

Abstract
Pulmonary embolism (PE) and deep vein thrombosis (DVT) are associated with considerable morbidity and mortality, mostly, in case of PE for its lack of sensitivity of its early detection. For as much as twenty-five percent of PE patients the primary clinical appearance is unexpected death. While PE is one of the most avertable causes of hospital associated deaths, its diagnostics can be extremely difficult. Newly increased interest in an inherited thrombophilic states has been provoked by the discovery of several common inherited abnormalities, i.e. the prothrombin (PT) gene G20210A, Factor V Leiden (FVL) mutation (Arg506Gln), hyperhomocystenemia and homocysteiuria, Wein-Penzing defect, Sticky Platelet Syndrome (SPS), Quebec platelet disorder (QPD) and Sickle Cell Disease (SCD). PE incidence rates increase exponentially with age for both men and women, as they might harbor more than one thrombophilic state. Although the impact of genetic factors on PE is to some extent documented with lacking taxonomy, its genetic testing as its prevention strategy fall short.
In this review thrombophilic states are divided into inherited or acquired, and only the inherited and newly documented are more closely followed. Factors are further grouped based on its thrombophilic taxonomy into; inherited defects of coagulation, inherited defects of fibrinolysis, inherited defects of enzymatic pathway in relation to development of VTE and PE and inherited defects of platelets in relation to PE. It was beyond the scope of this review to follow all inherited and newly recognized factors and its association to VTE and PE; however the overall taxonomy makes this review clinically valuable i.e. in relation to genetic testing as PE prevention.
Keywords: Inherited thrombophilic states, venous thromboembolism, pulmonary embolism
The distinct characteristic of hemostatic system is that it is an independent apparatus regulating blood fluidity while supports localized and temporary thrombus formation at place of vascular damage. In thrombophilia, clot forms inappropriately and thromboembolism usually occurs as a combination of inherited and acquired attributes. Hereditary thrombophilia is linked to a greater risk of later having venous rather than arterial thrombotic event. As thromboemboli form they tend to increment pulmonary vascular resistance thus increase the right ventricular afterload. If the right ventricle afterload is augmented severly failure of right ventricle develops with additional complicating factors that overall contribute to the pulmonary arterial constriction, which may later advance into hemodynamic collapse. In study made by Rossi et al in 2008,1 920 patients have been diagnosed with proximal deep vein thrombosis (DVT), with about 26% that had later symptoms of PE. Moreover the risk factors of proximal DVT or PE differed, based on the type of thrombophilia. Other recent reports describes as much as eighteen fold higher risk of early death that is documented in patients with symptomatic PE compared to patients with DVT alone. Furthermore, inherited thrombophilia is a common cause of VTE and is detectable in at least 30 to 40% of patients with DVT.1 In the scientific literature debate continues about what are the most frequent thrombophilic risk factors of VTE and moreover how to effectively separate thrombophilic risk factors of DVT from those of PE (Table 1). Although, isolated PE embraces higher relative risk for development of severe circulation collapse than isolated proximal calf or iliofemoral DVT, one can prevent its impact by early screening for major thrombophilic factors that predisposes venous stasis.
Table 1.
Thrombophilic deficiencies in relation to VTE and PE.
