 has been shown to disrupt this function (Newton et al 1996).
Transcriptional activation is a separate step from HS-inducible DNA-binding activity. In cells treated with sodium salicylate and other anti-inflammatory agents, HSF1 is bound to the promoter, but transcription of hsp70 genes does not occur, suggesting that a final barrier to transcriptional activation is present to prevent fortuitous induction of HS genes (Jurivich et al 1992). Oxidative injury is another condition where DNA-binding activity is induced but transcriptional activation is blocked (Bruce et al 1993). Although direct evidence is lacking, it seems likely that the NR is responsible for imposing this final regulatory checkpoint over transcriptional activity.
It is also evident that a different mechanism must determine the oligomerization state and DNA-binding ability of HSF1 as compared to transcriptional activation. This was illustrated when the overexpression of HSF1 in HeLa cells under non–heat shock conditions resulted in DNA binding but no transcriptional activation of the integrated HS reporter gene (hsp70/CAT) (Zuo et al 1995). Apparently, the titratable cellular factor hypothesized to regulate trimerization and DNA binding was depleted by overexpression of full-length HSF1, but the release of transcriptional repression, presumably controlled by the NR, was not affected.
Results of the current study suggest that the NR can interact with 1 or more TAFs within TFIID. This conclusion is based on the finding that the NR was able to pull down a complex of proteins containing TBP from HeLa nuclear extracts (Fig 6) but was not able to bind TBP directly in vitro (Fig 1B). Since the interaction between the NR and TFIID occurred in extracts from both control and heat-stressed cells, differential binding of the NR does not appear to be responsible for heat inducibility. Two models that account for AD1 and AD2 binding to TBP and the binding of the NR to TAFs are shown in Figure 10. In the first model, called the “constrained access model” (Fig 10A), the NR is folded under non–heat shock conditions and assumes a more open, or flexible, conformation with the onset of heat stress. During heat shock or other conditions where HS genes are normally induced, the unfolded conformation of the NR would allow contact between the C-terminal activation domains of HSF1 and TBP. This HSF1:TBP interaction may displace TBP binding by the H region of RNA polymerase II as proposed by Mason and Lis (1997). This displacement would facilitate release of RNA polymerase from the paused state existing initially at the promoter and prevent the H domain from re-establishing stable contact with TBP during subsequent rounds of reinitiation. According to the constrained access model, the NR would make contact with 1 or more TAFs when HSF1 is bound at the promoter. The interaction between the NR and TFIID would constrain the C-terminus of HSF1 but still permit interaction between HSF1 activation domains and TBP under suitable conditions. The timing and cellular location of NR unfolding is not specified, and the participation of other proteins or possible modification of the NR is not ruled out.
Fig 10.
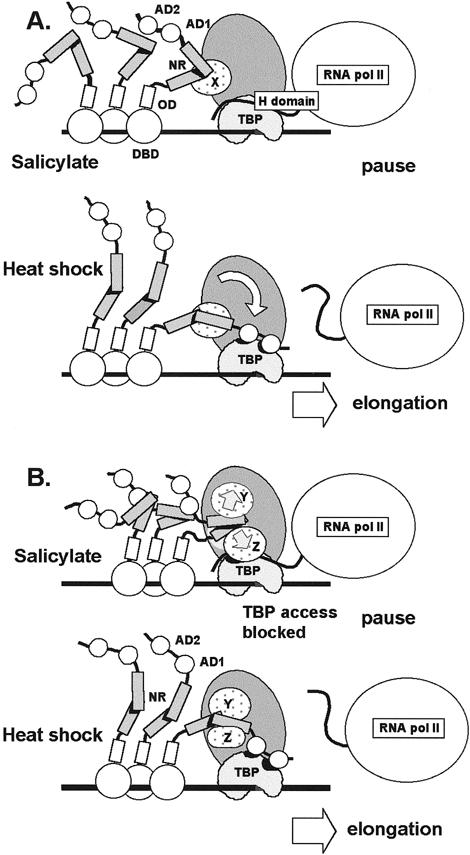
Model for the role of the negative regulatory region in controlling transcriptional activity of HSF1. The negative regulatory region (NR) binds to unknown TAFIIs (TAFIIX) within TFIID under basal and heat shock conditions. (A) Constrained access model. HSF1 is bound to the promoter of HS genes after treatment of cells with anti-inflammatory agents such sodium salicylate. Conformation of the NR constrains the C-terminal activation domains of hHSF1 (AD1 and AD2) from making productive contact with TBP. The H domain of paused RNA polymerase II interacts with TBP. After heat shock, the NR assumes a conformation that allows contact between AD1 and AD2 with TBP. The H domain is displaced, and