

Abstract
The GAF domain is a simple module widespread in proteins of diverse function including cell signalling proteins and transcription factors. Its structure, typically spanning 150 residues, has three tiers; a basal layer of two or more α-helices, a middle layer of β-pleated sheet and a top layer formed by segments of the polypeptide that connect strands of the β-sheet. In structures of GAF domains in complex with their effectors, these polypeptide segments envelop the ligand enclosing it in a cavity whose base is formed by the β-sheet, so that ligand binding and release must be accompanied by conformational rearrangements of the distal portion of the structure. Descriptions of binding are presently limited by the absence of a GAF domain for which both liganded and unliganded structures are known. Earlier, we solved the crystal structure of the GAF domain of CodY, a branched chain amino acid and GTP responsive regulator of the transcription of stationary phase and virulence genes in Bacillus, in complexes with isoleucine and valine. Here, we report the structure of this domain in its unliganded form, allowing definition of the structural changes accompanying ligand binding. The core of the protein and its dimerisation interface are essentially unchanged in agreement with circular dichroism spectroscopy experiments that show that the secondary structure composition is unperturbed by ligand binding. There is however, extensive refolding of the binding site loops, with up to 15 Å movements of the coiled segment linking β3 and β4, such that in the absence of the ligand, the binding pocket is not formed. The implications of these structural rearrangements for ligand affinity and specificity are discussed. Finally, saturation transfer difference NMR spectroscopy showed binding of isoleucine, but not GTP, to the GAF domain suggesting that the two cofactors do not have a common binding site.
Keywords: GAF domain, conformational change, transcription regulation, Bacillus subtilis, branched chain amino acids
Introduction
GAF domains are so-named because they were first identified in mammalian cGMP-regulated phosphodiesterases, Anabaena adenylyl cyclases and the transcription factor FhlA from E. coli 1. These domains are widely distributed having now been identified in >7,400 proteins2. Besides roles in intracellular signalling and transcription regulation, GAF domain proteins are found in systems involved in light detection in bacteria, fungi and plants, and in the response to oxidative stress in bacteria. The GAF domain structure comprises three layers 3 (Figure 1A), a basal layer of two or more α-helices, a middle layer of four or more strands which form a mixed β-pleated sheet, and a distal layer of more variable structure most often made up of two extended polypeptide segments that connect the strands of the β-sheet.
Figure 1.

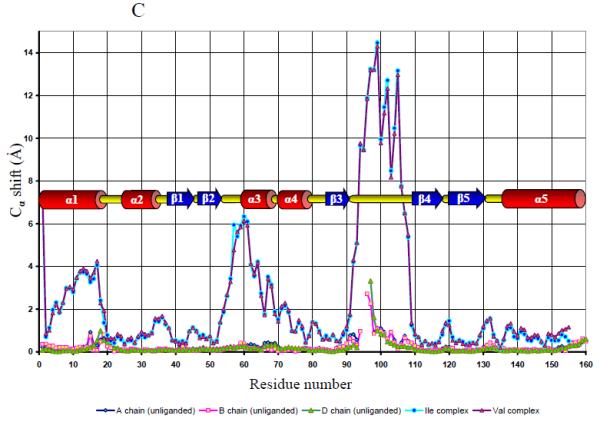
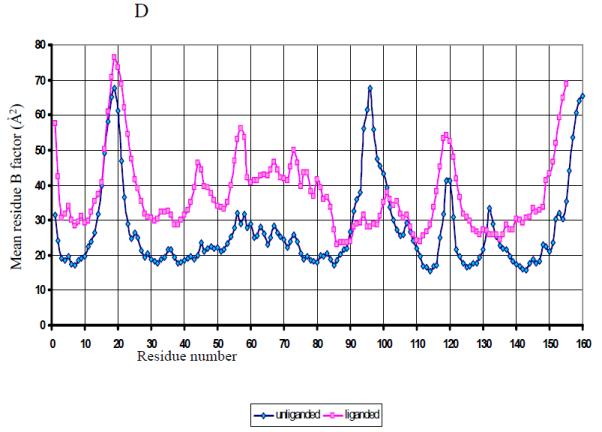
The structure of the unliganded GAF domain of CodY. A. Ribbon tracing of chain C from the asymmetric unit of the N-CodY crystal with the colouring ramped from the N-terminus in red to the C-terminus in magenta. The α-helices are labelled. B. The unliganded GAF domain dimer formed by chains C (cyan) and D (coral). The chain termini are labelled and distinguished by apostrophes. These panels were created using the program CCP4MG 39. C. A comparison of the four chains of the unliganded N-CodY structure with the single chains in the N-CodY-ILE and N-CodY-VAL structures. The Cα shift is plotted against residue number. Secondary structure elements as a function of residue number are indicated. D. A plot of mean residue temperature (B) factor versus residue number for the unliganded and liganded proteins. The mean residue B value is an average over all atoms of the residue and calculated using all four chains in the asymmetric unit for the unliganded GAF domain, and using the CodY-ILE and CodY-VAL chains for the liganded GAF domain.
GAF domains bind a range of ligands including linear and cyclic nucleotides, amino acids and porphyrin rings. In the majority of cases the nature of the ligand, if any, is unknown. In signal-transducing proteins such as DosT and the phytochrome Pr, the haem and bilin moieties are permanently bound cofactors 4; 5, while in the cell signalling system phosphodiesterases and adenylyl cyclases, and the transcription factors, effector binding is reversible. Methionine sulfoxide reductase from E. coli is the first example of a catalytically active GAF domain in which the ligand is chemically turned over 6. For a fourth category of GAF domain, those for which there is as yet no evidence of the ligand binding, the domains probably have purely structural roles3.