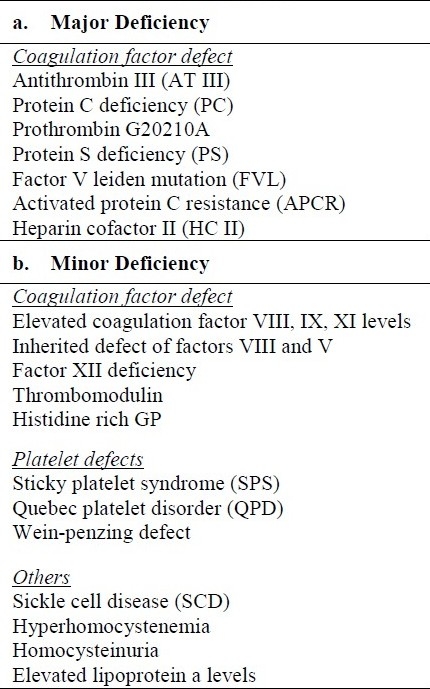
The word Thrombophilia was introduced into scientific literature in 1965 for a tendency to develop venous thrombosis in a Norwegian family, subsequently proven to have antithrombin deficiency (Figure 1).2 Since then twentytwo new antithrombin gene deficiencies were described, mostly in its heterozygous state.3 Later other important thrombophilias were described. Protein C deficiency was described first by Griffin et al in 1981.4 Intriguingly, the report describes 22 years old male that suffered recurrent thrombophlebitis and later was diagnosed with PE and treated with doses of Coumarin. It was year 1978, and it was most likely the first described link between the inherited thrombophilia and PE. The protein Scongenital deficiency was in 1984 described by Comp et al5 in seventeen years old white male. He was hospitalized at age 14, and later venographically documented to have lower extremity DVT with clots in both calf and left thigh. At age 15 he suffered an episode of multiple PE resulting from documented lower extremity DVT. Other family members with protein C and S deficiencies were later identified and treated for VTE or PE; in some cases soon after deceased, postmortem diagnosed with PE.
Figure 1.

Trombophilia timeline (major deficiencies).
In 1982 Tollefsen et al6 first described HCII. Since then in 15 families HCII inherited deficiency was documented the defect that was later recognized to be transmitted from generation to generation as an autosomal dominant. In many cases affected individuals had reduced plasma levels of active HCII and (or) its antigen, which transpired in seven families into DVT/PE.7
One of the leading hereditary thrombophilia, factor V Leiden (FVL), was first reported in 1993 by two independent groups of European investigators that explained a novel defect in the hemostatic pathway, based on resistance to activated protein C.8,9 Family history of the patients revealed a high incidence of thrombosis and multiple episodes of DVT, suggesting an inherited cause of the disease.8 No other coagulation abnormalities and deficiencies were noted on examination. Moreover, patient, his brother and his uncle and aunt developed VTE later in their lives, making diagnosis of this congenital disease more obscure.
In 1996 Poort et al became one of the first in identification of genes involved in inheritable thrombophilic defect of prothrombin and its role in VTE.10 An association was found between the presence of the 20210A allele and elevated prothrombin levels with threefold increased risk of VTE.10
The exceptionally well controlled hemostatic system maintains a subtle equilibrium among a hemorrhagic and a prothrombotic state by not only regulating its members (Platelets, RBC's, WBC's), but as well through the receptor activation and inhibition within its area (vascular endothelium in organs). Thus blood coming through the organ constantly monitors the hemostasis, thrombosis and fibrinolysis while receiving the upto date information about the organ conditions. Pathologic thrombotic states develop when the equilibrium is misbalanced e.g. by platelet abnormal aggregation, deficiencies of a natural anticoagulants, coagulation factors or its resistance to inactivation by a natural anticoagulants, and by an excess plasma levels of fibrinolytic inhibitors.
The morbidity and mortality in patients with hypercoagulable states are primarily due to VTE and PE. In many cases no specific clinical symptoms or signs directly attributable to thrombophilic disorders are presented on physical exam. In some paradoxical cases distinct hypercoagulable state will result in an obvious thrombosis and VTE, but not all those with thrombosis will present a particular hypercoagulable state. Thus thorough blood analysis could be of interest in future testing (receptor activity of its members, presence of quantitative and qualitative change of its adhesion molecules immediately after they passed through the lung vasculature). As various adhesion molecules substantially differs between pulmonary microvessels11 these parameters can provide additional information to better assess the organ coagulation or thrombotic state to later react and partially correct the hypercoagulable state (Table 2). Additionally, vascular lumen targeting by stably expressed, non-internalized determinants such as Inter-Cellular Adhesion Molecule-1 (ICAM) or Platelet-Endothelial-Cell Adhesion Molecule 1 (PECAM-1) linked to for example low-molecularweight single-chain prourokinase plasminogen activator (lmw-scuPA) with a single-chain variable fragment (scFv) of a PECAM-1 could better access thrombus or prevent thrombus extension in PE.12,13
Table 2.
