

Abstract
A significant enrichment of CD4+Foxp3+ T cells (Tregs) is frequently observed in murine and human carcinomas. As Tregs can limit effective anti-tumor immune responses, thereby promoting tumor progression, it is important that the mechanisms underpinning intra-tumoral accumulation of Tregs are identified. Due to evidence gathered mostly in vitro, the conversion of conventional T cells (Tconv) into Tregs has been proposed as one such mechanism. We assessed the contribution of conversion in vivo by analyzing the TCR repertoires of Tconv and Treg cells in carcinogen-induced tumors in mice. Our results indicate that the TCR repertoires of Treg and Tconv cells within tumor-infiltrating lymphocytes (TILs) are largely distinct. Indeed, the cell population with the greatest degree of repertoire similarity with tumor-infiltrating Tregs was the Treg population from the tumor draining lymph node. These findings demonstrate that conversion of Tconv cells does not contribute significantly to the accumulation of tumor-infiltrating Tregs; rather, Tconv and Treg cells arise from different populations with unique TCR repertoires. Enrichment of Tregs within TILs most likely, therefore, reflects differences in the way that Treg and Tconv cells are influenced by the tumor microenvironment. Elucidating the nature of these influences may indicate how the balance between tumor-infiltrating Treg and Tconv cells can be manipulated for therapeutic purposes.
Keywords: Conversion, methylcholanthrene, regulatory T cell, TCR repertoire, tumor immunology
Introduction
There is evidence that T cells can infiltrate tumors and limit their progression (1). In turn, it is thought that successful tumor cells in patients with cancer are those that evolve strategies to avoid the attention of the immune response, either through down-modulation of recognition molecules or indirectly through immune subversion. Data from experiments performed using mouse models directly support this hypothesis (reviewed in 2). One mechanism through which the immune response to tumors might be subverted is by enrichment of regulatory T cells (Tregs) within the tumor tissue and tumor draining lymph nodes.
The normal functions of Tregs are to maintain immune homeostasis, prevent autoimmunity and limit immunopathology (reviewed in 3). However, many groups have reported, in both studies of mouse models and patients with cancer, that tumor development is often associated with an enrichment of Tregs in peripheral blood, local lymph nodes and tumor tissue (reviewed in 4). Using the chemical carcinogen 3-methylcholanthrene (MCA), we previously examined the impact of Tregs on tumor immunosurveillance (5). We found that Tregs are significantly enriched in MCA-induced tumors (fibrosarcomas) compared to lymphoid tissue (approximately 50% of CD4+ T cells in tumors express Foxp3 compared to approximately 15% in lymph nodes, p<0.0001) and that even a partial and transient depletion of these cells results in a marked reduction in tumor incidence. Along with other studies, this observation supports the hypothesis that tumors can utilize Tregs for their own advantage by promoting Treg activity (6-9). As well as their potential for limiting the effectiveness of tumor immunosurveillance, it is likely that Tregs also represent a significant obstacle to successful immunotherapy (reviewed in 10). It is important, therefore, to understand the factors that lead to the enrichment of Tregs during tumor development to enable the development of inhibitory strategies.
The majority of Tregs that express the transcription factor Foxp3 (termed naturally occurring Tregs) are generated in the thymus as a distinct cell lineage (11-13). Foxp3+ Tregs may also be generated in the periphery through the conversion of conventional Foxp3− T cells (Tconv) into Foxp3+ Tregs; these have been termed adaptive (or induced) Tregs (reviewed in 14). Experiments using mice with restricted TCR repertoires and limited studies in humans have suggested that the TCR repertoires of Tregs and Tconv cells are largely distinct, overlapping by approximately 10 – 20% (11, 12, 15, 16). It has been suggested that Tconv cells with TCRs overlapping with those of the Treg repertoire represent cells that recognize self-antigens (11). Adaptive Tregs that have arisen by conversion of Tconv cells in the periphery may also contribute to the observed overlap in the TCR repertoires. The role and significance of adaptive Tregs in vivo, however, is unclear as the majority of the peripheral Treg repertoire is also represented within the thymic Treg repertoire (11). Furthermore, studies of diabetogenic NOD mice showed no overlap between the TCR repertoire of Tregs and Tconv, indicating a lack of conversion in autoimmune disease (17). However, studies showing that tumors can facilitate conversion of Tconv into Treg in vitro (18) and, more recently, in vivo, imply that conversion contributes significantly to the accumulation of tumor-infiltrating Tregs (19, 20). Whilst informative, the tumor models described here often use tumor cell lines and track the fate of adoptively transferred T cells, but omit interactions between the immune system and the tumor during the early stages of tumor development; the reciprocal influences of the immune system and the tumor during these stages are likely to have a significant impact on the nature of their relationship during the period of tumor outgrowth. Here, we utilize the MCA tumor induction model described above, which takes these early interactions into account, to analyze the TCR repertoires of Tconv and Treg cells in order to assess definitively the contribution of conversion to Treg enrichment in tumors.
Materials and Methods
Mice
Mice expressing GFP under the control of the Foxp3 promoter (Foxp3-GFP mice) were obtained from Professor Alexander Rudensky, University of Washington and have been described previously (21). Mice were housed in accordance with UK Home Office regulations under specific pathogen-free conditions.