CodY is a highly conserved protein found in low G + C Gram positive bacteria 7. In B. subtilis, it has been shown to be a negative regulator of up to 200 genes encoding extracellular degradative enzymes, transporter proteins, catabolic enzymes, factors involved in genetic competence, antibiotic synthesis, chemotaxis and sporulation 8. In pathogenic Gram positive bacteria, CodY has a role in regulating virulence gene expression 7. At a number of promoters, it has been shown that in the presence of branched chain amino acids and/or GTP, CodY from B. subtilis binds to the regulatory region of the gene/operon to prevent transcription. CodY's function therefore is to turn off during vegetative growth the expression of genes required for adaptations to nutrient limitation. CodY repression is relieved when the concentration of these cofactors drops as the cells enter stationary phase. CodY also plays a role in central metabolism, repressing the metabolism of acetyl CoA through the Krebs cycle while stimulating conversion of pyruvate and acetyl CoA to lactate and acetate 9.
X-ray analysis of crystals of CodY fragments showed that the protein consists of an amino terminal GAF domain spanning residues 1-155 and a C-terminal winged helix-turn-helix (wHTH) domain encompassing residues 168-259 10. The wHTH domain is expected to mediate binding to DNA since it contains a recognisable DNA binding element, the helix-turn-helix, mutations in which impair DNA binding 11. The GAF domain is a dimer with the interface formed by the basal three α-helical bundle layers of each subunit. The ligand binding pockets are distal to the dimer interface and in separate structures are seen to contain isoleucine and valine. In these complexes, the branched chain amino acid is enclosed in a pocket whose base is formed by the β-sheet of the GAF domain and whose sides are formed by a pair of extended loops emerging from the sheet to wrap around the bound amino acid. It has been assumed that the GAF domain also binds GTP though there is limited direct evidence to support this assertion.
The overall fold of the GAF domain of CodY, its mode of dimerisation and the nature of the ligand binding site are fully consistent with the collective observations derived from analysis of some 20 or so GAF domain structures that have been reported to date3; 4; 5; 6; 12; 13; 14; 15; 16; 17; 18; 19; 20. Where bound, the ligands are consistently located in a pocket on the distal face of the β-sheet, clasped by two prominent connecting elements of structure emerging from the sheet. The ligands are extensively buried, with the GAF domain in a closed state that is evidently stabilised by ligand binding. The corresponding ‘open’ states and the ligand-dependent conformational changes accompanying binding are currently poorly defined because the unliganded GAF domain structures determined to date are of domains which are not known to bind a ligand. In the present study, we present the structure of the unliganded GAF domain of CodY allowing a conformational description of the mechanism of ligand binding to be derived. To complement this study we have explored the influence of ligand binding on the circular dichroism spectrum of the GAF domain. Finally, to determine whether GTP binds to the GAF domain of CodY, we carried out saturation transfer difference NMR spectroscopy.
Results and Discussion
Structure of the unliganded GAF domain of CodY
In efforts to grow crystals of guanine nucleotide complexes of a GAF domain fragment of CodY (N-CodY) encompassing residues 1-167, we obtained from solutions containing ammonium sulphate, sodium citrate, polyethylene glycol 400 and 20 mM GMP (or cGMP) crystals of what turned out to be unliganded protein 21. These crystals were strongly diffracting and data were collected to 1.7 Å spacing on beamline ID23-1 at ESRF, Grenoble (Table 1). We were able to solve their structure by molecular replacement using the N-CodY-ILE coordinate set (2b18) as a search model 10. The asymmetric unit of the unliganded GAF domain crystals contains four molecules (A-D) in the form of two dimers, AB and CD. The electron density maps allowed complete tracing of residues Met1 to Glu160 of chains B and C, with four and three residues missing from chains A and D, respectively, in a disordered segment between residues 93 and 98. The C-terminal residues 161-167 and the residues of the N-terminal histidine tag are similarly missing. In all four molecules, residues 11-18, comprising two turns of helix α1, were modelled in two conformations.
Table 1.
|
Data collection |
|
|---|---|
| Crystal | CodY-GAF domain (1-167) |
| X-ray source | ID23-1, ESRF |
| Wavelength (Å) | 0.97570 |
| Resolution range (Å) | 50.00 - 1.74 |
| Space group | P4322 |
| Unit-cell parameters (Å) |
a = b = 90.2, c = 205.6 α = β = γ = 90° |
| Number of unique reflections, | 85,808 / 8,202 |
| overall/outer shella | |
| Completeness (%), | 98.0 / 95.2 |
| overall/outer shella | |
| Redundancy, overall/outer | 7.2 / 3.7 |
| shella | |
| I /σ(I), overall/outer shella | 22.1 / 1.7 |
| Rmergeb (%), overall/outer | 8.3 / 61.2 |
| shella | |
|
Refinement and model statistics
|
|
| Resolution range (Å) | 31.89 – 1.74 |
| R-factorc (R-freed) | 0.187 (0.216) |
| Reflections (working/free) | 79,437 / 4,191 |
| Outer shell R-factorc (R-freed) | 0.279 (0.319) |
| Outer shell reflections (working/free) |
4,191 / 248 |
| Molecules/asymmetric unit | 4 |
| Number of protein non hydrogen atoms |
10930 |
| Number of water and small molecules atoms |
1528 |
| R.m.s. deviation from targetf | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 1.076 |
| Average B-factor (Å2) | 23.7 |
| Ramachandran plotg | 91.1/8.9/0.0/0.0 |
The outer shell corresponds to 1.74 – 1.80 Å.
Rmerge = ΣhklΣi|Ii - <I> |/ΣhklΣi <I> where Ii is the intensity of the ith measurement of a reflection with indexes hkl and <I> is the statistically weighted average reflection intensity.
R-factor = Σ∥Fo| - | Fc∥/Σ|Fo| where Fo and Fc are the observed and calculated structure factor amplitudes, respectively.
R-free is the R-factor calculated with 5 % of the reflections chosen at random and omitted from refinement.
Outer shell for refinement corresponds to 1.740 – 1.785 Å.
Root-mean-square deviation of bond lengths and bond angles from ideal geometry.
Percentage of residues in most-favoured/additionally allowed/generously allowed/disallowed regions of the Ramachandran plot.