Several Cell Adhesion Molecules (CAM's) activated in patients with DVT and PE.

To understand how thrombophilic states lead to pathologic thrombosis and later VTE and PE, thrombophilia has to be linked to normal hemostasis. In hemostasis, two equally important processes are interconnected, the primary and the secondary hemostasis. While mostly described as isolated events, both occur concomitantly. Primary hemostasis consists of platelet adhesion, activation, and aggregation. Platelets adhere to the vascular subendothelium by its attachment to von Willebrand factor (vWf) molecules exposed after vascular injury. Platelet activation along with activation of other plasma proteins and blood cells leads to more blood stasis. Stasis leads to platelet adhesion and release of its granules, which brings more coagulation factors into play with pronounced platelet-plasma local activity. The Platelet specific GP Ib-V-IX glycoprotein complex is implicated in interaction of Platelets and vessel wall in the course of binding to collagen through vWf. GP Ib-V-IX is important as well in initiation of platelets adhesion and aggregation under high shear conditions. Through integrin α2 β1 and GP VI platelets become rigidly adhered. Release of vasoactive agents such as serotonin, adenosine diphosphate (ADP), prostaglandins, and thromboxane A2 (TXA-2) engage more circulating platelets by activating i.e. ADP receptors P2Y1 or P2Y12 or TXA2 receptors TP-α and β and further act via integrin αIIb β3 (GP IIb/IIIa). Following extra platelets recruitment, platelets are linked by fibrinogen through their surface GP IIb/IIIa receptors to form the unstable plug, which is further modified. GP IIb/IIIa receptor is essential for platelet responses including aggregation and for later clot retraction through it's binding to vitronectin, fibrinogen, fibrin, fibronectin, vWf and soluble CD 40. Simultaneously, inactive protease zymogens are converted to active serine proteases, which result in thrombin and covalently cross-linked fibrin creation. Thrombin has central role in conversion of clottable fibrinogen into fibrin by releasing two fibrinopeptides A and B in thrombogenesis. On cellular level, thrombin binds to GPCR Protease-Activated Receptors PARs (PAR-1,3 and 4) and promotes numerous other effects, i.e. release of inflammatory mediators, chemotaxis, proliferation and growth factors (GF) release, smooth muscle contractions, central stress fiber formation, EC contractions and barrier dysfunction, gap formations, barrier disruption, endothelial release of NO and vWf. The activation of thrombin receptor on platelets results in release of platelet factor V and its exteriorization thus provides surface for other enzyme(s) to bind and promote coagulation cascade. PAR-1 activated by thrombin has been shown to stimulate tyrosine phosphorylation of the GF receptors, activating MAPK cascade, stimulating transactivation of GF receptors, promoting cell survival, and enhancing mitogenesis. Thrombin can further downstream signal through proteolytic cleavage and generation of a new ligand SFLLRN that interacts with the extracellular loop-2 of the receptor.14 It induces EC barrier dysfunction through multiple signaling pathways and cytoskeletal targets, including Rhokinase (RhoK)15 and the Ca2+ /calmodulin-dependent Myosin Light Chain Kinase (MLCK).16,17 Thus thrombin role in inherited thrombophylic states is multifaceted.
Inherited defect of coagulation factors and PE
Gene mutation which results in gaining function of a procoagulant factors are rarer than those that results of loosing the anticoagulant factor. In case of clinical expression of prothrombin thrombophilia many individuals are either heterozygous or homozygous for the G20210A allele, and may or may not develop thrombosis. The mutation was found on chromosome 11 in about 1-2% of asymptomatic and 3-7% of thrombosis patients. Heterozygotes that develop thrombotic complications remain asymptomatic until adulthood and a few have recurrent thromboembolism before the age of 30. It is predictable that increased levels of prothrombin upregulates the coagulation cascade including activation and upregulation of blood cells. It is still uncertain however, whether its heterozygosity increases the risk of recurrent VTE after the first episode.