Tumor induction
Foxp3-GFP mice were injected subcutaneously in the left hind leg with 400μg of 3-methylcholanthrene (MCA; Sigma-Aldrich) in 100μl of olive oil under general anesthetic. Mice were monitored for tumor development weekly for up to 6 months. Tumor-bearing mice were culled when their tumors were between 1cm and 2cm in diameter, typically within 100-150 days after MCA injection.
Antibody staining and cell sorting
Single cell suspensions of tumor, spleen, and inguinal lymph nodes were prepared by filtering tissues through 70μm cell strainers (BD). The inguinal lymph node from the tumor (left) side of the mouse was taken as the tumor draining lymph node and the contralateral inguinal lymph node was considered to be a non-draining lymph node. For cell sorting, cells were stained with the following fluorescently labeled monoclonal antibodies: anti-CD4 Pacific Blue (BD), anti-CD25 PE (eBioscience), anti-CD44 APC (BD) and anti-CD62L PE-Cy7 (eBioscience). Cell sorting was performed using a MoFlo cell sorter (Dako Cytomation) or a customized 20-parameter FACSAria II flow cytometer (BD). CD4+GFP+ lymphocytes were sorted as Tregs and CD4+GFP− lymphocytes were sorted as Tconv. Where indicated, Tconv were further purified into antigen-experienced (CD44hiCD62Llo) Tconv and naïve (CD44loCD62Lhi) Tconv. Post-sort purity was >98% in all cases.
Immunohistochemistry
After removal, tumors were fixed in 10% neutral buffered formalin (NBF) and embedded in paraffin wax. 5μm thick sections were dewaxed and antigen retrieval was performed in 10mM sodium citrate buffer pH6. Sections were equilibrated in PBS prior to blocking peroxidase activity with Peroxidase Suppressor (ThermoScientific). Non-specific antibody binding was blocked by incubating sections with 2.5% horse serum (Vector). TGFβ was detected using rabbit anti-mouse TGFβ (sc-146, Santa Cruz Biotechnology, Santa Cruz, USA) followed by anti-mouse/rabbit Immpress and visualized using Vector SG. Foxp3 was detected subsequently using rat anti-Foxp3 (FJK-16, eBioscience) followed by anti-rat Immpress and visualized using Impact DAB (VectorLabs, Burlingame, USA). Equivalent concentrations of rabbit IgG and rat IgG2a were used as control antibodies. Sections were dehydrated and mounted in DPEX. Photomicrographs were taken using a NIKON microscope and digital camera.
TCR clonotyping
For analysis of specific segments of the TCR repertoire, RNA was extracted from sorted cells using a column-based RNA extraction kit (RNeasy micro, Qiagen). cDNA was reverse transcribed from 30-200ng of total RNA using Superscript III (Invitrogen) with random hexamers. TCR sequences were amplified by PCR using either a TRBV13-2 specific primer (5′-GGTGACATTGAGCTGTAAT-3′) paired with a common (TRBC) primer (5′-CACTGATGTTCTGTGTGACAG-3′) or using a TRBV13-2 specific primer (5′-GGTGACATTGAGCTGTAATCAGAC-3′) paired with a TRBJ2-5 specific primer (5′-TAACACGAGGAGCCGAGTG-3′). Agarose gel purified PCR products were cloned into pCR®2.1-TOPO® plasmid vector (Invitrogen). The PCR products were then transformed into chemically competent E. coli (TOP10; Invitrogen) and grown on selective LB plates (100μg/ml ampicillin with IPTG and X-gal for blue/white screening). Up to 96 colonies per sort were picked and DNA sequenced (Beckman Coulter Genomics). The TRB gene usage and CDR3 amino acid composition was established using IMGT/V-QUEST software. For analysis of the total TCR repertoire, an unbiased template-switch anchored RT-PCR was used as described previously (22).
Statistical analysis
The level of similarity between the different TCR repertoires was measured using the Morisita-Horn (MH) similarity index. This unitless index ranging from 0 to 1 takes into account the number of shared sequences between two repertoires as well as the contribution of those shared sequences to each repertoire. The EstimateS software package was used to calculate the MH values (23).
Results
Enrichment of CD4+Foxp3+ T cells in tumor and tumor draining lymph node
We have reported that Foxp3+ cells are enriched (approximately 40-50% of CD4+ T cells in tumors express Foxp3) within the CD4+ TILs isolated from MCA-induced fibrosarcomas (5). This preliminary observation was replicated and extended in this study, confirming that there is a significant accumulation of Tregs in the tumor (p = 0.0002; Figure 1A). Strikingly, mice partially depleted of Tregs showed a significant reduction in tumor incidence (p = 0.0004; Figure 1B). Given the clear relevance of Tregs in this model, we utilized MCA-induced tumors to determine whether conversion of conventional CD4+Foxp3− cells into CD4+Foxp3+ cells accounted for Treg enrichment within TILs. This possibility was considered likely, as TGFβ is readily detectable in MCA-induced tumors (Figure 1C) and has been shown to induce Foxp3 expression in CD4+CD25− cells (24, 25). We surmised that if Treg enrichment in tumors is due to the conversion of CD4+Foxp3− cells into CD4+Foxp3+ cells, then the degree of overlap between their TCR repertoires would be significantly higher in the tumor compared to other lymphoid tissues where Treg enrichment is not observed. Thus, we compared the extent of TCR repertoire overlap in Treg and Tconv populations isolated from tumor, spleen, non-draining inguinal lymph node and draining inguinal lymph node. For this purpose, we purified the CD4+ T cells from tumor-bearing Foxp3-GFP TCR transgenic mice. This was important as the Tregs and Tconv cells could not be distinguished by CD25 expression. Whilst around 80 – 95% of tumor-infiltrating CD4+Foxp3+ cells express CD25, CD25 expression is also observed on approximately 15% of the corresponding CD4+Foxp3− population (Figure 1D).