Each chain has a GAF domain fold consisting of a central 5-stranded β-sheet (strand order β2–β1–β5–β4–β3), below which is a three-helix bundle consisting of two N- and one C-terminal α-helices, and above which are two extended loop segments, β2–β3, which contains the two short α-helices α3 and α4, and β3–β4 which is a meandering segment of polypeptide without obvious secondary structure (Fig. 1A). 148 equivalent Cα residues in the four chains can be overlaid pair-wise to give root mean squared positional deviations (rmsΔ) in the range 0.2 – 0.4 Å (Fig. 1C). The largest deviations (~ 2 Å) between molecules are located in the β3–β4 loop flanking the residues that are disordered in some of the chains. This region is also associated with high mean residue temperature factors (Fig. 1D). The AB and CD dimers are very similar with 295 equivalent Cα atoms superimposing to give an rmsΔ of 0.4 Å. The dimer interface is α-helical with helices α1 and α5 packing against their partners in the opposing subunits (Fig. 1B).
Structural changes upon ligand binding
When the structure of the unliganded GAF domain was compared to that of the isoleucine or valine liganded forms of the protein 10, extensive structural changes were seen to accompany ligand binding and release (Fig. 1C, Fig. 2A and B). Overlapping of 143-147 equivalent Cα atoms in these structures gave pair-wise rmsΔ values of 3.2 - 3.5 Å. As shown in Figure 2A, the structural changes are unevenly distributed with closer superposition of the chains in the β-sheet and helix bundle (α1, α2 and α5) regions. In contrast, the distal segments of the polypeptide between β2 and β3 and especially those between β3 and β4 undergo significant rearrangement (Fig. 1C and 2B). The largest deviations are at residues 57-61 and 92-108, located in the regions that form the ligand-binding site. In contrast to the disorder evident in the unliganded protein, residues 93-98 are well ordered in N-CodY-ILE and N-CodY-VAL. There are also differences associated with residues 11-18, which exhibit alternate conformations in the unliganded domain but a single conformation in the isoleucine- and valine-bound structures.
Figure 2.




Comparison of the structures of unliganded N-CodY and N-CodY-ILE. A and B; Orthogonal stereo views of the superposed structures of unliganded N-CodY (Chain C, light blue) and N-CodY-ILE (green). The chains were superimposed using the SSM superpose option in CCP4MG with the α3 and β4 helices of the β2–β3 loop excluded from the calculation. The ILE ligand in N-CodY-ILE is shown as spheres coloured by atom (C, green; N, blue; O, red). In B the α-helical subdomain has been omitted for clarity. The strands of the β-sheet are labelled. C and D; View as in B illustrating the altered juxtaposition of the ligand binding residues Arg61, Tyr75 and Thr96 and the alternative salt-bridging partners of Arg61, Glu101 and Asp104. E. Stereo superposition of the chains of unliganded N-CodY and N-CodY-ILE with residues Thr96-Phe106 coloured by residue with ramping from Thr96 (red) to Phe 106 (magenta). The residue side chains are in thick cylinder representation for the unliganded N-CodY and in thin cylinder representation for N-CodY-ILE. The ILE ligand is in ball-and-stick and residues 57 to 81 have been omitted for clarity. The large rearrangement of the β3–β4 segment is evident. F and G; Surface rendering of unliganded N-CodY and N-CodY-ILE respectively illustrating the absence of the isoleucine binding cavity in the former structure. The surface is coloured according to electrostatic potential with blue, positive and red, negative. All images were created in CCP4MG39.
In the unliganded protein, helix α3 and the loop segment preceding it have moved towards and into the vacant amino acid binding site (Fig. 2B; Supplementary Movie M1). Meanwhile, there is a dramatic twisting of the β3–β4 segment that transforms the conformation of this loop. The change in conformation is facilitated by the rotations of main chain bonds at the well-conserved Gly109 residue at the start of β4. Residues 102-107 are swept into the binding site to occupy volume otherwise occupied by residues 96-100 and the isoleucine ligand. The average displacement in Cα positions across this residue range (96 to 107) is 10 Å. (Figs 1C and 2G).
The side chain of Arg61, which in N-CodY-ILE is extended to form an ionic interaction with the ligand's carboxylate, adopts a more compact conformation in the unliganded form as helix α3 moves towards the vacant amino acid binding pocket (Fig. 2 C & D). In the absence of the ligand, its guanidinium group packs against the face of the aromatic ring of Tyr75, which also forms interactions with the ligand side chain in the complex. Arg61 and Tyr75 each form polar interactions with the carboxylate group of Asp104, which has moved 13 Å from its surface location in N-CodY-ILE to form salt-bridging interactions with NεH and Nη2H2 of the arginine, and a charge-dipole interaction with the phenolic –OH of the tyrosine (Fig. 2 C & D). These three side chains effectively fill what was the opening to the branched chain amino acid binding pocket. Arg61 forms a second salt-bridge in the liganded protein with Glu101, which orients the guanidinium group for its interaction with the isoleucine carboxylate. The Glu101 carboxylate has undergone a 15 Å displacement from its position in the complex to a surface location away from the binding site.
Meanwhile, Thr96, whose main chain carbonyl makes a charge-dipole interaction with the ligand's amino group, has moved away from the ligand-binding site to a peripheral surface location. The Cαs of residues 96 to 100, which contribute multiple contacts to the bound isoleucine in the liganded structure, have moved by between 8 and 13 Å in the unliganded molecule. The structural changes accompanying ligand binding also involve rearrangement of the local hydrophobic core. Leu105 which has substantial solvent exposure (123 Å2) in the liganded protein, becomes buried following a 16 Å movement of its side chain in the unliganded protein, where it occupies volume alongside that occupied by the ILE in the complex (Fig. 2G). Phe98 and Phe106, which are substantially buried in both structures, experience movements of similar magnitude across the face of the β-sheet, the result being that these two residues reside in alternate hydrophobic environments (Fig. 2G).
