

Summary
Paired receptors are families of membrane proteins containing similar extracellular regions but differing in their potential for signalling with one type able to give inhibitory signals and the other activating. Inhibitory receptors could be good targets for pathogens to restrict immune responses against them. Here we suggest that, activating members may have evolved to counterbalance pathogens utilising the inhibitory pathway. Thus, if a pathogen utilises any part of the inhibitory receptor to down-regulate responses against itself, it may, because of similarities in structure, also bind the activating receptor and give an opposing signal. We evaluate recent structural data on SIRPα (signal regulatory protein) and LILRB1 (Leukocyte immunoglobulin-like receptor subfamily B member 1) showing evidence of pathogen pressure in non ligand binding regions of these receptors together with data on pathogen binding to PIR’s (paired Ig-like receptor) to provide support for this theory.
Introduction
The term ‘paired receptor’ is commonly used to describe families of membrane receptors that have very similar extracellular regions but different transmembrane and cytoplasmic regions. Indeed the latter are so different that they can give opposite signals (Lanier, 2001). One type can give inhibition through immunoreceptor tyrosine-based inhibition motifs (ITIM) in the cytoplasmic region. The other can activate through signalling proteins like DAP12 that contain immunoreceptor tyrosine-based activating motifs (ITAM), that are associated with the receptor via interactions through their transmembrane regions (Dietrich et al., 2000; Lanier, 2005). Paired receptors are often expressed by NK cells, others are restricted to myeloid cells but some are found on other leukocytes and also neuronal cells (Lanier, 2005). Paired receptors include SIRP, CD200R, KIR, Ly49, CD300, DCIR, PIR, PILR, TREM, LILR, Siglecs etc with many alternative names summarised in (Yamada and McVicar, 2008).
If the outcomes of engagement of paired receptors are so different and the extracellular regions so similar, then if their ligands are the same, then one gets the confusing situation of two outcomes for the presence of the same ligand. Often a cell will express both inhibitory and activating members. In most cases ligands for the inhibitory receptors are known and the activating receptors bind more weakly or not at all with quantitative data available for several pairs e.g. CD94-NKG2 (Vales-Gomez et al., 1999), CD200R (Hatherley et al., 2005), SIRP (Barclay and Brown, 2006), PILR (Tabata et al., 2008).
The inhibitory receptors generally interact with self proteins and provide a mechanism to limit cell activity as shown in NK cells (Lanier, 2005) and myeloid cells (Barclay and Brown, 2006). The roles of the activating receptors are less clear especially those on cells other than NK cells? Many of the paired receptor families are evolving rapidly, indicative of pressure from pathogens (Vilches and Parham, 2002). Although paired receptors on NK cells are heavily involved in the recognition of pathogen infected cells, others such as CD200R and SIRPα are involved in the control of myeloid cell activity (Barclay and Brown, 2006; Foster-Cuevas et al., 2004). How might pathogens drive this evolution? The targeting by pathogens of inhibitory receptors involved in cell regulation is clearly a sensible strategy from the pathogen’s point of view. We suggest a mechanism for paired receptors by which activating receptors have evolved to interact with those pathogens that target inhibitory receptors i.e. the activating receptors act as a counterbalance. Thus for paired receptors such as SIRP, if a pathogen targets the inhibitory receptor, it is probable that the pathogen also binds the activating receptor because of its similar extracellular regions, and hence nullifies the inhibitory effect (Hatherley et al., 2008). We discuss recent structural data on the SIRP family and LILRB1 together with pathogen binding data for other paired receptors with respect to this model.
The structure of SIRPα
SIRPα (also known as SHPS-1, BIT, CD172a (van den Berg et al., 2005)) is the inhibitory member of the SIRP family, SIRPβ the activating form associating with DAP12 and SIRPγ a third form that does not signal (Barclay and Brown, 2006). The N-terminal immunoglobulin superfamily (IgSF) domain of SIRPα (d1) interacts with the single IgSF domain of CD47, a widely distributed membrane protein. X-ray crystallography structures have been determined for the SIRP family members and CD47 (Hatherley et al., 2008; Hatherley et al., 2007; Nakaishi et al., 2008). SIRPα binds CD47 through loops in a manner analogous to binding of antigen by immunoglobulins and the T cell receptor and the failure of SIRPβ to bind to CD47 is due to subtle differences in these loops (Hatherley et al., 2008).