Diagnosis of prothrombin thrombophilia requires DNA analysis of F2, the gene encoding prothrombin. In a study by Reuner, PE as complication of VTE was associated with prothrombin G20210A allele mutation.18 PE was related to DVT of the lower limbs in 37%. In 74% PE with or without combination of DVT was the first event of thromboembolism, whereas the remaining 26% of patients had already thromboembolic events before.
Other very frequent hereditary risk for VTE and PE, described predominantly in Caucasians, involves an Activated Protein C (APC)-cleavage site mutation (Arg506Gln) that is the major target for factor Va.19 Factor V levels are controlled by thrombindependent activation, and APC-dependent inactivation. Thrombin cleaves factor V resulting in the generation of an aminoterminal heavy chain and a carboxyterminal light chain. These chains form a dimer (factor Va) that coordinates a calcium ion and, together with factor Xa and anionic phospholipid, form the prothrombinase complex, which promotes prothrombin (II) conversion to thrombin (IIa) on the platelet surface.20 Thus patients with Factor V Leiden (FVL) are resistant to factor Va inactivation and in consequence have a longer factor Va halflife in plasma with increased thrombin generation. Leiden factor is an autosomal dominant condition, with about 4-7% of population identified as heterozygotes, 0.06-0.25% are homozygotes.21 In general population FVL resistance to APC has an incidence about 4.8%, accounting for 40% to 50% of cases of inherited thrombosis, while e.g. PT 20210A mutation occurs in about 2.7% of normal patients, with the frequency increasing to 7.1% to 16% in affected patients.22,23 Although the FVL is the most common cause of inherited thrombosis the overall risk for development of PE is less with FVL mutation than with the prothrombin G20210A mutation or antithrombin deficiency for yet unknown reason. Recently, another mutation (Factor V Cambridge) was described in the activated protein APC cleavage sites of human factor V distinct from the FVL R506Q mutation.24 These mutations affect the APC cleavage site at Arg306 in the heavy chain of activated FV. It is still not clear what role these mutation plays in VTE and PE.
Another Protein C deficiency (autosomal recessive disorder) and its coinheritance with deficiency of FVL in heterozygous carriers results in a high degree of penetrance.25 Protein C is a glycoprotein, synthesized in the liver as a zymogen, which circulates in the blood. The inherited heterozygous state and its deficiency are most frequently associated with DVT and PE. A significant number of patients with protein C deficiency remain asymptomatic. Two types of protein C are described. While type I, is associated with its quantitative deficiency, the type two refers to defect of its molecular structure. Acquired protein C deficiency occurs transiently in certain clinical circumstances.26 On molecular level, APC controls thrombin generation and reduces apoptosis. Furthermore, it inactivates factors Va and VIIIa and creates cytoprotective PAR-1 downstream signaling. Thrombin bound to thrombomodulin activates protein C bound to Endothelial protein C receptor (EPCR). APC binding to EPCR both modifies and stimulates PAR-1 signaling. Recently APC's systemic anticoagulant activity and its beneficial antiinflammatory effects were more closely revisited for its ability to down regulate generation of thrombin (known for its proinflammatory properties). Lately, separate activities from its anticoagulant potential were studied as an alteration of gene expression profiles, antiapoptotic activity and endothelial barrier stabilization.