As a result of the rearrangements described above, the isoleucine-binding cavity no longer exists in the unliganded GAF domain. There is no opening into the unliganded protein at the location corresponding to the isoleucine binding pocket in the liganded form on the top face of the protein (Fig 2 E and F). Analysis of the GAF domain structure using the programme VOIDOO 22 showed that the unliganded N-CodY possesses a cavity whose entrance is bounded by the β2-loop-α3 segments (Fig 3A). This cavity has a glycerol molecule at its entrance and four water molecules 163, 167, 240 and 243 with which the –OH of Thr111 defines a hydrogen-bonding chain running approximately parallel to the plane of the β-sheet and perpendicular to the direction of its strands. Residues Phe40, Ile53, Gln56-Asn59, Met62, Lys63, Leu66, Leu105, Thr111, Ile 127 and Ser129 define this cavity.
Figure 3.

The cavity structure of the GAF domain of CodY. A) The unliganded protein showing the glycerol at the entrance to a cavity containing four water molecules which are shown as red spheres. These species together with Thr111, which is also shown, form a polar network. B) The isoleucine liganded protein with the bound ILE and the extended cavity containing four water molecules. The cavity structures were calculated using the program VOIDOO 22 and displayed as a molecular surface in the program PYMOL40.
In N-CodY-ILE, the side chain of Ile53 occludes the glycerol-binding site. Examination of the cavity structure in N-CodY-ILE shows that the BCAA binding pocket extends beyond the ILE ligand and deeper into the protein before turning around the side chain of Met62 towards the β2–α3 edge of the protein where it opens to the solvent as illustrated in Figure 3B. Part of the channel overlaps with the cavity described above for the unliganded protein and is lined by side chains of Asn38, Phe40, Ile57, Met62, Leu66, Phe71, Phe98 and Ser129, with residues Gln55, Glu58, Asn59, Leu105 and Phe106 surrounding the opening. The latter part of the channel is partially filled by a line of four water (12, 117, 91, 56) molecules. However the side entrance to the cavity in N-CodY-ILE is located on the opposite side of the β2–α3 segment from the pocket opening in the unliganded domain. Thus, access to this cavity would be facilitated by movements of the β3–β4 loop allowing entry and escape of ligands.
There appear to be no striking additional differences in the quaternary structures of the unliganded and liganded GAF domain dimers which can be overlaid with an rmsΔ of 3.9 – 4.0 Å for 280 equivalent Cα atoms. The dimer interface is formed by the packing of helices α1 and α5 in a parallel four-helix bundle in the unliganded GAF domain, resulting in the burial of 2100 Å2 of accessible surface area (Fig. 1B). The subunit interactions and the buried surface area are essentially identical in the liganded dimer.
The structural changes observed in unliganded N-CodY correlate well with earlier protease susceptibility studies which showed that BCAAs protected CodY from trypsin cleavage at Lys64, Arg69 and Arg130 and from chymotrysin cleavage at Tyr95 and Tyr145 10. Four of these residues are in conformationally sensitive locations; Tyr95 is in the most disordered segment of the polypeptide in unliganded N-CodY, Lys64 and Arg69 are situated on the mobile wings and Arg130 is in the β5–α5 loop.
Structure-Affinity Implications
The concentration of isoleucine is reported to vary from 10 mM in the well-fed B. subtilis cell to 0.5 mM during starvation 7. The function of CodY as a sensor of the nutritional status of the cell demands that its affinity for its cognate ligands matches that of their ambient concentration and this is consistent with Kd values, inferred indirectly from DNA binding experiments, in the millimolar range23. Thus, CodY has moderate to low affinity relative to leucine-isoleucine-valine binding protein, which serves as a receptor for the selective uptake of branched chain amino acids in bacteria, and isoleucyl tRNA synthetase which have Kd values in the micromolar range24; 25. Despite the differences in affinity, the structures of BCAA complexes of these proteins show similarity in the pattern of protein-ligand interactions with fulfilment of the hydrogen bonding / electrostatic potential of the ligand's amino and carboxylate groups and burial of the side chains in hydrophobic pockets 24; 25.
The structure presented here shows that in the absence of ligand, the BCAA binding pocket in CodY is not formed and that ligand binding must be accompanied by substantial structural reorganisation. There is structural disorder in the β3–β4 segment of the unliganded GAF domain as evidenced by (i) the high mean residue B-values for residues 90-100 (Fig. 1D) and (ii) the absence of interpretable electron density for residues 94-98 in two of the molecules of the asymmetric unit. Moreover, there are large displacements of residues 90-110 upon ligand binding in all four molecules (Fig. 1C). Disordered/unstructured regions of proteins that gain defined structure upon ligand binding are often associated with high specificity, low affinity binding. The underlying reasoning is that the strong favourable enthalpic protein-ligand interactions required to develop specificity are offset by unfavourable entropic changes, which lower affinity, as disordered regions of the protein refold around the ligand. In N-CodY, there is likely to be an additional enthalpic penalty associated with the dismantling of the packed structure of the β2–β3 and β3–β4 segments prior to their refolding in establishing the ligand bound conformation. This is reminiscent of the use of ligand binding energy to disrupt T-state stabilising interactions which lower ligand affinity in allosteric proteins 26.
Ligand binding in GAF domains
The structural data show that amino acid binding to the GAF domain of CodY is associated with drastic but localised changes in structure. These changes are confined to the distal wings that embrace the ligand and involve changes in tertiary but not secondary structure. This observation is consistent with what is known of the effects of ligand binding in other GAF domains. cGMP binding of the first (GAF A) of the tandem GAF domains of phosphodiesterase 5 (PDE5) was examined by CD spectroscopy which showed that the secondary structure content was similar irrespective of the presence of the ligand. In contrast, the broadening of GAF A associated NMR resonances in the cGMP-free form suggested that this domain is conformationally more flexible in the absence of the ligand 15. Qualitatively similar observations were made when the same techniques where used to study cGMP binding to the GAF-A domain of PDE6. In this system, CD spectroscopy experiments were additionally used to show that the presence of cGMP stabilises the domain, increasing the melting temperature associated with unfolding by 20 °C 14.