Polymorphisms in human SIRPα and ligand binding
SIRPα shows extensive polymorphism with 10-12 amino acid differences in domain 1 but only 0-2 differences in domains 2 and 3 between three mouse strains (Sano et al., 1999) and even more differences between the NOR (non-obese-resistant) and NOD (non-obese diabetic) mice (20 differences in domain 1 (Takenaka et al., 2007)). In humans, 37 different individuals showed 9 different SIRPα domain 1 sequences (Takenaka et al., 2007) (Figure 1) making this one of the most polymorphic genes in the immune system after MHC and KIR antigens (Vilches and Parham, 2002). In contrast SIRPβ and SIRPγ lack extensive polymorphism in domain 1 and the two alleles of SIRPβ have extensive differences throughout the sequence. Most researchers studying SIRPα have used one or other of two sequences (1 and 2 in Figure 1A). We have shown that both proteins bind CD47 with the same affinity (KD = ~1 μM (Hatherley et al., 2008) and unpublished data). The majority (14 out of 17) of the polymorphic residues in SIRPα d1 are strikingly distant from the contact site for CD47 (Figure 1A). However the three residues near the contact site are unlikely to affect binding as they are present in one or other of the two sequences which bind CD47 equally. The 5 residues not in either of the two standard sequences are distant from the binding site (shown in magenta in Figure 1A and B). Given this and the similarity of structures between the SIRPs away from the binding site (see (Hatherley et al., 2008)) and the failure of mutants outside of the binding site to affect CD47 binding (Hatherley et al., 2007; Liu et al., 2007), it is unlikely the polymorphisms in human SIRPα will markedly affect binding to CD47. CD47 itself lacks extensive polymorphisms.
Figure 1.


Polymorphisms and structure of SIRPα. (A) Alignments of the amino acid sequences of domain 1 of the two commonly studied SIRPα sequences (accession numbers CAA71403 and NP_542970 for sequence 1 and 2 respectively) together with polymorphisms identified in (Takenaka et al., 2007). The positions of polymorphic residues are indicated by giving the residue for each sequence and indicating those that differ from SIRPα (1) by green or for those different from either SIRPα (1) or SIRPα (2) by magenta. The residues that form contacts with CD47 are highlighted in red and the positions of the beta strands and one helical region are shown above the alignment. (B) The positions of the polymorphic residues are mapped onto the SIRPα domain 1 - CD47 extracellular domain co-crystal structure (Hatherley et al., 2008). The sidechains of the polymorphic residues are indicated with spheres using green and magenta to distinguish the polymorphic residues as in Figure 1A.
So what is driving the extensive SIRPα d1 polymorphism? In both human and mouse SIRPα d1 (see above) the majority of polymorphisms are non-synonomous ((Sano et al., 1999); ENSEMBL database) indicating selective pressure. The sidechains of the polymorphic residue (16 out of 17) are out-pointing and hence are likely to be involved in interactions with other molecules whereas inpointing residues might be expected to have affects on the folding of the domain. This is consistent with evolutionary pressure through interactions in this region. One possibility is that these variants have been selected not to react with particular pathogens (i.e. avoiding down regulation of the myeloid cell). Another comparable example is the Nkrp1 lectin-like paired receptor where the receptor but not the ligand shows extensive polymorphism and indeed it has recently been suggested this is to avoid virus decoys targeting this receptor (Carlyle et al., 2008). Thus, two mechanisms are involved in avoiding down regulation of activity – a high degree of polymorphism in the inhibitory receptor together with evolution of an activating receptor. One important concept is that the pathogen does not necessarily need to bind to the ligand binding site or indeed, domain, to perturb the SIRPα and give a signal – thus the sequence conservation in the remainder of the extracellular region is relevant (there is about 90% sequence identity between the extracellular regions of SIRPα, SIRPβ and SIRPγ) (Figure 2). There are no binding data for pathogens to SIRPs but there are data for other paired receptors (see below).