Protein S autosomal dominant disorder increases risk of heterozygotes having VTE complications. APC's cofactor Protein S, works in the cleavage of FVa and FVIIIa. APC form a complex with Protein S on negatively charged phospholipid membranes, while protein S increases the affinity of APC for the membrane approximately 10-fold.27 The APC cofactor activities of FV and protein S are synergistic and result in efficient management of activity of the tenase complex. Heeb et al projected APC-independent anticoagulant activity of protein S in 1993.28 Proposed APC activity was through its direct interactions with FVa/ FVIIIa and/or FXa. Later it was suggested that the direct anticoagulant activity of protein S is most likely due to competition for phospholipids.29
Study of Protein S gene defects revealed more point mutations than deletions.27 Non-bound protein C levels in healthy individuals present approximately 30-40% of total protein S,27 with only free protein S that is capable of acting as a cofactor in the protein C system. There are a total of three known deficiencies of protein C. Types one and three are reduction of its free and total protein levels, and its free protein reduction respectively. Type II deficiency is a reduction in its cofactor activity. Large population of protein S deficient patient has type I deficiency with about 6% prevalence. In patients with protein S deficiency, 74% develop DVT, about 72% develop superficial thrombophlebitis and 38% develop PE.30
Another factor that has a pivotal role in assembly of APC is Thrombomodulin (TM) with its gene mutation that constitutes a potential risk for thrombosis. Ohlin et al in 1995 identified TM mutation as a heterozygous exchange of T for G at nucleotide position 1456, which predicted Asp468 with Tyr in a Ser/Thrrich domain and led to PE.31 Formation of the thrombin-TM complex limits the procoagulant and cellular activating functions of thrombin and results in thrombin-mediated catalytic transformation of protein C into APC, an effect that is facilitated bythe EPCR.
Antithrombin is the main inhibitor of thrombin and other serine proteases i.e. ff. IXa, Xa, XIa, and XIIa. Its deficiency, usually noted in the period of an early child development, is rare autosomal recessive condition and is associated with an increased prothrombogenicity. Antithrombin III (ATIII) is additional inherited risk factor of VTE and PE. AT III deficiency, an autosomal dominant disorder, in heterozygosity leads to an increased risk of VTE and PE, characteristically appearing in young adulthood.
Its deficiency can be divided into type I, which is a reduction of functional antithrombin, while in II type AT III molecule is functionally abnormal.32 Acquired deficiencies of AT III are commonly due to presence of antiphospholipid (AP) antibodies (eg, lupus anti-coagulans), where ATIII is used at augmented rates due to intense activation of the coagulation pathway. Additional mechanisms (no or low synthesis of AT III) might include chronic liver disease and loss of protein due to ascites and (or) nephrotic syndrome. Recently retrospective study of patients with AT III deficiency concluded that the relative risk of symptoms of PE increases 2.4 with 95% confidence interval, to those with no history of this inherited defect.33
Another inherited factor that plays role in thrombophilic states leading to some extent to predisposition to VTE and PE is heparin cofactor II (HCII) deficiency. Unlike in AT III, a mouse with HC II-deficiency is born and later undergoes normal fetal development and does not reveal any inclination to thrombosis or other irregularities.34 Further blood testing also indicated normal hematopoesis, and liver and kidney function.34 Synthesis of HC II (a serine protease inhibitor) take place in liver, while later it circulates in plasma in concentration 1 Hmol/L with half-life of 2 to 3 days.35 Next to thrombin inhibition HC II has no other reported anti-protease(s) activity in coagulation or fibrinolysis.36 Rate at which HCII inhibits thrombin increases more than 1000-fold in the presence of heparin, heparan sulfate, or dermatan sulfate.37 In comparison with antithrombin, higher concentrations of heparin or heparan sulfate are necessary to activate HCII. The inherited deficiency of HC II is rare. In 1992, Jobin et al37 documented that in families with HC II inherited deficiency could be directly linked to predisposition of the individual to DVT and PE. Its inherited deficiency contributes to thrombotic risk only when combined with other factor shortages.33,37
In between non-autosomal inherited risk of VTE and PE belong elevated coagulation factors VIII (F VIII). Prevalence of an elevated plasma level of f. VIII is approximately 20 percent among patients with VTE.38 Most f. VIII circulates as a complex with vWF.39 After its activation by thrombin, f. VIIIa dissociates from vWF to form a complex with f. IXa, which increases the rate of the activation of f X. Patients with high level of f VIII (15 00 IU/L) had 5-fold increase of thrombotic risk when compared to patients with lower levels.40 Higher levels of f. VIII could enhance risk of VTE through excess of thrombin formation and (or) via induction of acquired APC resistance. In study by Kyrle,41 the relative risk of recurring venous thrombosis was higher in patients with intermittent venous thromboembolism. Moreover, in those patients elevated plasma levels of f. VIII were noted as compared to those without thrombosis recurrence.41
Inherited deficiency of other coagulation factor f. XI is autosomal disorder mainly associated with bleeding; first recognized in 1953. Factor XI deficiency is called Plasma Thromboplastin Antecedent (PTA) deficiency; or Rosenthal syndrome, or hemophilia C. Deficient patients do not require treatment or prophylaxis. The mean level of f. XI in confirmed PE patients was 112%, whereas this level in the patients without PE was 104% (P = 0.026).42 Higher levels of F XI (levels above 114%) were independently associated with a 2.4-fold increase risk of PE.42
In 1991, Lammle et al43 described homozygous factor XII deficiency in association with an increased risk for VTE. Later, Girolami et al concluded that the role played by FXII deficiency in the pathogenesis of VTE is minor.44 In plasma FXII is the precursor of a transglutaminase that cross-links fibrin, thereby altering its properties, including resistance to fibrinolysis.