elongation and reinitiation proceed. (B) TFIID distortion model. Access to TBP by activators is determined by TAFs that mask TBP when in contact with the NR under non–heat shock conditions (eg, salicylate treatment). The NR changes conformation in the transition from basal to heat shock conditions. Under non–heat shock conditions, NR contacts with TFIID, causing TBP to be inaccessible to contact with activation domains. Under heat shock, the altered conformation of the NR makes contacts with TFIID that do not result in TBP masking. TAFs are designated X, Y, and Z. The contribution of additional proteins or HSF1 modifications in formation of the active and inactive conformations of the NR is unknown
Under certain non–heat shock conditions, such as those existing after treatment of cells with sodium salicylate or after oxidative injury (Jurivich et al 1992; Bruce et al 1993), the NR would interact with TFIID, but transcriptional activation would not occur. The folded conformation of the NR and the constraints on positioning imposed on the C-terminal activation domains by NR:TAF interactions would prevent the activation domains from contacting TBP. The folded conformation of the NR and the binding of the NR to TFIID would act together to block fortuitous activation of HS genes by a mechanism distinct from that involved in the regulation of HSF1 oligomerization and DNA binding.
When viewed in the context of a single HSF1 monomer, the constrained access model is consistent with the observations of protein-protein interactions; however, the question of access to TBP by other monomer subunits of the HSF1 trimer and by other trimers bound to the promoter is not addressed. The constrained access model predicts that the NR must be folded and bound to a TAF in order to block TBP access; neither condition alone would necessarily prevent activator contact with TBP. A better model would provide a mechanism for blocking TBP access to multiple HSFs in view of the relatively large number of HSF monomers potentially bound to a typical HS promoter containing multiple HSE sites.
One possibility is that folding of the NR alone is sufficient to mask the adjacent C-terminal activation domains and prevent TBP access. This simple masking model, however, does not accommodate the possibility that the NR can act in trans, and the role of NR binding to TFIID is left unexplained. The possibility that the NR may act in trans is considered here since preliminary experiments suggest that coexpression of GAL4 DBD-NR fusion protein with GAL4 DBD-AD1+2 inhibits transcription (not shown).
These concerns are resolved in the TFIID distortion model presented in Figure 10B, which provides a mechanism to prevent TBP access for multiple HSFs and accommodates trans-regulation by the NR. Here, the NR is postulated to make 2 different types of contact with the TAFs of TFIID. In the inhibitory mode, the NR assumes a conformation (possibly folded) that distorts TFIID by altering the positioning, or conformation, of TAFs so that activation domains are denied access to TBP. This alteration in TFIID structure would not affect the interaction of prebound H domain but would block HSF activation domains from displacing H domain contacts with TBP. Under induction conditions, the NR would change its conformation (unfold or become more flexible) so that TFIID binding would not cause TAF blockage of TBP access. Alternatively, the NR could mask access to the target site on TBP while attached to the TAFs without making direct contact with TBP. Under HS conditions, NR-TFIID interactions may stabilize TFIID binding to the promoter for multiple reinitiations. The TFIID distortion model predicts that under noninducing conditions, the binding of a single NR to TFIID can inhibit promoter activity mediated by activation domains located on the same protein (cis-regulation) or those present on other HSF1 proteins bound to the promoter.