Figure 1. CD4+Foxp3+CD25+ regulatory T cells are enriched within TGFβ containing MCA-induced tumors and tumor draining lymph nodes.
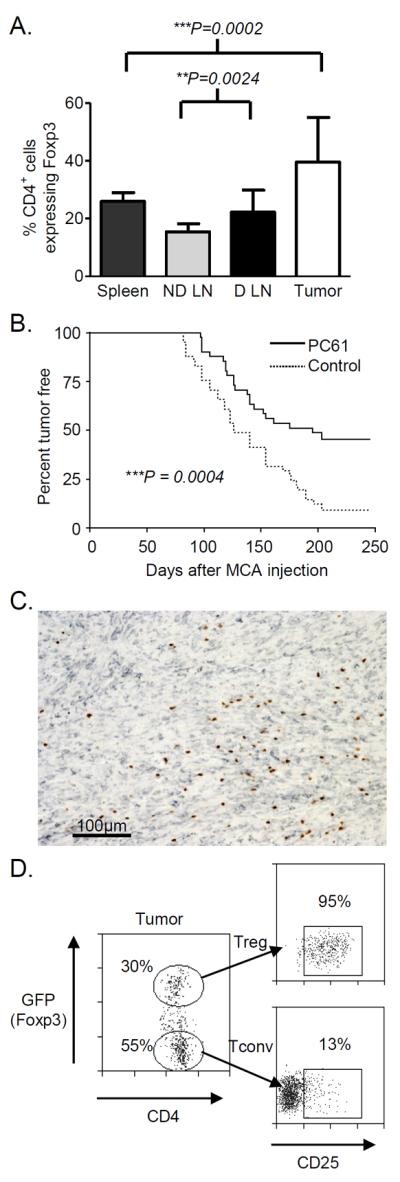
Spleen, tumor, non-draining and draining inguinal lymph nodes were excised from mice with MCA-induced tumors. Cells were analyzed for CD4, CD25 and Foxp3 expression by flow cytometry. (A) The percentages of CD4+ lymphocytes expressing Foxp3 are shown as mean +/− SD. Statistical significance was assessed using the paired T test (tumor and spleen n = 23, lymph nodes n = 11; 5 independent experiments). (B) Mice (n = 41) were given 500μg of anti-CD25 (PC61) or control antibody prior to MCA injection and monitored for tumor development. Statistical analysis was performed using the log rank method. (C) Serial sections from MCA-induced tumors were stained with antibodies specific for TGFβ (grey) and Foxp3 (brown). The stain shown is representative of 6 MCA tumors from 3 independent experiments. (D) Representative flow cytometry plots showing CD4+GFP(Foxp3)+ T cells infiltrating an MCA tumor and the gating strategy used for sorting. Gated cells were analyzed for CD25 expression. The percent of gated cells expressing CD25 is shown.
TCR repertoires of CD4+Foxp3− and CD4+Foxp3+ T cells in tumor-bearing mice
We aimed to analyze the repertoire of CD4+Foxp3− cells and CD4+Foxp3+ by sequencing individual TCRs expressed by T cells present within the different anatomical locations described above. For this purpose, we focused our analysis on a representative Vβ chain subset. Initially CD4+Foxp3− and CD4+Foxp3+ cells from MCA tumor-bearing mice were screened for T cell receptor β chain variable domain (TRBV) subset usage with antibodies specific for TRBV 2, 13-1/2, 13-3, 15 and 16 (Supplementary Figures 1 & 2). We found no statistically significant difference in Vβ subset usage between Treg and Tconv cells sorted from MCA tumors. Both cell populations appeared to have a broad range of TRBV gene usage with no skewing towards any particular subset within the tumor or the spleen of tumor-bearing mice. These findings were confirmed at the molecular level in tumor, spleen and non-draining inguinal lymph node using an unbiased template-switch anchored RT-PCR (Supplementary Figure 3). Given that many thousands of unique clonotypes can comprise a single TRBV family, we expanded the investigation to include high resolution clonotypic analysis of one Vβ subset. For this detailed analysis, the TRBV13-2 subset was selected as it was well represented (up to 30%) within the total Vβ TCR repertoire.