To provide a direct correlation of these observations with ligand binding in the GAF domain of CodY, we measured the CD spectrum of N-CodY in the presence and absence of isoleucine. As shown in Figure 4A, the spectra are very similar, consistent with the observations from the crystal structures that the secondary structure type and content is insensitive to the presence or absence of the ligand. CD spectroscopy is not able to reveal either the rigid body movements of helices α3 and β4, or the refolding of β3–β4 coiled segment in CodY. The extensive refolding of the loop structure seen here, may recur in the phosphodiesterase GAF domain systems upon cGMP/cAMP binding. However, as the phosphodiesterase GAF domains have Kds for cyclic nucleotides in the low nanomolar range14, the structural changes would not be needed as a mechanism for lowering the ligand affinity.
Figure 4.



Spectroscopic analysis of ligand binding to the GAF domain of CodY. A. CD spectra of CodY(1-155) (blue curve) and CodY(1-155) in the presence of 5 mM isoleucine (green curve). The profiles suggest that the secondary structure of CodY(1-155) does not change significantly upon binding of isoleucine. Analysis of these spectra using CDNN predicted the α–helical content to be 45 % and the β–sheet content to be 12% in the presence and absence of Ile. These predictions are in good agreement with the secondary structure composition observed in the crystal structure. B. STD NMR spectroscopy. Upper spectrum – 1H NMR reference spectrum of 10 μM CodY(1-155) with 1 mM isoleucine. Lower spectrum – STD spectrum of 10 μM CodY(1-155) with 1 mM isoleucine. Peaks in the STD spectrum correspond to isoleucine peaks in the 1H NMR reference spectrum. The spectrum shows saturation of the free isoleucine, resulting from interaction with CodY(1-155). C. Upper spectrum – 1H NMR reference spectrum of 10 μM CodY(1-155) with 1 mM GTP. The region of the spectrum shown is highlighted in the top panel. Lower spectrum – STD spectrum of 10 μM CodY(1-155) with 1 mM GTP. No peaks can be seen in the STD spectrum, showing no saturation of GTP hence no evidence of reversible interaction of GTP with CodY(1-155).
We superimposed structures of the CodY and PDE GAF domains, and compared the distal wing conformations in an attempt to determine whether the unliganded CodY GAF domain is systematically more similar to unliganded PDE GAF domains than it is to liganded PDE GAF domains. It was apparent that the GAF domains of CodY and the PDEs are too structurally divergent in the β2–β3 and β3–β4 segments for this comparison to be meaningful.
GTP binding in CodY
A fascinating characteristic of CodY is its capacity to respond to cofactors as different as branched chain amino acids and a nucleoside triphosphate. For the branched chain amino acids, the cofactor-binding site is clearly defined by the crystal structures of the N-CodY-ILE and N-CodY-VAL complexes, but the nature of the GTP binding site is unknown. GTP binding in CodY has eluded detailed biophysical analysis as a consequence of its low affinity and the strong absorbance of GTP in the UV range. Direct evidence of GTP binding was obtained by UV-induced cross-linking, although the site of GTP attachment was not mapped 27. It is well established that GTP has a marked effect on CodY-mediated transcriptional repression from various promoters in vitro. Moreover GTP has a specific effect on the binding of CodY to those CodY target promoters that have been analysed in DNase I foot-printing studies 28. GTP, as well as dGTP, enhance the protection conferred by CodY on the ilvB promoter, but the nature and extent of the CodY footprint is unaffected by GDP, ATP, CTP, TTP, ppGpp or pppGpp. Unlike ILE, GTP does not alter the pattern of partial proteolysis of CodY with a series of proteases, indicating that GTP binding does not mediate the same conformational changes as ILE 28. We extended these experiments to the isolated GAF domain and again observed an altered pattern of proteolysis with ILE but not with GTP (V. Colledge unpublished observations). These data point to differences in the mode of binding of the two ligands.
To explore whether GTP binds to the GAF domain of CodY, we performed saturation transfer difference (STD) NMR spectroscopy, a method well suited for monitoring the binding of low affinity ligands to proteins. In an STD experiment two data sets are collected, in the first a selective pulse is used to saturate the protein resonances at a particular frequency. The whole protein is rapidly saturated through fast spin diffusion. The frequency of the saturation pulse is chosen to avoid direct saturation of the free ligand, however ligand molecules bound to the protein behave as though they are part of the protein and are saturated indirectly through the spin diffusion process. During the saturation period, chemical exchange between the bound ligand and free ligand pools results in partial saturation of the free ligand pool. The second experiment is identical to the first, except that the saturation frequency is many kHz away from any proton resonances and neither the protein nor the ligand are saturated. A difference spectrum is obtained by subtracting the first spectrum from the second. Ligand peaks in the difference spectrum are a measure of the saturation of the free ligand pool and provide evidence of binding 29. We first monitored the binding of isoleucine to CodY(1-155) as shown in Figure 4B. The upper spectrum is a reference 1H NMR of 10 μM CodY(1-155) in the presence of 1 mM isoleucine showing the distinctive peaks associated with the small molecule ligand. The lower STD spectrum contains a set of peaks that correspond with peaks in the reference spectrum allowing these to be assigned to isoleucine. Since saturation transfer from protein to ligand can take place only in ligands that have been bound to the protein, these data are consistent with the binding of isoleucine to CodY(1-155). The same experiment was next performed with 10 μM CodY(1-155) and 1 mM GTP. The upper trace in Figure 4C shows part of the NMR spectrum with peaks corresponding to the GTP resonances. The STD spectrum however is featureless in this region, showing that the GTP ligand is not bound reversibly in this experiment.
While peaks in the difference spectrum for isoleucine are evidence of binding, the absence of peaks in the GTP difference spectrum is not necessarily evidence that GTP does not bind. If GTP binds strongly and there is little or no exchange between the bound and free ligand pools, then no peaks will be observed in the difference spectrum. The possibility of tight binding of GTP was tested through a competition experiment, in which STD data were first collected for CodY(1-155) in the presence of 1 mM isoleucine. 1 mM GTP was then added to the sample and a new STD experiment was carried out. If GTP is a strong binder, then it will displace the isoleucine from CodY(1-155), reducing or even removing the BCAA signals in the difference spectrum. In contrast, we found that the STD spectrum for CodY(1-155) with both isoleucine and GTP (data not shown) present was unchanged from that of CodY(1-155) and isoleucine alone, strongly suggesting that GTP does not bind to the GAF domain of CodY. This conclusion is supported by a recent site-directed mutagenesis study30.