The finding that HSF1 has affinity for TBP, TFIIB, and TFIIA-γ is consistent with previous studies related to the function of HS promoters. Numerous reports have shown that in Drosophila and in humans, RNA polymerase II exists in a paused state at the 5′ end of HS genes under control conditions (Rougvie and Lis 1988; Rasmussen and Lis 1993; Brown et al 1996; Li et al 1996). Prior to HS and the binding of HSF1 to the HS promoter, RNA polymerase has initiated transcription and has synthesized up to 45 nucleotides of transcript before entering a stably paused state (Rasmussen and Lis 1993). It follows that HSF is not involved in the initial recruitment of general transcription factors to the promoter. Instead, assembly of the PIC is dependent on the GAGA factor, the presence of a TATAA motif, and sequences downstream near the start of transcription (Lee et al 1992; Shopland et al 1995). The CTD is hypophosphorylated when RNA polymerase is in the paused state, which is consistent with the prediction that it is bound to TBP (Xia and Voellmy 1997). Release of the pause requires the binding of HSF1 to the promoter and is proposed to involve the displacement of the H domain from TBP by HSF (Lis and Wu 1993; Xiao et al 1994). After elongation from the paused state, the only general factors remaining at the promoter are TFIID and TFIIA (Sandaltzopoulos and Becker 1998). Cell-free systems using reconstituted chromatin have provided evidence that HSF1 also plays an important role in reinitiation (Sandaltzopoulos and Becker 1998). One pathway critical to efficient reinitiation may be the recruitment of holoenzyme through contacts between HSF1 AD1 (and possibly the OD) and TFIIB. It also seems likely that the demonstrated affinity between HSF1 and general transcription factors TBP and TFIIA-γ may function in vivo to stabilize the prebound TBP-DNA-TFIIA complex present at the promoter between rounds of reinitiation.
In summary, our studies have identified 3 members of the PIC (TBP, TFIIB, and TFIIA-γ) that may act as targets of contact for hHSF1 in the activation of transcription. Interactions between C-terminal activation domains AD1 and AD2 with TBP may be critical for releasing the paused RNA polymerase tethered to TBP. Competition between the acidic H-domain of RNA polymerase II and HSF1 proposed by Lis and colleagues (Usheva et al 1992; Lis and Wu 1993; Mason and Lis 1997) may be regulated by the constraining influence of the NR domain, which binds to unidentified components of TFIID. According to the proposed model, conformational changes in the NR are critical for access to TBP by HSF1 activation domains AD1 and AD2. In addition to releasing paused polymerase, contacts between HSF1 and TFIIB, TBP, and TFIIA-γ may also facilitate reinitiation during multiple rounds of transcription. At present, this view of HSF1 function is based only on evidence for protein-protein interactions and is only one of several possible scenarios. Confirmation of the predictions inherent in the proposed model of HSF1 function requires further experimentation.
Acknowledgments
Research support was provided in part by USDA-CSRS grant 95371001617. This publication is Florida Agricultural Experiment Station Journal Series number R-07570. We express our appreciation to Dr. R.C. Patel for providing the mammalian 2-hybrid vectors, Dr. Robert Roeder for providing a TBP cDNA clone, Dr. Stephan G.E. Roberts and Dr. Michael R. Green for providing VP16 and TFIIB clones, and Dr. Carl Wu and Dr. Robert Kingston for providing cDNA clones for HSF1 and HSF2. We are indebted to Dr. James B. Flanegan and members of his laboratory for providing HeLa nuclear extracts. We also thank the Interdisciplinary Center for Biotechnology Research (ICBR) for DNA sequencing and Dr. Eva Czarnecka-Verner for a critical reading of this manuscript.