Tissue factor pathway inhibitor (TFPI) is a protein that contains 276 amino acids. It is synthesized mainly by the vascular endothelium. More than half of the amount of TFPI in the blood vessels is normally bound to the vascular endothelium. This pool may be mobilized to circulating blood. Full length TFPI that is, TFPI bound to the vascular endothelium has a much stronger anticoagulant effect than truncated forms of TFPI that is, TFPI found in lipo-proteins.45 TFPI is an important regulator of the extrinsic pathway through its ability to interact with the blood coagulation F VIIa-TF complex and the activated F X. Inherited deficiency of TFPI increases the activity of prothrombinase complex that in turn activates more readily thrombin and thus increases chances of VTE and PE. Clear relationship between genotype, TFPI levels, and the risk of DVT and PE is still missing.
Inherited defects of fibrinolysis and PE
Congenital tissue plasminogen activator (t-Pa) deficiency, plasminogen deficiency, increased plasminogen activator inhibitor (PAI), inherited dysfibrinogenemia, and factor f. XIII deficiency can be incorporated into inherited defects called Dysfibrinolysis, because all have a close relation to the control of fibrin.
Absent or dysfunctional plasminogen dysfibrinolysis, with symptoms that usually starts early in life, are a rare autosomal dominant disorder.46 Frequently, VTE or PE represents dysfunction of plasminogen, in cases when plasminogen levels drops to less than forty percent of the normal values. Inherent deficiency of t-Pa and congenital increases of PAI are extremely infrequent. Mostly, data obtainable on the association of PAI-1 and the risk of thrombosis is conflicting. Inherited dysfibrinogenemias are relatively rare conditions where an abnormality in the fibrin molecule results in defective fibrin clot arrangement. Because of the circulating mixture of normal and abnormal fibrinogen in variety of dysfibrinogenemias, the assessment of the risks is complicated since most of the affected individuals are heterozygous for the mutation. The majority of diagnosed individuals are asymptomatic, whereas about thirty percent of dysfibrinogenemias are associated with bleeding and less than ten are associated with thrombotic tendencies. Typically, they are discovered accidentally in routine coagulation tests. Dysfibrinogenemias are classified by the city or place of discovery. Those with thrombotic risk includes for example Bergamo II, Haifa I, Baltimore I, Bicetre II, Giessen IV, Vlissingen I, Melun I, Kaiserslautern I, Bologna I, Cedar Rapids I, Barcelona III.