Figure 2.
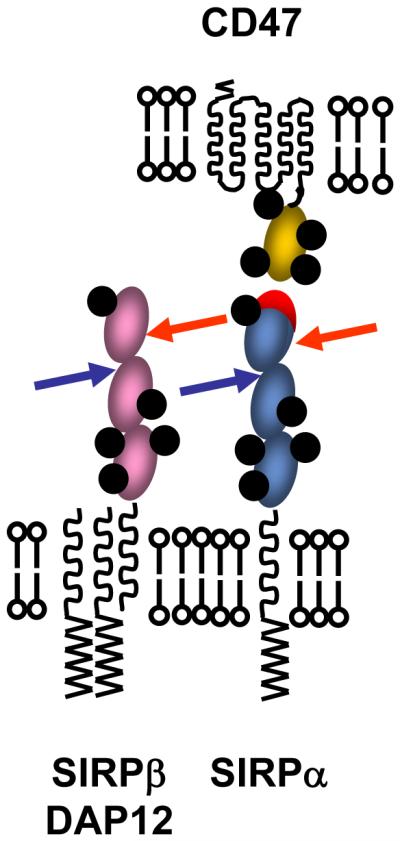
Sites distant from ligand binding regions of inhibitory receptors are attractive targets for pathogens. The cartoon shows SIRPα (inhibitory) together with its ligand CD47 with its binding face indicated in red. The arrows indicate how pathogens might target the regions of SIRPα not involved in CD47 binding, and then react with equivalent sites in the activating SIRPβ. N-linked glycosylation sites are shown in black but the area covered by carbohydrate would be much larger in 3D (Barclay et al., 1997). The close proximity of the membranes in immunological synapses when SIRPα engages CD47 would also hinder access by pathogens.
Pathogen binding to paired receptors
The concept that pathogens could bind paired receptors has been established with extensive functional data available for the Ly49 NK receptors found in mice. Resistance of B6 mice to mouse cytomegalovirus (mCMV) was found to be conferred by the expression of Ly49H, an activating receptor that binds to the mCMV MHC-like protein m157. This m157 protein also binds the inhibitory Ly49I receptor found in a susceptible mouse strain (129) but does not bind the Ly49I receptor found in B6 mice (Arase et al., 2002; Cheng et al., 2008; Smith et al., 2002). Arase et al suggested that whilst the inhibitory receptors have evolved for immunoregulation and preventing autoimmunity, the activating receptor may have evolved to recognise pathogen encoded ligands (Arase et al., 2002). Given that pathogens evolve faster than hosts one might imagine that the pathogen could easily evade these activating receptors. Our suggestion discussed above that pathogens might bind both types of paired receptors and the activating receptors act as a counterbalance, would fit with the data in this case and provide a general rationale for at least some paired receptors. With regard to Ly49, genetic divergence has led to strains with either inhibitory or activating receptors giving dramatic functional differences.
A variety of bacterial strains can bind both the inhibitory mouse paired Ig-like receptor (PIR-B) and at least one activating counterpart (PIR-A1) (Nakayama et al., 2007); a different repertoire of bacteria binds PIR-B (inhibitory) orthologs in humans, namely leukocyte immunoglobulin-like receptor subfamily B1 (LILRB1) and LILRB3 (or ILT2 and ILT5) (Nakayama et al., 2007). For the LILRB1 it seems likely that the bacterial binding site is distinct from the ligand binding as monoclonal antibody can block the former and not affect the latter (Nakayama et al., 2007). Another example of pathogen binding is the involvement of the inhibitory receptor PILRα in the uptake of herpes simplex virus (Satoh et al., 2008); however no data were given on the activating counterpart.