In agreement with previous reports (11, 12), we observed that the TCR repertoires of Treg and Tconv in the spleen and the tumor non-draining lymph node of tumor-bearing mice were largely distinct (Figure 2A and 2B). In the spleen, out of 68 different TRBV13-2 Treg clonotypes, only 1 overlapped with the Tconv repertoire; this accounted for only 1.3% of the Treg TCR repertoire. Similarly, none of the 73 different Treg clonotypes overlapped in the non-draining lymph node. Thus, the TCR repertoires of Tconv and Treg cells are generally distinct. Interestingly, the lack of overlap between the Tconv and Treg TCR repertoires was also reflected in the tumor draining lymph node (Figure 2C), where less than 2.5% of the Treg TCR repertoire overlapped with the Tconv TCR repertoire, as well as in the tumor (Figure 2D), where only 2 out of 77 different Treg clonotypes overlapped with Tconv TCRs. These 2 clonotypes only accounted for around 3% of the Treg TCR repertoire; thus, more than 95% of the tumor-infiltrating Treg TCR repertoire was distinct compared with the co-localized Tconv TCR repertoire. Similar patterns were also observed in analyses of other mice, indicating no statistically significant increase in overlap between the two T cell repertoires in tumors compared to the other sites (Supplementary Figures 4 and 6).
Figure 2. TRBV13-2 TCR repertoires of Tconv and Treg are non-overlapping in tumor-bearing mice.

Tconv (CD4+Foxp3−) and Treg (CD4+Foxp3+) sorted cells were analyzed by TCR clonotyping. CDR3 amino acid sequences of the TRBV13-2 subset were determined and used to identify individual TCRs for each T cell subset from the (A) spleen, (B) non-draining lymph node, (C) draining lymph node and (D) tumor of mice with MCA-induced tumors. Each graph displays the different TCR sequences observed (x axis) within the Treg (filled & above) and Tconv (open & below) repertoires, and the frequency (y axis) of each sequence within that subset. The total number of TCR sequences analyzed is displayed as n=, with the number of unique TCR sequences observed in parentheses. The proportion of the Treg repertoire that shared sequences with the Tconv repertoire is also shown. The data are representative of two independent experiments in two mice. LN, lymph node.
To quantify the similarity between the TCR repertoires from the CD4+Foxp3− and CD4+Foxp3+ subsets, we used the Morisita-Horn similarity index. The levels of similarity between the TRBV13-2 TCR repertoires of Tconv and Treg were consistently low (MH < 0.04) throughout all four tissues measured (Figure 3A). The highest level of similarity was actually observed between the Treg repertoires from draining lymph node and tumor (Figure 3B). The Treg repertoire from the tumor was on average ~20% (MH = 0.19) similar to the tumor draining lymph node Treg repertoire and ~12% similar to the spleen Treg repertoire. In contrast, the Tconv and Treg repertoires from the tumor were only 3.8% (MH = 0.038) similar. This suggests that the Treg cells within the tumor are more likely to have derived from the draining lymph node Treg pool or vice versa than the tumor-infiltrating Tconv population.
Figure 3. Low levels of similarity between the TRBV13-2 TCR repertoires of Tconv and Treg cell subsets from various tissues.

The TRBV13-2 TCR sequences were determined by clonotyping and used to identify individual TCRs. (A) Similarity between each TCR repertoire was measured using the Morisita-Horn index. This index measures similarity based on the number of shared sequences between two populations as well as how well represented those overlapping sequences are within the two populations. A value between 0 (no similarity) and 1 (identical) was calculated by comparing the TCR repertoire from each population and colored according to the scale shown. Values used are mean similarity taken from two independent experiments in two mice. (B) The TCR repertoires of Tregs from tumor and Tregs from draining lymph node were compared. Different TCR sequences observed (x axis) within the tumor Treg (filled & above) and draining lymph node Treg (open & below) repertoires, and the frequency (y axis) of each sequence within that subset, are displayed. The total number of TCR sequences analyzed is displayed as n=, with the number of unique TCR sequences observed in parentheses. The proportion of the tumor Treg repertoire that shared sequences with the draining lymph node Treg repertoire is shown. LN, lymph node.
TRBV13-2/TRBJ2-5 TCR repertoires in tumor-bearing mice
Due to the high clonotypic diversity observed within the total TRBV13-2 repertoire across all tissues, it is possible that some TCR sequences may have been missed in the initial TRBV13-2 analysis. To focus our clonotypic examination in more detail, we performed TCR sequencing on a single T cell receptor β chain joining (TRBJ) segment within the TRBV13-2+ population. As for the Vβ usage analysis, we observed no significant skewing towards any particular Jβ subset within the Treg or Tconv repertoires from the spleen, tumor or lymph nodes using either an unbiased template-switch anchored RT-PCR (Supplementary Figure 3) or a specific TRBV13-2/TRBC PCR (Supplementary Figure 5). Therefore, as above, we selected the relatively common TRBJ2-5 gene as a representative subset for the deeper analysis.
In agreement with our findings from the TRBV13-2 TCR repertoire analysis, the TRBV13-2/TRBJ2-5 TCR repertoires of Tconv and Treg were generally non-overlapping within all tissues including the tumor (Figure 4). No shared TCR sequences were found within the tumor or the non-draining lymph node. Furthermore, only 1 overlapping sequence was found within the spleen and draining lymph node repertoires. Therefore, the tumor-infiltrating Treg cells had a distinct TRBV13-2/TRBJ2-5 TCR repertoire compared to Tconv cells. This further supports the notion that tumor-infiltrating Tregs do not arise through the conversion of Tconv cells. Conversely, when the TRBV13-2/TRBJ2-5 TCR repertoires of Tregs from the draining lymph node and Tregs from the tumor were compared, a high degree of overlap (33%) was clearly observed (Figure 5A). This similarity between the Treg subsets was highlighted when analyzed using the Morisita-Horn similarity index (Figure 5B). The high similarity (MH = 0.16) between the TCR repertoires of the tumor-infiltrating Tregs and draining lymph node Tregs suggests, as previously, that the tumor Tregs and the draining lymph node Tregs derive from the same population that is distinct from the Tconv pools residing in the same locations.