Mechanistic Implications
It is evident from the structures described above that isoleucine and valine binding to the GAF domain of CodY is associated with large changes in conformation on the protein surfaces distal to the dimer interface. In the full-length molecule, these ligand binding events must be communicated to the DNA binding surfaces which are located in the C-terminal wHTH domain. The wHTH domain could experience the effects of BCAA binding either 1) by direct interaction of the wHTH with the conformationally sensitive distal surfaces of the GAF domain or 2) by propagation of the structural changes through the GAF domain to the C-terminal helix (α5) triggering movements that alter relative positions of the wHTH domains in the CodY dimer.
For ligand binding to alter the conformation of helix α5, the structural changes must be propagated across the β-sheet to the helix bundle (Figure 1). The dual conformations evident in residues 11-18 of all four chains in the unliganded N-CodY structure, but absent in CodY(1-155)-ILE and CodY(1-155)-VAL argue that the effects of ligand binding are experienced at the dimer interface. A further notable observation in this regard is that Gln132 in the loop preceding α5 is a Ramachandran outlier in N-CodY-ILE and N-CodY-VAL but not in any of the unliganded protein chains. Steric strain in well refined crystal structures usually has functional significance 31 suggesting that the altered main chain conformation at this residue may play a mechanistic role. Along with the residues of the binding site forming-loop β3–β4, the residues around Gln132 stand out as having higher mean B values in the unliganded GAF domain chains than they do in the liganded chains (Fig 1D). Future work will address the structural basis of DNA recognition by CodY and its modulation by branched chain amino acids and GTP.
Materials and methods
Protein preparation
An 530 bp PCR-amplified DNA fragment encoding the GAF domain of CodY spanning residues 1-167 with an N-terminal His6-tag was cloned using a ligation-independent technique into the plasmid pET-YSBLIC 32. The encoded N-CodY fragment described here is 12 residues longer than the fragment whose structure was solved in earlier work. The extra residues constitute a highly charged segment 156-REKAEEIEEEAR-167 10. The recombinant CodY-GAF domain was overproduced in E. coli BL21 and purified by Ni2+-chelation and gel filtration chromatography as described elsewhere33.
Crystallisation and X-ray data collection
Crystals of CodY-GAF domain were grown in hanging drops containing a 1:1 ratio of 20 mg/ml protein in 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 20 mM GMP (or cGMP) and 57 % saturated ammonium sulphate in 100 mM sodium citrate, pH 5.6, and 2 % PEG400. Crystals were cryocooled in a stream of liquid nitrogen-gas after a brief soak in mother liquor containing 25 % (v/v) glycerol. The crystals belonged to a primitive tetragonal space group, with unit-cell parameters a = b = 90.2 Å, c = 205.6 Å, α = β = γ = 90°. X-ray diffraction data were collected to 1.74 Å resolution on beamline ID23-1 of the European Synchrotron Radiation Facility (ESRF), Grenoble, and processed using the HKL2000 package 34. The estimated solvent content indicated four molecules in the asymmetric unit.
Structure solution and refinement
The structure of the unliganded form of N-CodY was solved by molecular replacement using the program MOLREP 35 with the coordinates of N-CodY-ILE (PDB ID: 2B18) from which residues 90 – 110 were truncated, as a search model. The search was complicated by (i) significant conformational differences between the liganded and unliganded forms of the protein (rmsΔ in Cα positions was 3.3 Å between individual subunits with a relative orientational difference of 14° in the dimers), (ii) the presence of four molecules in the asymmetric unit related by translational pseudosymmetry with a translational vector c/2 and (iii) two possible space groups (P41/322) with alternative origins in each. Perfect twinning tests showed no signs of twinning suggesting that the point group assignment was correct. Molecular replacement calculations in both space groups were performed. Numerous searches in P4322 using a single protomer as the search model gave no high-contrast solution and no low-contrast solution with reasonable packing. One of many searches in P4122 resulted in a low-contrast solution, in which protomers formed dimers topologically similar to dimers of the liganded structure and initial refinement with REFMAC 35 led to a significant drop in the Rfree factor to 0.38. The electron density was good enough to allow correction of the model with COOT 36 but further refinement did not provide any further improvement in the crystallographic statistics. To validate the space group assignment, the refined P4122 structure was expanded into P1 and rigid body refinement was performed. The potential internal symmetry within the P1 refined structure was examined by overlapping the structure with transformed structures after application of potential symmetry operations in the programme LSQKAB 37. This analysis revealed P4322 as the true space group. The P1 structure was transformed into P4322 by removing redundant molecules and further refined. The electron density maps were generally straightforward to interpret, a notable exception being residues between 93 and 98 which are not defined in chains A and D. Nevertheless the maps can be interpreted in this region for chains B and C (Supplementary Fig. S1A). A simulated annealing omit map38, with the β3–β4 loop (residues 91-111 of chain C) omitted from the model, clearly reveals the course of the polypeptide chain throughout this loop (Supplementary Fig. S1B). Details of the diffraction data and refinement statistics are given in Table 1.
The coordinates and structure factors have been deposited in the Protein Data Bank under the accession code 2GX5.
CD Spectroscopy
Circular dichroism (CD) data were acquired on a Jasco J810 spectrophotometer at 20°. Wavelength measurements were made using 6 μM CodY(1-155) in 50 mM phosphate buffer, pH 7.5, 50 mM NaCl in the presence and absence of 5mM Ile. A 1 mm path-length quartz cuvette was used. A total of 10 scans were collected for each sample, at a speed of 200 nm/min. Data were analysed using CDNN software.