Factor XIII is an enzyme that catalyzes the formation of intermolecular epsilon (K-glutamyl)-lysine covalent cross-links in fibrin. It also participates in other physiologic processes, including clot retraction, cell migration, and wound healing. There are four common forms of f. XIII, resulting in amino acid changes at Val34Leu, Pro564Leu, Val650Ile, and Glu651Gln. The Val34Leu polymorphism results from a G100T coding alteration, and this alteration causes a change in amino acid structure in the A polypeptide chain 3 amino acids from the thrombin cleavage site, which occurs at Arg37-Gly38. Recent evidence has related this polymorphism to risk of VTE. f. XIII disorder is inherited as an autosomal dominant trait with variable severity. As a result, variation of f. XIII activity could affect pulmonary thrombotic risk.47
Inherited defects of enzymatic pathway in relation to development of VTE and PE
Homocysteine is an important contributing factor in thrombus formation, vascular injury, and vascular disease. Its metabolism is a vitamin B6-dependent while goes through transsulfuration which links cystathionine β-synthase (CBS), and a folate and vitamin B12-dependent pathway, involving the enzymes methylenetetrahydrofolate reductase (MTHFR) and methionine synthase. Mudd et al uncovered the innate defect in cobalamin transport and its metabolism in 1964.48 In hyperhomo-cysteinemia plasma homocysteine levels are elevated, which indicates that its metabolism has been compromised, thus excess homocysteine is disposed into blood. Vascular tissue is exposed to its harmful effects. Several molecular mechanisms underlying prothrombotic actions of homocysteine are incompletely understood and involve oxidative stress, DNA hypomethylation, and proinflammatory effects.49 Further abnormalities of platelets, vascular endothelium, and soluble factors involved in blood coagulation, or possibly even lipoproteins might be contributory. Rees et al observed homocysteine effects as cytotoxic on vascular endothelium resulting in intimal thickening with presence of lipidladen macrophages, and smooth muscle cell proliferation.50
Homocysteinuria is a heterogenous group of diseases caused by inherited defects of homocystein, cobalamin and folate metabolism. Two different forms of the disease can be distinguished on the basis of responsiveness to the treatment with large dosages of vitamin B6.51 Congenital homocysteinuria belongs to defects where homozygous inheritance of the gene encoding (CBS) and is characterized higher urine homocysteine levels. Homocysteinuria should be considered as risk factor in VTE patients in early thirties, who had arterial and/or venous thrombotic events. Yap et al reported PE in 3 patients with hyperhomocysteinemia where CBS deficiency was treated chronically.52 Further reported treatment regimens designed to lower plasma homocysteine, significantly reduced cardiovascular risk in CBS deficiency, pointing out the importance of managing mild hyperhomocysteinemia, commonly found in patients with vascular disease.52
Other congenital thrombophilic metabolic condition is based on lipid turnover. Innate high Lipoproteina (Lp a) concentrations are associated with VTE and PE.53 Since the major protein component of Lp (a) is apolipoprotein (a), which acts as a competitive inhibitor of the tissue Plasminogen activator (t-Pa) and is structurally close to plasminogen, it was proposed by von Depka et al54 that Lp (a) competes with plasminogen for fibrin binding leading to impaired fibrinolysis. Additionally, Lp (a) is a significant regulator of synthesis of plasminogen activator inhibitor (PAI-1) by endothelium.
Inherited defects of platelets in relation to PE
In between inherited platelet thrombophilic defects belongs Quebec platelet disorder (QPD), which is a rare, autosomal dominant disease. QPD was characterized at first by a deficiency of platelet associated factor V.55 This defect is further characterized by slower onset of bleeding following hemostasis and consumption of platelet PAI-1. In addition, amplified generation of plasmin in platelets is accompanied by lysis of stored α-granule pro-teins, including Factor V.56 In literature only reported PE associated to QPD is report by McKay in 2004, characterizing woman with QPD after a Caesarian section, as she was taking a fibrinolytic inhibitor to treat a postoperative hematoma.57
Another autosomal dominant platelet disorder is called Sticky Platelet Syndrome (SPS). In platelet rich plasma SPS characterizes hyperaggregability of platelets with adenosine diphosphate (ADP) and epinephrine.58 Platelets become hyperactive upon ADP or epinephrine release, however normal levels of platelet factor 4 (PF4) and beta-thromboglobulin in plasma suggests that they are not activated at all times.59 Hyperactivity of platelets or its state of increased activation is typical for the Weinpenzing defect. It is a rare deficiency of the lipoxygenase metabolic pathway resulting in elevation of thromboxane (TXA) levels. As one of the end products of arachidonic acid metabolism, TXA2 a potent proaggregating and vasoconstrictor agent, is involved in the early pathogenesis of PE.60 TXA2 dominates early vasomotor response, accounting for the early hemodynamic impairment, while prostacyclin is more active in later course.