Figure 4. TRBV13-2/TRBJ2-5 TCR repertoires of Tconv and Treg cells are non-overlapping in a tumor-bearing mouse.

Tconv and Treg populations from a tumor-bearing mouse were sorted by flow cytometry and analyzed by TCR clonotyping. The CDR3 amino acid sequences of TCRs from the TRBV13-2/TRBJ2-5 subset were used to identify individual TCRs from the (A) spleen, (B) non-draining lymph node, (C) draining lymph node and (D) tumor. Each graph is displayed as described in the legend for Figure 2. LN, lymph node.
Figure 5. Overlap between the Treg TCR repertoires from draining lymph node and tumor.

The TRBV13-2/TRBJ2-5 TCR sequences were determined by clonotyping and used to identify individual TCRs. (A) The TCR repertoires of Tregs from tumor and Tregs from draining lymph node were compared and displayed as described in legend for Figure 3B. (B) Similarity between each TCR repertoire was measured using the Morisita-Horn index and displayed as described in the legend for Figure 3A. LN, lymph node.
TCR repertoires of antigen-experienced Tconv and Treg cells in tumor-bearing mice
TCR engagement is necessary for the conversion of Tconv cells into Tregs in vitro. Therefore, it is likely that Tconv cells must similarly encounter antigen in vivo before they are converted into Tregs. Accordingly, we hypothesized that if conversion of Tconv cells into Tregs was an ongoing process in tumors, an overlap between the TCR repertoires would most likely be observed within the antigen-experienced population. To test this hypothesis, CD4+Foxp3− cells were purified as an antigen-experienced (CD44hiCD62Llo) Tconv population (Figure 6A) and analyzed by TCR clonotyping. Similar to our previous analyses of the whole Tconv population, the TCR repertoires of antigen-experienced Tconv cells and Tregs were non-overlapping in all tissues including the tumor (Figure 6B-D). In addition we observed no overlap between CD44loCDL62hi (naive) T conv cells and Tregs purified from the tumor (data not shown). Collectively these data further support the premise that conversion of tumor-infiltrating Tconv cells into Tregs is infrequent and does not account for the large enrichment of Tregs in tumors. The TCR repertoires of antigen-experienced Tconv cells and Tregs from the spleen, draining lymph node and tumor were each compared using the Morisita-Horn similarity index (Figure 6E). This analysis confirmed the previous results. No overlap was observed between Tregs and Tconv cells, and the greatest degree of overlap was present between the same type of T cell, most notably the antigen-experienced Tconv populations, recovered from the tumor and the tumor draining lymph node (Figure 6E). Collectively, these data demonstrate that migration of T cells between the tumor and lymphoid tissue occurs, but that these cells remain largely unconverted.
Figure 6. TRBV13-2 TCR repertoires of antigen-experienced Tconv and Treg cells are non-overlapping in a tumor-bearing mouse.

Antigen-experienced (CD44hiCD62Llo) Tconv cells and naïve (CD44loCD62Lhi) Tconv cells (A) from a tumor-bearing mouse were sorted by flow cytometry together with Tregs and analyzed by TCR clonotyping. The CDR3 amino acid sequences from the TRBV13-2+ subset were used to identify individual TCRs from the spleen, draining lymph node and tumor. The TCR repertoires of CD44hiCD62Llo Tconv and Tregs from the spleen (B), draining lymph node (C) and tumor (D) are displayed as described in the legend for Figure 2. (E) Similarity between each TCR repertoire was measured using the Morisita-Horn index; data are displayed as described in the legend for Figure 3A. LN, lymph node.
Discussion
Our study was undertaken to understand further the mechanisms underlying the enrichment of Tregs in tumors. We assessed the contribution of conversion of Tconv cells into Tregs in tumor-bearing mice through a detailed dissection of the TCR repertoires from CD4+Foxp3− and CD4+Foxp3+ cells in lymphoid tissue and TILs in mice expressing a normal TCR repertoire. The degree of overlap between the TCR repertoires expressed by both populations was lower than that reported by other groups using TCR-transgenic mice (11, 12). This disparity is expected given the curtailed TCR repertoires found within such mice. In these models, a limited number of TCR structural combinations pass thymic selection resulting in restricted peripheral diversity and, accordingly, an increased chance of identifying recurring overlapping clonotypes. The T cell dynamics during a natural tumor response in a TCR intact animal are considerably more complex considering the full 2×106 (25×106 in humans) receptor pool available in mice (26, 27). In this study, we used non-TCR transgenic mice to investigate whether the degree of repertoire overlap between the two T cell populations was significantly different in cells isolated from the tumor compared to lymphoid tissue. In agreement with the findings of others, we observed that the TCR repertoires of Tconv and Treg cells are largely distinct in the lymphoid compartments. However, we also observed that this is the case for tumor-isolated Tconv and Treg cells, indicating that conversion of Tconv does not make a major contribution to the accumulation of tumor-infiltrating Tregs, even in tumors where TGFβ is readily detected. In support of this conclusion, the greatest degree of repertoire similarity was observed between corresponding T cell populations from the draining lymph node and the tumor. These data imply that T cells may be primed in the draining lymph node or enter the node via draining lymphatics (Figure 6E).