NMR Spectroscopy
STD NMR experiments were carried out in 50 mM phosphate buffer, pH 7.5, 50 mM NaCl, 10 % (v/v) 2H2O. Spectra were acquired on a Bruker 700-MHz spectrometer using the Bruker pulse sequence stddiffesgp.2 at 25°. CodY(1-155) at 10 μM was incubated with ligand (Ile or GTP) at 1 mM. The sample was saturated at 6.2 ppm (for Ile) or 0 ppm (for GTP) with a 7 second saturation time. 256 scans were acquired. The saturation transfer spectrum was subtracted from the reference spectrum to obtain a difference spectrum. For the competition experiment, 1 mM Ile was incubated with 10 μM CodY(1-155) and the experiment performed as above with saturation at −1 ppm. 1 mM GTP was then added to the sample and the experiment was repeated.
Supplementary Material
Acknowledgements
We are grateful to Dr Jen Potts and Dr Andrew Leach for advice on the spectroscopic experiments, to Dr Johan Turkenberg and staff at the ESRF, Grenoble for help with data collection and to Prof Guy Dodson for advice on the manuscript. This work was supported by Grants from the BBSRC (BBS/B1213X to AJW) and the Wellcome Trust (082829/Z/07/Z to AJW) and from the US National Institute of General Medical Sciences (GM042219 to ALS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ACCESSION NUMBERS:
Coordinates and structure factors have been deposited in the Protein Data Bank with accession number 2GX5.
References
- 1.Avarind L, Ponting CP. The GAF domain: an evolutionary link between diverse phototransducing proteins. Trends Biochem. Sci. 1997;22:458–459. doi: 10.1016/s0968-0004(97)01148-1. [DOI] [PubMed] [Google Scholar]
- 2.Schultz JE. Structural and biochemical aspects of tandem GAF domains. Handb Exp Pharmacol. 2009;191:93–1109. doi: 10.1007/978-3-540-68964-5_6. [DOI] [PubMed] [Google Scholar]
- 3.Ho Y-WJ, Burden LM, Hurley JH. Structure of the GAF domain, a ubiquitous signalling motif and a new class of cyclic GMP receptor. EMBO J. 2000;20:5288–5299. doi: 10.1093/emboj/19.20.5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Essen L, Mailliet J, Hughes J. The structure of a complete phytochrome sensory module in the Pr ground state. Proc Natl Acad Sci USA. 2008;105:14709–14714. doi: 10.1073/pnas.0806477105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Podust LM, Ioanoviciu A, Ortiz de Montellano PR. 2.3 Å X-ray Structure of the Heme-Bound GAF Domain of Sensory Histidine Kinase DosT of Mycobacterium tuberculosis. Biochemistry. 2008;47:12523–12531. doi: 10.1021/bi8012356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin Z, Johnson LC, Weissbach H, Brot N, Lively MO, Lowther WT. Free methionine-(R)-sulfoxide reductase from Escherichia coli reveals a new GAF domain function. Proc Natl Acad Sci USA. 2007;104:9597–9602. doi: 10.1073/pnas.0703774104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sonenshein AL. CodY, a global regulator of stationary phase and virulence in Gram-positive bacteria. Curr. Opin. Microbiol. 2005;8:203–207. doi: 10.1016/j.mib.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Molle V, Nakaura Y, Shivers RP, Yamaguchi H, Losick R, Fujita Y, Sonenshein AL. Additional targets of the Bacillus subtilis global regulator CodY identified by chromatin immunoprecipitation and genomw-wide transcript analysis. J. Bacteriol. 2003;185:1911–1922. doi: 10.1128/JB.185.6.1911-1922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shivers RP, Dineen SS, Sonenshein AL. Positive regulation of Bacillus subtilis ackA by CodY and CcpA: establishing a potential heirarchy in carbon flow. Mol. Microbiol. 2006;62:811–822. doi: 10.1111/j.1365-2958.2006.05410.x. [DOI] [PubMed] [Google Scholar]
- 10.Levdikov VM, Blagova E, Joseph P, Sonenshein AL, Wilkinson AJ. The structure of CodY, a GTP- and isoleucine-responsive regulator of stationary phase and virulence in Gram-positive bacteria. J. Biol. Chem. 2006;281:11366–11373. doi: 10.1074/jbc.M513015200. [DOI] [PubMed] [Google Scholar]
- 11.Joseph P, Ratnayake-Lecamwasam M, Sonenshein AL. A region of Bacillus subtilis CodY protein required for interaction with DNA. J. Bacteriol. 2005;187:4127–4139. doi: 10.1128/JB.187.12.4127-4139.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez SE, Wu AY, Glavas NA, Tang X-B, Turley S, Hol WGJ, Beavo JA. The two GAF domains in phosphodiesterase 2A have distinct roles in dimerization and in cGMP binding. Proc. Natl. Acad. Sci. U. S. A. 2002;99:3260–13265. doi: 10.1073/pnas.192374899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez SE, Bruder S, Schultz A, Zheng N, Schultz JE, Beavo JA, Linder JU. Crystal structure of the tandem GAF domains from a cyanobacterial adenylyl cyclase: modes of ligand binding and dimerization. Proc. Natl. Acad. Sci. U. S. A. 2005;102:3082–3087. doi: 10.1073/pnas.0409913102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinez SE, Heikaus CC, Klevit RE, Beavo JA. The structure of the GAF A domain from phosphodiesterase 6C reveals determinants of cGMP binding, a conserved binding surface, and a large cGMP dependent conformational change. J. Biol. Chem. 2008;283:25913–25919. doi: 10.1074/jbc.M802891200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heikaus CC, Stout JR, Sekharan MR, Eakin CM, Rajagopal P, Brzovic PS, Beavo JA, Klevit RE. Solution structure of the cGMP binding GAF domain from phosphodiesterase 5: insights into nucleotide specificity, dimerization, and cGMP-dependent conformational change. J. Biol. Chem. 2008;283:22749–22759. doi: 10.1074/jbc.M801577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Handa N, Mizohata E, Kishishita S, Toyama M, Morita S, Uchikubo-Kamo T, Akasaka R, Omori K, Kotera J, Terada T, Shirouzu M, Y. S. Crystal structure of the GAF-B domain from human phosphodiesterase 10A complexed with its ligand, cAMP. J. Biol. Chem. 2008;283:19657–19664. doi: 10.1074/jbc.M800595200. [DOI] [PubMed] [Google Scholar]