REFERENCES
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, and Struhl K. eds. 1991 Current Protocols in Molecular Biology, vol. 1. Wiley Interscience, Boston. [Google Scholar]
- Benjamin LR, Gilmour DS. Nucleosomes are not necessary for promoter-proximal pausing in vitro on the Drosophila hsp70 promoter. Nucleic Acids Res. 1998;26:1051–1055. doi: 10.1093/nar/26.4.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SA, Imbalzano AN, Kingston RE. Activator-dependent regulation of transcriptional pausing on nucleosomal templates. Genes Dev. 1996;10:1479–1490. doi: 10.1101/gad.10.12.1479. [DOI] [PubMed] [Google Scholar]
- Brown SA, Kingston RE. Disruption of downstream chromatin directed by a transcriptional activator. Gene Dev. 1997;11:3116–3121. doi: 10.1101/gad.11.23.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SA, Weirich CS, Newton EM, Kingston RE. Transcriptional activation domains stimulate initiation and elongation at different times and via different residues. EMBO J. 1998;17:3146–3154. doi: 10.1093/emboj/17.11.3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce JL, Price BD, Coleman CN, Calderwood SK. Oxidative injury rapidly activates the heat shock transcription factor but fails to increase levels of heat shock proteins. Cancer Res. 1993;53:12–15. [PubMed] [Google Scholar]
- Chiang CM, Roeder RG. Cloning of an intrinsic human TFIID subunit that interacts with multiple transcriptional activators. Science. 1995;267:53–536. doi: 10.1126/science.7824954. [DOI] [PubMed] [Google Scholar]
- Colgan J, Wampler S, Manley JL. Interaction between a transcriptional activator and transcription factor IIB in vivo. Nature. 1993;362:549–553. doi: 10.1038/362549a0. [DOI] [PubMed] [Google Scholar]
- Ge H, Roeder RG. Purification, cloning, and characterization of a human coactivator, PC4, that mediates transcriptional activation of class II genes. Cell. 1994;78:513–523. doi: 10.1016/0092-8674(94)90428-6. [DOI] [PubMed] [Google Scholar]
- Giardina C, Lis JT. Polymerase processivity and termination on Drosophila heat shock genes. J Biol Chem. 1993;268:23806–23811. [PubMed] [Google Scholar]
- Giardina C, Pérez-Riba M, Lis JT. Promoter melting and TFIID complexes on Drosophila genes in vivo. Genes Dev. 1992;6:2190–2200. doi: 10.1101/gad.6.11.2190. [DOI] [PubMed] [Google Scholar]
- Green M, Schuetz TJ, Sullivan EK, Kingston RE. A heat shock-responsive domain of human HSF1 that regulates transcription activation domain function. Mol Cell Biol. 1995;15:3354–3362. doi: 10.1128/mcb.15.6.3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan KL, Dixon JE. Eukaryotic proteins expressed in E. coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D. 1988 Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Hisatake K, Ohta T, Takada R, Guermah M, Horikoshi M, Nakatani Y, Roeder RG. Evolutionary conservation of human TATA-binding-polypeptide-associated factors TAFII31 and TAFII80 and interactions of TAFII80 with other TAFs and with general transcription factors. Proc Natl Acad Sci USA. 1995;92:8195–8199. doi: 10.1073/pnas.92.18.8195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman A, Sinn E, Yamamoto T, Wang J, Roy A, Horikoshi M, Roeder RG. Highly conserved core domain and unique N terminus with presumptive regulatory motifs in a human TATA factor (TFIID) Nature. 1990;346:387–390. doi: 10.1038/346387a0. [DOI] [PubMed] [Google Scholar]
- Jurivich DA, Sistonen L, Kroes RA, Morimoto RI. Effect of sodium salicylate on the human heat shock response. Science. 1992;255:1243–1245. doi: 10.1126/science.1546322. [DOI] [PubMed] [Google Scholar]