Previous investigations, despite showing that conversion of conventional T cells into Tregs is possible, do not demonstrate the extent to which conversion might occur in a natural context and whether it accounts for the large enrichment of Tregs often seen in tumors (18-20). Exemplifying this, a more recent study of melanoma patients implied that peripheral conversion of CD4+CD25− T cells into Tregs in response to tumor antigens leads to Treg enrichment in these patients (28). Whilst this study demonstrates that conversion is possible, the extent to which this mechanism contributes to the enrichment of Tregs in tumours was not addressed. Our data do not exclude conversion of Tconv cells as a mechanism contributing to Treg enrichment in tumors but do demonstrate, in the case of a spontaneously developing tumor, that the majority of tumor-infiltrating Tregs do not arise through conversion. We found no significant difference in similarity between the TCR repertoires of Tconv cells and Tregs from tumor compared to normal lymphoid tissue (Supplementary Figure 6). Importantly, on average, more than 96% of the tumor Treg repertoire did not overlap with the TCR repertoire of Tconv cells from the tumor or tumor draining lymph node (Supplementary Figure 6). Indeed, the cell population with the greatest degree of repertoire similarity with tumor-infiltrating Tregs was the Treg population from the tumor draining lymph node. In addition, we observed a significant overlap in the repertoires of antigen-experienced Tconv cells in the tumor and tumor draining lymph node (Figure 6E and Supplementary Figure 7). These data strongly suggest that migration of both Tconv cells and Tregs between these two sites occurs without conversion.
It could be argued that conversion of Tconv cells into Tregs is instantaneous in the tumor environment. Should this be the case, it may not be possible to detect Tconv cells and Tregs with overlapping repertoires in the tumor and an analysis such as ours would therefore fail to detect conversion. In vitro studies, however, argue against this scenario as it takes between 1 and 4 days for detectable levels of Foxp3 to be induced in Tconv cells (29). Our own in vivo data also argue against the possibility of instant conversion. In this scenario, one would expect to observe a significant overlap not only between antigen-experienced Tconv cells in the tumor draining lymph node and Tconv cells in the tumor (as shown in Figure 6E and Supplementary Figure 7A) but also between antigen-experienced Tconv cells in the tumor draining lymph node and Tregs in the tumor. No such overlap was observed (Figure 6E and Supplementary Figure 7B).
We selected the MCA tumor model for this work as several reports support a role for the immune system, including Tregs, in influencing disease progression (5, 30). In accordance with our findings, a recent study of T cell responses in patients with colorectal carcinoma demonstrated that Treg and Tconv cells tend to recognize distinct antigens (31). Our findings are also similar to those from a study that analyzed the degree of conversion in self antigen-specific T cells in the context of a mouse model of type I diabetes (17). The TCRs of Tregs and Tconv cells isolated from the pancreas and pancreas-draining lymph nodes of BDC2.5/NOD TCR transgenic mice were distinct, thereby indicating no role for conversion in response to a pancreatic autoantigen. Similarly, a more recent study of the TCR repertoire and T cell specificity in experimental allergic encephalomyelitis (EAE) concluded that, despite sharing autoantigen specificity, effector T cells and Tregs had largely distinct TCR repertoires, thereby similarly suggesting that conversion between these two populations was limited (32). Thus, the combined results of these studies imply that tolerance in the context of autoimmunity and tumor immunity is achieved without substantial conversion of conventional T cells into Tregs.
In contrast, a role for conversion may exist in preventing immune responses to some foreign antigens such as those of commensal bacteria in the gut (induced tolerance). In the intestine, the immune system must resist infection from occasional pathogens whilst maintaining tolerance to commensal flora and dietary antigens. The generation of Tregs by conversion from Tconv cells has been suggested as an important mechanism for establishing this tolerance to persistent antigens in the gut (33). Recent data suggest that the induction of Foxp3 in gut-associated lymphocytes involves the conserved non-coding DNA element CNS1 (34).
The mechanisms that underlie Treg cell enrichment in tumors remain unclear. Our data indicate that the TCR repertoires of tumor-infiltrating Tconv and Treg cells are largely distinct, implying that conversion of Tconv cells is not a significant cause of Treg enrichment in tumors. It is possible that Treg enrichment reflects the type of antigens expressed by tumor cells. Evidence suggests that the Treg population contains a higher number of cells that recognize self-antigens compared to Tconv cells (35-37). Thus, in the tumor environment, Tregs may receive stronger antigen-driven signals than conventional T cells. It also remains possible that Tregs are preferentially recruited or retained in the tumor environment and/or that the tumor microenvironment promotes Treg cell proliferation and survival. It is known that Tregs exhibit a higher turnover than Tconv under homeostatic conditions and that tumors can license dendritic cells (DCs) to promote the proliferation of Tregs in lymph nodes, possibly through the production of TGFβ (38). Thus, strong antigenic signals within the tumor microenvironment accompanied by the secretion of cytokines such as IL-2 and TGFβ may serve to further promote Treg cell proliferation and survival. In support of this, it has previously been found that the majority of CD25+CD4+ Tregs infiltrating transplanted MCA-derived cell lines emanate from the recruitment and proliferation of naturally occurring CD25+ Tregs (39).