Evolution of paired receptors
There are data indicating the fast evolution of paired receptors both in terms of gene numbers and sequence diversity. Although the evolution of KIRs (killer cell immunoglobulin-like receptor) is tied to MHC Class I (the ligand) divergence (Vilches and Parham, 2002), recent data on the LILRB1 family support the concept that evolutionary pressures may act on sites other than the ligand binding as suggested above for SIRPα. The N-terminal two IgSF domains of LILRB1 form a binding site for MHC Class I antigens. LILRB1, like SIRPα, is polymorphic and 4 out of the 5 amino differences are in the ligand binding domains; three of these have no affect on ligand binding affinity as tested with a variety of MHC Class I ligands and the polymorphic residues are shown by X-ray crystallography to be outside the binding site (Kuroki et al., 2005). This parallels the observation discussed above for the SIRPs where there is evidence for pressure of change in the inhibitory receptor away from the ligand binding site and in support of the counterbalance theory.
It is likely that activating receptors evolved from the inhibitory receptor and they show much shorter half lives in evolution with greater variation in gene numbers (Abi-Rached and Parham, 2005; Carlyle et al., 2008; Vilches and Parham, 2002; Wilson et al., 2006). The advantage of greater numbers of activating receptors in protecting against more pathogens is balanced by greater risks for autoimmunity.
Another safeguard against unwanted membrane receptor signalling may be the utilisation of synapse like structures. It is notable how many receptors including many paired receptors are likely to span around four IgSF domains compatible with these interactions occurring at an immunological synapse (Barclay and Brown, 2006; Tsai and Discher, 2008; Wright et al., 2000). If an inhibitory receptor is only active in the immunological synapse, for instance due to the close proximity of other receptors and kinases to ensure phosphorylation of the cytoplasmic region, then it is likely to be inaccessible to pathogens which would be excluded from the immunological synapse because of their size. However once a virus has infected a cell, virally encoded proteins can be synthesised and reach the surface and subvert immune responses against the infected cell. This appears to be the case in Kaposi’s sarcoma virus that has acquired a CD200 homologue that can mimic the host protein by interacting with the inhibitory CD200R to down regulate macrophage activity (Foster-Cuevas et al., 2004). Nevertheless, for initial virus infections and bacterial infections, the immunological synapse may have a general, underappreciated role to prevent subversion of receptors once they have been activated in this specialised contact region.
Conclusion
The theory that the activating partner of a paired receptor might act as a counterbalance to pathogens that are utilising the inhibitory receptor, is strengthened by three sets of recent data; first, structural data on SIRP and LILRB1 recognition establish the basis of ligand binding, second genetic studies on SIRPs and other paired receptors reveal the extent of polymorphisms outside of the ligand binding domain, indicating evolutionary pressure for change in these regions and third, the concept that pathogens might bind regions outside the ligand binding area is strengthened by recent data showing direct binding of pathogens to paired receptors. The prevalence of paired receptors on myeloid cells would fit with involvement of pathogen recognition given that cells of the innate system will be the first line of attack against pathogens. Many paired receptors may use the activating receptor as a counterbalance in this way. In the counterbalance model the pathogen may evolve to bind the inhibitory receptor but have to accept the down side of hitting the activating receptor. It is notable that the number of activating receptors seems to vary in individuals (and rodent strains) and obviously a particular selection of inhibitory and/or activating members may be adventitious in the continuous battle between pathogen and host as observed with the Ly49 discussed above.
Acknowledgements
This work was supported by the MRC and the Wellcome Trust. We are grateful to Marion H Brown for helpful comments.