- 17.Vannini A, Volpari C, Gargioli C, Muraglia E, Cortese R, De Francesco R, Neddermann P, Di Marco S. The crystal structure of the quorum sensing protein TraR bound to its autoinducer and target DNA. EMBO J. 2002;21:4393–4401. doi: 10.1093/emboj/cdf459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang R-G, Pappas T, Brace JL, Miller PC, Oulmassov T, Molyneaux JM, Anderson JC, Bashkin JK, Winans SC, Jochmimiak A. Structure of a bacterial quorum-sensing transcription factor complexed with pheromome and DNA. Nature. 2002;417:971–974. doi: 10.1038/nature00833. [DOI] [PubMed] [Google Scholar]
- 19.Wagner JR, Brunzelle JS, Forest KT, Vierstra RD. A light-sensing knot revealed by the structure of the chromophore-binding domain of phytochrome. Nature. 2005;438:325–331. doi: 10.1038/nature04118. [DOI] [PubMed] [Google Scholar]
- 20.Asen I, Djuranovic S, Lupas AN, Zeth K. Crystal Structure of SpoVT, the Final Modulator of Gene Expression during Spore Development in Bacillus subtilis. J. Mol. Biol. 2009;386:962–975. doi: 10.1016/j.jmb.2008.10.061. [DOI] [PubMed] [Google Scholar]
- 21.Blagova EV, Levdikov VM, Tachikawa K, Sonenshein AL, Wilkinson AJ. Crystallization of the GTP-dependent transcriptional regulator CodY from Bacillus subtilis. Acta Crystallogr. Sect. D. 2003;59:155–157. doi: 10.1107/s0907444902018358. [DOI] [PubMed] [Google Scholar]
- 22.Kleywegt GJ, Jones TA. Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Cryst. 1994;D50:178–185. doi: 10.1107/S0907444993011333. [DOI] [PubMed] [Google Scholar]
- 23.Belitsky BR, Sonenshein AL. Genetic and biochemiscal analysis of CodY-binding sites in Bacillus subtilis. J. Bacteriol. 2008;190:1224–1236. doi: 10.1128/JB.01780-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nureki O, Vassylyev DG, Tateno M, Shimada A, Nakama T, Fukai S, Konno M, Hendrickson TL, Schimmel P, Yokoyama S. Enzyme structure with two catalytic sites for double-sieve selection of substrate. Science. 1998;280:578–582. doi: 10.1126/science.280.5363.578. [DOI] [PubMed] [Google Scholar]
- 25.Trakhanov S, Vyas NK, Leucke H, Kristensen DM, Ma J, Quiocho FA. Ligand-free and -bound strucutres of the binding protein (LivJ) of the Escherichia coli ABC Leucine/Isoleucine/Valine transport system. Biochemistry. 2005;44:6597–6608. doi: 10.1021/bi047302o. [DOI] [PubMed] [Google Scholar]
- 26.Perutz M, Wilkinson AJ, Paoli M, Dodson G. The stereochemical mechanism of the cooperative effects in hemoglobin revisited. Annu Rev Biophys Biomol Struct. 1998;27:1–34. doi: 10.1146/annurev.biophys.27.1.1. [DOI] [PubMed] [Google Scholar]
- 27.Ratnayake-Lecamwasam M, Seror P, Wong KW, Sonenshein AL. Bacillus subtilis CodY represses early stationary-phase genes by sensing GTP levels. Genes Dev. 2001;15:1093–1103. doi: 10.1101/gad.874201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Handke L, Shivers R, Sonenshein AL. Interaction of Bacillus subtilis CodY with GTP. J. Bacteriol. 2008;190:798–806. doi: 10.1128/JB.01115-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayer M, Meyer B. Characterisation of ligand binding by saturation transfer difference NMR spectroscopy. Angew.Chem. Int. Ed. 1999;12:1784–1788. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1784::AID-ANIE1784>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 30.Villapakkam AC, Handke LD, Belitsky BR, Levdikov VM, Wilkinson AJ, Sonenshein AL. Genetic and biochemical analysis of the interaction of Bacillus subtilis CodY with branched chain amino acids. doi: 10.1128/JB.00818-09. Manuscript in preparation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herzberg O, Moult J. Analysis of the steric strain in the polypeptide backbone of protein molecules. Proteins. 1991;11:223–229. doi: 10.1002/prot.340110307. [DOI] [PubMed] [Google Scholar]
- 32.Fogg MJ, Wilkinson AJ. High-throughput approaches to crystallisation and crystal structure determination. Biochem. Soc. Trans. 2008;36:771–775. doi: 10.1042/BST0360771. [DOI] [PubMed] [Google Scholar]
- 33.Levdikov VM, Blagova EV, Brannigan JA, Cladiere L, Antson AA, Isupov MN, Seror SJ, Wilkinson AJ. The crystal structure of YloQ, a circularly permuted GTPase essential for Bacillis subtilis viability. J. Mol. Biol. 2004;340:767–782. doi: 10.1016/j.jmb.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 34.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Meth. Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 35.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum- likelihood method. Acta Crystallogr. Sect. D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Collaborative Computational Project, N The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 38.Brunger AT, Adams PD, Clore GM, Delano WL, Gros P, GrosseKunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software system for macromolecular structure determination. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 39.Potterton L, McNicholas S, Krissinel E, Gruber J, Emsley P, Cowtan K, Murshudov GN, Cohen S, Perrakis A, Noble M. Developments in the CCP4 molecular graphcs project. Acta Crystallogr. Sect. D. 2004;60:2288–2294. doi: 10.1107/S0907444904023716. [DOI] [PubMed] [Google Scholar]
- 40.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific LLC.; Palo Alto, CA, USA: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.