Tregs clearly play a role in the progression of some types of tumors and, on this basis, targeting these cells for immunotherapeutic purposes is attractive. Conversion of conventional T cells into Tregs has been proposed as a means of Treg cell enrichment within TILs and hence as a mechanism that could be targeted in the immunotherapeutic setting. Our study, however, indicates that conventional T cells entering the tumor remain largely unconverted. It is more likely, therefore, that the enrichment of Tregs within TILs reflects differences in the way Tregs and conventional T cells are influenced by the tumor microenvironment. Understanding the nature of these influences should reveal mechanisms through which the balance between tumor-infiltrating Tregs and conventional T cells can be altered for the purpose of halting tumor progression.
Supplementary Material
Acknowledgments
This work was supported by a Medical Research Council Senior Fellowship (G117/488) awarded to Awen Gallimore and a University Award from the Wellcome Trust (086983). David A. Price is a Medical Research Council Senior Clinical Fellow. Katherine K. Wynn is funded by a Wellcome Trust Value In People Award. Funding for Cristina Ferreira, Yogesh Singh and Julian Dyson was provided by the Medical Research Council and the Biotechnology and Biological Sciences Research Council.
The authors wish to thank Dr John Miles and Dr R. James Matthews for invaluable advice and discussion.
Footnotes
The authors declare that they have no conflicts of interest.
References
- 1.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–4. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 2.Dunn G, Old L, Schreiber R. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 3.Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–62. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- 4.Betts G, Clarke S, Richards H, Godkin A, Gallimore A. Regulating the immune response to tumours. Adv Drug Deliv Rev. 2006;58:948–61. doi: 10.1016/j.addr.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 5.Betts G, Twohig J, Van den Broek M, Sierro S, Godkin A, Gallimore A. The impact of regulatory T cells on carcinogen-induced sarcogenesis. Br J Cancer. 2007;96:1849–54. doi: 10.1038/sj.bjc.6603824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999;163:5211–8. [PubMed] [Google Scholar]
- 7.Jones E, Dahm-Vicker M, Simon A, Green A, Powrie F, Cerundolo V, et al. Depletion of CD25+ regulatory cells results in suppression of melanoma growth and induction of autoreactivity in mice. Cancer Immun. 2002;2:1–12. [PubMed] [Google Scholar]
- 8.Turk M, Guevara-Patiño J, Rizzuto G, Engelhorn M, Sakaguchi S, Houghton A. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J Exp Med. 2004;200:771–82. doi: 10.1084/jem.20041130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu P, Lee Y, Liu W, Krausz T, Chong A, Schreiber H, et al. Intratumor depletion of CD4+ cells unmasks tumor immunogenicity leading to the rejection of late-stage tumors. J Exp Med. 2005;201:779–91. doi: 10.1084/jem.20041684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 11.Hsieh C, Zheng Y, Liang Y, Fontenot J, Rudensky A. An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nat Immunol. 2006;7:401–10. doi: 10.1038/ni1318. [DOI] [PubMed] [Google Scholar]
- 12.Pacholczyk R, Ignatowicz H, Kraj P, Ignatowicz L. Origin and T cell receptor diversity of Foxp3+CD4+CD25+ T cells. Immunity. 2006;25:249–59. doi: 10.1016/j.immuni.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 13.Fazilleau N, Bachelez H, Gougeon M, Viguier M. Cutting edge: size and diversity of CD4+CD25high Foxp3+ regulatory T cell repertoire in humans: evidence for similarities and partial overlapping with CD4+CD25- T cells. J Immunol. 2007;179:3412–6. doi: 10.4049/jimmunol.179.6.3412. [DOI] [PubMed] [Google Scholar]
- 14.Bluestone J, Abbas A. Natural versus adaptive regulatory T cells. Nat Rev Immunol. 2003;3:253–7. doi: 10.1038/nri1032. [DOI] [PubMed] [Google Scholar]
- 15.Scheinberg P, Melenhorst J, Hill B, Keyvanfar K, Barrett A, Price D, et al. The clonal composition of human CD4+CD25+Foxp3+ cells determined by a comprehensive DNA-based multiplex PCR for TCRB gene rearrangements. J Immunol Methods. 2007;321:107–20. doi: 10.1016/j.jim.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong J, Obst R, Correia-Neves M, Losyev G, Mathis D, Benoist C. Adaptation of TCR repertoires to self-peptides in regulatory and nonregulatory CD4+ T cells. J Immunol. 2007;178:7032–41. doi: 10.4049/jimmunol.178.11.7032. [DOI] [PubMed] [Google Scholar]
- 17.Wong J, Mathis D, Benoist C. TCR-based lineage tracing: no evidence for conversion of conventional into regulatory T cells in response to a natural self-antigen in pancreatic islets. J Exp Med. 2007;204:2039–45. doi: 10.1084/jem.20070822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu V, Wong L, Jang T, Shah A, Park I, Yang X, et al. Tumor evasion of the immune system by converting CD4+CD25- T cells into CD4+CD25+ T regulatory cells: role of tumor-derived TGF-beta. J Immunol. 2007;178:2883–92. doi: 10.4049/jimmunol.178.5.2883. [DOI] [PubMed] [Google Scholar]
- 19.Valzasina B, Piconese S, Guiducci C, Colombo M. Tumor-induced expansion of regulatory T cells by conversion of CD4+CD25- lymphocytes is thymus and proliferation independent. Cancer Res. 2006;66:4488–95. doi: 10.1158/0008-5472.CAN-05-4217. [DOI] [PubMed] [Google Scholar]
- 20.Zhou G, Levitsky H. Natural regulatory T cells and de novo-induced regulatory T cells contribute independently to tumor-specific tolerance. J Immunol. 2007;178:2155–62. doi: 10.4049/jimmunol.178.4.2155. [DOI] [PubMed] [Google Scholar]
- 21.Fontenot J, Rasmussen J, Williams L, Dooley J, Farr A, Rudensky A. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–41. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 22.Price D, Brenchley J, Ruff L, Betts M, Hill B, Roederer M, et al. Avidity for antigen shapes clonal dominance in CD8+ T cell populations specific for persistent DNA viruses. J Exp Med. 2005;202:1349–61. doi: 10.1084/jem.20051357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colwell R. EstimateS: Statistical estimation of species richness and shared species from samples. Version 7.5. 2005. User’s Guide and application published at: http://purl.oclc.org/estimates.