- Kim TK, Hashimoto S, Kelleher RJ, Flanagan PM, Kornberg RD, Horikoshi M, Roeder RG. Effects of activation-defective TBP mutations on transcription initiation in yeast. Nature. 1994;369:252–255. doi: 10.1038/369252a0. [DOI] [PubMed] [Google Scholar]
- Klemm RD, Goodrich JA, Zhou S, Tjian R. Molecular cloning and expression of the 31 kDa subunit of the human TFIID reveals interactions with VP16 and TFIIB that mediate transcriptional activation. Proc Natl Acad Sci USA. 1995;92:5788–5792. doi: 10.1073/pnas.92.13.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HS, Kraus KW, Wolfner MF, Lis JT. DNA sequence requirements for generating paused polymerase at the start of hsp70. Genes Dev. 1992;6:284–295. doi: 10.1101/gad.6.2.284. [DOI] [PubMed] [Google Scholar]
- Li B, Weber JA, Chen Y, Greenleaf AL, Gilmour DS. Analyses of promoter-proximal pausing by RNA polymerase II on the hsp70 heat shock gene promoter in a Drosophila nuclear extract. Mol Cell Biol. 1996;16:5433–5443. doi: 10.1128/mcb.16.10.5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis JT, Wu C. Protein traffic on the heat shock promoter: parking, stalling, and trucking along. Cell. 1993;74:1–20. doi: 10.1016/0092-8674(93)90286-y. [DOI] [PubMed] [Google Scholar]
- Mason PB Jr, Lis JT. Cooperative and competitive protein interactions at the hsp70 promoter. J Biol Chem. 1997;272:33227–33233. doi: 10.1074/jbc.272.52.33227. [DOI] [PubMed] [Google Scholar]
- Montecucco A, Savini E, Weighardt F, Rossi R, Ciarrocchi G, Villa A, Biamonti G. The N-terminal domain of human DNA ligase I contains the nuclear localization signal and directs the enzyme to sites of DNA replication. EMBO J. 1995;14:5379–5386. doi: 10.1002/j.1460-2075.1995.tb00222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai A, Morimoto RI. Characterization of a novel chicken heat shock transcription factor, heat shock factor 3, suggests a new regulatory pathway. Mol Cell Biol. 1993;13:1983–1997. doi: 10.1128/mcb.13.4.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton EM, Knauf U, Green M, Kingston RE. The regulatory domain of human heat shock factor 1 is sufficient to sense heat stress. Mol Cell Biol. 1996;16:839–846. doi: 10.1128/mcb.16.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien T, Lis JT. Rapid changes in Drosophila transcription after an instantaneous heat shock. Mol Cell Biol. 1993;13:3456–3463. doi: 10.1128/mcb.13.6.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozer J, Moore PA, Bolden AH, Lee A, Rosen CA, Lieberman PM. Molecular cloning of the small subunit of human TFIIA reveals functions critical for activated transcription. Genes Dev. 1994;8:2324–2335. doi: 10.1101/gad.8.19.2324. [DOI] [PubMed] [Google Scholar]
- Patel RC, Stanton P, McMillan NMJ, Williams BRG, Sen GC. The interferon-inducible double-stranded RNA-activated protein kinase self-associates in vitro and in vivo. Proc Natl Acad Sci USA. 1995;92:8283–8287. doi: 10.1073/pnas.92.18.8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabindran SK, Haroun RI, Clos J, Wisniewski J, Wu C. Regulation of heat shock factor trimer formation: role of a conserved leucine zipper. Science. 1993;259:230–234. doi: 10.1126/science.8421783. [DOI] [PubMed] [Google Scholar]
- Rasmussen E, Lis JT. Short transcripts of the ternary complex provide insight into RNA polymerase II elongational pausing. J Mol Biol. 1995;252:522–535. doi: 10.1006/jmbi.1995.0517. [DOI] [PubMed] [Google Scholar]
- Rasmussen EB, Lis JT. In vivo transcriptional pausing and cap formation on three Drosophila heat shock genes. Proc Natl Acad Sci USA. 1993;90:7923–7927. doi: 10.1073/pnas.90.17.7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SGE, Green MR. Activator-induced conformational change in general transcription factor TFIIB. Nature. 1994;371:717–720. doi: 10.1038/371717a0. [DOI] [PubMed] [Google Scholar]