References
- Abi-Rached L, Parham P. Natural selection drives recurrent formation of activating killer cell immunoglobulin-like receptor and Ly49 from inhibitory homologues. J. Exp. Med. 2005;201:1319–1332. doi: 10.1084/jem.20042558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science. 2002;296:1323–1326. doi: 10.1126/science.1070884. [DOI] [PubMed] [Google Scholar]
- Barclay AN, Brown MH. The SIRP family of receptors and immune regulation. Nat. Rev. Immunol. 2006;6:457–464. doi: 10.1038/nri1859. [DOI] [PubMed] [Google Scholar]
- Barclay AN, Brown MH, Law SKA, McKnight AJ, Tomlinson MG, van der Merwe PA. Leucocyte Antigens Factsbook - second edition. Academic Press; London: 1997. [Google Scholar]
- Carlyle JR, Mesci A, Fine JH, Chen P, Belanger S, Tai LH, Makrigiannis AP. Evolution of the Ly49 and Nkrp1 recognition systems. Semin Immunol. 2008 doi: 10.1016/j.smim.2008.05.004. doi:10.1016/j.smim.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Cheng TP, French AR, Plougastel BF, Pingel JT, Orihuela MM, Buller ML, Yokoyama WM. Ly49h is necessary for genetic resistance to murine cytomegalovirus. Immunogenetics. 2008;60:565–573. doi: 10.1007/s00251-008-0313-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich J, Nakajima H, Colonna M. Human inhibitory and activating Ig-like receptors which modulate the function of myeloid cells. Microbes Infect. 2000;2:323–329. doi: 10.1016/s1286-4579(00)00294-x. [DOI] [PubMed] [Google Scholar]
- Foster-Cuevas M, Wright GJ, Puklavec MJ, Barclay AN. Human Herpesvirus-8 K14 protein mimics CD200 in down-regulating macrophage activation through CD200 receptor. J Virol. 2004;78:7667–7676. doi: 10.1128/JVI.78.14.7667-7676.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatherley D, Cherwinski HM, Moshref M, Barclay AN. Recombinant CD200 Protein Does Not Bind Activating Proteins Closely Related to CD200 Receptor. J. Immunol. 2005;175:2469–2474. doi: 10.4049/jimmunol.175.4.2469. [DOI] [PubMed] [Google Scholar]
- Hatherley D, Graham SC, Turner J, Harlos K, Stuart DI, Barclay AN. Paired receptor specificity explained by structures of signal regulatory proteins alone and complexed with CD47. Mol. Cell. 2008;31:266–277. doi: 10.1016/j.molcel.2008.05.026. [DOI] [PubMed] [Google Scholar]
- Hatherley D, Harlos K, Dunlop DC, Stuart DI, Barclay AN. The structure of the macrophage signal regulatory protein alpha (SIRPalpha) inhibitory receptor reveals a binding face reminiscent of that used by T cell receptors. J. Biol. Chem. 2007;282:14567–14575. doi: 10.1074/jbc.M611511200. [DOI] [PubMed] [Google Scholar]
- Kuroki K, Tsuchiya N, Shiroishi M, Rasubala L, Yamashita Y, Matsuta K, Fukazawa T, Kusaoi M, Murakami Y, Takiguchi M, et al. Extensive polymorphisms of LILRB1 (ILT2, LIR1) and their association with HLA-DRB1 shared epitope negative rheumatoid arthritis. Hum Mol Genet. 2005;14:2469–2480. doi: 10.1093/hmg/ddi247. [DOI] [PubMed] [Google Scholar]
- Lanier LL. Face off--the interplay between activating and inhibitory immune receptors. Curr. Opin. Immunol. 2001;13:326–331. doi: 10.1016/s0952-7915(00)00222-3. [DOI] [PubMed] [Google Scholar]