- 24.Chen W, Jin W, Hardegen N, Lei K, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–86. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fantini M, Becker C, Monteleone G, Pallone F, Galle P, Neurath M. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25- T cells through Foxp3 induction and down-regulation of Smad7. J Immunol. 2004;172:5149–53. doi: 10.4049/jimmunol.172.9.5149. [DOI] [PubMed] [Google Scholar]
- 26.Casrouge A, Beaudoing E, Dalle S, Pannetier C, Kanellopoulos J, Kourilsky P. Size estimate of the alpha beta TCR repertoire of naive mouse splenocytes. J Immunol. 2000;164:5782–7. doi: 10.4049/jimmunol.164.11.5782. [DOI] [PubMed] [Google Scholar]
- 27.Arstila T, Casrouge A, Baron V, Even J, Kanellopoulos J, Kourilsky P. A direct estimate of the human alphabeta T cell receptor diversity. Science. 1999;286:958–61. doi: 10.1126/science.286.5441.958. [DOI] [PubMed] [Google Scholar]
- 28.Fourcade J, Sun Z, Kudela P, Janjic B, Kirkwood J, El-Hafnawy T, et al. Human tumor antigen-specific helper and regulatory T cells share common epitope specificity but exhibit distinct T cell repertoire. J Immunol. 2010;184:6709–18. doi: 10.4049/jimmunol.0903612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Selvaraj R, Geiger T. A kinetic and dynamic analysis of Foxp3 induced in T cells by TGF-beta. J Immunol. 2007;178:7667–77. doi: 10.4049/jimmunol.178.12.7667. [DOI] [PubMed] [Google Scholar]
- 30.Koebel C, Vermi W, Swann J, Zerafa N, Rodig S, Old L, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450:903–7. doi: 10.1038/nature06309. [DOI] [PubMed] [Google Scholar]
- 31.Bonertz A, Weitz J, Pietsch D, Rahbari N, Schlude C, Ge Y, et al. Antigen-specific Tregs control T cell responses against a limited repertoire of tumor antigens in patients with colorectal carcinoma. J Clin Invest. 2009;119:3311–21. doi: 10.1172/JCI39608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu X, Nguyen P, Liu W, Cheng C, Steeves M, Obenauer J, et al. T cell receptor CDR3 sequence but not recognition characteristics distinguish autoreactive effector and Foxp3(+) regulatory T cells. Immunity. 2009;31:909–20. doi: 10.1016/j.immuni.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coombes J, Siddiqui K, Arancibia-Cárcamo C, Hall J, Sun C, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–64. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng Y, Josefowicz S, Chaudhry A, Peng X, Forbush K, Rudensky A. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010;463:808–12. doi: 10.1038/nature08750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Romagnoli P, Hudrisier D, van Meerwijk J. Preferential recognition of self antigens despite normal thymic deletion of CD4(+)CD25(+) regulatory T cells. J Immunol. 2002;168:1644–8. doi: 10.4049/jimmunol.168.4.1644. [DOI] [PubMed] [Google Scholar]
- 36.Hsieh C, Liang Y, Tyznik A, Self S, Liggitt D, Rudensky A. Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity. 2004;21:267–77. doi: 10.1016/j.immuni.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 37.Pacholczyk R, Kern J, Singh N, Iwashima M, Kraj P, Ignatowicz L. Nonself-antigens are the cognate specificities of Foxp3+ regulatory T cells. Immunity. 2007;27:493–504. doi: 10.1016/j.immuni.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghiringhelli F, Puig P, Roux S, Parcellier A, Schmitt E, Solary E, et al. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J Exp Med. 2005;202:919–29. doi: 10.1084/jem.20050463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bui J, Uppaluri R, Hsieh C, Schreiber R. Comparative analysis of regulatory and effector T cells in progressively growing versus rejecting tumors of similar origins. Cancer Res. 2006;66:7301–9. doi: 10.1158/0008-5472.CAN-06-0556. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.