- Roberts SGE, Ha I, Maldonado E, Reinberg D, Green MR. Interaction between an acidic activator and transcription factor TFIIB is required for transcriptional activation. Nature. 1993;363:741–744. doi: 10.1038/363741a0. [DOI] [PubMed] [Google Scholar]
- Rougvie AE, Lis JT. The RNA polymerase II molecule at the 5′ end of the uninduced hsp70 gene in D. melanogaster is transcriptionally engaged. Cell. 1988;54:795–804. doi: 10.1016/s0092-8674(88)91087-2. [DOI] [PubMed] [Google Scholar]
- Sandaltzopoulos R, Becker PB. Heat shock factor increases the reinitiation rate from potentiated chromatin templates. Mol Cell Biol. 1998;18:361–367. doi: 10.1128/mcb.18.1.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi YH, Kroeger PE, Morimoto RI. The carboxyl-terminal transcription domain of heat shock factor 1 is negatively regulated and stress responsive. Mol Cell Biol. 1995;15:4309–4318. doi: 10.1128/mcb.15.8.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shopland LS, Hirayoshi K, Fernandes M, Lis JT. HSF access to heat shock elements in vivo depends critically on promoter architecture defined by GAGA factor, TFIID, and RNA polymerase II binding sites. Genes Dev. 1995;9:275–2769. doi: 10.1101/gad.9.22.2756. [DOI] [PubMed] [Google Scholar]
- Stringer KF, Ingles CJ, Greenblatt J. Direct and selective binding of an acidic transcriptional activation domain to the TATA-box factor TFIID. Nature. 1990;345:783–786. doi: 10.1038/345783a0. [DOI] [PubMed] [Google Scholar]
- Usheva A, Maldonado E, Goldring A, Lu H, Houbavi C, Reinberg D, Aloni Y. Specific interaction between the nonphosphorylated form of RNA polymerase II and the TATA-binding protein. Cell. 1992;69:871–881. doi: 10.1016/0092-8674(92)90297-p. [DOI] [PubMed] [Google Scholar]
- Weber JA, Taxman DJ, Lu Q, Gilmour DS. Molecular architecture of the hsp70 promoter after deletion of the TATA box or the upstream regulation region. Mol Cell Biol. 1997;17:3799–3808. doi: 10.1128/mcb.17.7.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. Heat shock transcription factors: structure and regulation. Annu Rev Cell Dev Biol. 1995;11:441–469. doi: 10.1146/annurev.cb.11.110195.002301. [DOI] [PubMed] [Google Scholar]
- Xia W, Voellmy R. Hyperphosphorylation of heat shock transcription factor 1 is correlated with transcriptional competence and slow dissociation of active factor trimers. J Biol Chem. 1997;272:4094–4102. doi: 10.1074/jbc.272.7.4094. [DOI] [PubMed] [Google Scholar]
- Xiao H, Friesen JD, Lis JT. A highly conserved domain of RNA polymerase II shares a functional element with acidic activation domains of upstream transcription factors. Mol Cell Biol. 1994;14:7507–7516. doi: 10.1128/mcb.14.11.7507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan C-X, Czarnecka-Verner E, Gurley WB. Expression of human heat shock transcription factors 1 and 2 in HeLa cells and yeast. Cell Stress Chaperones. 1997;2:263–275. doi: 10.1379/1466-1268(1997)002<0263:eohhst>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo J, Baler R, Dahl G, Voellmy R. Activation of the DNA-binding ability of human heat shock transcription factor 1 may involve the transition from an intramolecular to an intermolecular triple-stranded coiled-coil structure. Mol Cell Biol. 1994;14:7557–7568. doi: 10.1128/mcb.14.11.7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo JR, Rungger D, Voellmy R. Multiple layers of regulation of human heat shock transcription factor 1. Mol Cell Biol. 1995;15:4319–4330. doi: 10.1128/mcb.15.8.4319. [DOI] [PMC free article] [PubMed] [Google Scholar]