- Lanier LL. NK cell recognition. Annu. Rev. Immunol. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- Liu Y, Tong Q, Zhou Y, Lee HW, Yang JJ, Buhring HJ, Chen YT, Ha B, Chen CX, Yang Y, Zen K. Functional elements on SIRPalpha IgV domain mediate cell surface binding to CD47. J. Mol. Biol. 2007;365:680–693. doi: 10.1016/j.jmb.2006.09.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaishi A, Hirose M, Yoshimura M, Oneyama C, Saito K, Kuki N, Matsuda M, Honma N, Ohnishi H, Matozaki T, et al. Structural insight into the specific interaction between murine SHPS-1/SIRP alpha and its ligand CD47. J. Mol. Biol. 2008;375:650–660. doi: 10.1016/j.jmb.2007.10.085. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Underhill DM, Petersen TW, Li B, Kitamura T, Takai T, Aderem A. Paired Ig-like receptors bind to bacteria and shape TLR-mediated cytokine production. J. Immunol. 2007;178:4250–4259. doi: 10.4049/jimmunol.178.7.4250. [DOI] [PubMed] [Google Scholar]
- Sano S, Ohnishi H, Kubota M. Gene structure of mouse BIT/SHPS-1. Biochem. J. 1999;344(Pt 3):667–675. [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Arii J, Suenaga T, Wang J, Kogure A, Uehori J, Arase N, Shiratori I, Tanaka S, Kawaguchi Y, et al. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell. 2008;132:935–944. doi: 10.1016/j.cell.2008.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HR, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, Iizuka K, Furukawa H, Beckman DL, Pingel JT, et al. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. USA. 2002;99:8826–8831. doi: 10.1073/pnas.092258599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata S, Kuroki K, Wang J, Kajikawa M, Shiratori I, Kohda D, Arase H, Maenaka K. Biophysical characterization of O-glycosylated CD99 recognition by paired Ig-like type 2 receptors. J. Biol. Chem. 2008;283:8893–8901. doi: 10.1074/jbc.M709793200. [DOI] [PubMed] [Google Scholar]
- Takenaka K, Prasolava TK, Wang JC, Mortin-Toth SM, Khalouei S, Gan OI, Dick JE, Danska JS. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat. Immunol. 2007;8:1313–1323. doi: 10.1038/ni1527. [DOI] [PubMed] [Google Scholar]
- Tsai RK, Discher DE. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J. Cell Biol. 2008;180:989–1003. doi: 10.1083/jcb.200708043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vales-Gomez M, Reyburn HT, Erskine RA, Lopez-Botet M, Strominger JL. Kinetics and peptide dependency of the binding of the inhibitory NK receptor CD94/NKG2-A and the activating receptor CD94/NKG2-C to HLA-E. EMBO J. 1999;18:4250–4260. doi: 10.1093/emboj/18.15.4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg TK, van Beek EM, Buhring HJ, Colonna M, Hamaguchi M, Howard CJ, Kasuga M, Liu Y, Matozaki T, Neel BG, et al. A nomenclature for signal regulatory protein family members. J. Immunol. 2005;175:7788–7789. doi: 10.4049/jimmunol.175.12.7788. [DOI] [PubMed] [Google Scholar]
- Vilches C, Parham P. KIR: diverse, rapidly evolving receptors of innate and adaptive immunity. Annu. Rev. Immunol. 2002;20:217–251. doi: 10.1146/annurev.immunol.20.092501.134942. [DOI] [PubMed] [Google Scholar]
- Wilson MD, Cheung J, Martindale DW, Scherer SW, Koop BF. Comparative analysis of the paired immunoglobulin-like receptor (PILR) locus in six mammalian genomes: duplication, conversion, and the birth of new genes. Physiol Genomics. 2006;27:201–218. doi: 10.1152/physiolgenomics.00284.2005. [DOI] [PubMed] [Google Scholar]
- Wright GJ, Puklavec MJ, Willis AC, Hoek RM, Sedgwick JD, Brown MH, Barclay AN. Lymphoid/neuronal cell surface OX2 glycoprotein recognizes a novel receptor on macrophages implicated in the control of their function. Immunity. 2000;13:233–242. doi: 10.1016/s1074-7613(00)00023-6. [DOI] [PubMed] [Google Scholar]
- Yamada E, McVicar DW. Paired receptor systems of the innate immune system. Curr Protoc Immunol. 2008 doi: 10.1002/0471142735.ima01xs81. Chapter 1, Appendix 1X. [DOI] [PMC free article] [PubMed] [Google Scholar]