Loss of normal membrane phospholipid asymmetry present in a subpopulation of RBCs in Sickle Cell Disease (SCD) results from the substitution of Glu by Val at the sixth β-globin chain position of RBCs. Hemoglobin is identified as hemoglobin S (HbS). Other, forms include sickle-hemoglobin disease (HbSC), sickle beta-plus- and sickle beta-zero-thalassemia (HbS/β+), (HbS/β0). These other SCD forms are heterozygous states with one HbS mutated allele and another copy of abnormal hemoglobin allele. Partial or total degradation of globin chains leads to asymmetrical distribution of RBC's membrane phospholipids. One of the important members is the Phosphatidylserine (PS), which is translocated to the edge of RBC's membrane. Procoagulation changes later develop due to the enhanced aggregation of PS, which exposes RBC's to amplify adherence to EC's, and their capacity to enhance thrombin generation through the assembly of the prothrombinase complex.61 The enhanced thrombin generation leads to activation of all other procoagulant factors facilitating consequential hypercoagulable state development. SCD patients exhibit high plasma levels of markers of thrombin generation, reduction of natural anticoagulant proteins, anomalous activation of the fibrinolytic system, and increased TF expression.62 Reported twenty-four out of 83 patients (29%) developed PE, DVT, or portal vein thrombosis after 10 years of observation.63
The congenital deficiency of Histidine rich glycoprotein (HRG) was revealed in a number of individuals with thrombophilia.64 In two separate families spontaneous DVT and in one occasion death due to PE was described, directly linking genetically transmitted high levels of HRG to thrombotic symptoms.65 It is synthesized in liver, found in platelets and mega-karyocytes. HRG is released after platelet thrombin stimulation. By reducing the binding of plasminogen to fibrin, it may represent a physiologic equivalent of antifibrinolytic amino acids, resulting in an antifibrinolytic effect.
Prevalence of inherited thrombophilic states in relation to VTE and PE
Inherited defects of the blood coagulation play a significant role in pathogenesis of VTE. Lowest prevalence is noted in general popula-tion, compared to individuals with single and (or) recurrent VTE (Table 3). Due to the fact that pulmonary embolus is being detected in approximately 50% of patients with documented venous thrombosis in the lower ex-tremities, strong relationship between DVT and PE is warranted. Carriers of two defects seem to be at a higher risk for thrombosis than their relatives with a single defect.
Table 3.
Inherited thrombophilia and its relation to VTE

In patients presenting isolated PE compared to those with PE associated with DVT the prevalence of APCR and (or) FVL has been found to be different.66 Moreover, isolated DVT was more common than DVT plus PE in patients with inherited FVL thrombophilia (22% vs 17%), which contrasted to PT 20210A caused thrombophilia, where DVT + PE developed with higher prevalence than isolated DVT (15% vs 12%) reported by Rossi et al.1 In contrast,1 Okumus et al67 found similar prevalence for FVL and for PT 20210A in Turkish patients with isolated PE and patients with PE plus DVT. The same investigators later analyzed that hereditary thrombophilic risk factors are the most prevalent for later development of isolated DVT, PE, and DVT + PE.68 The PC, PS, ATIII activities, homocysteine levels, FVL and PT 20210A mutations were evaluated in patient and control group. Except FVL and PC, no other factors were significantly different in predisposing patient to PE, DVT or its combination. The combination of thrombophilic risk factors was significantly higher in patients with DVT + PE patient's group as compared to patients with isolated PE or isolated DVT.
Inherited thrombophilic factors and its risk stratification differ between men and woman. Since VTE occurs infrequently during pregnancy, issues concerning its natural history, prevention and therapy stay unresolved. In the report by De Stefano et al69 inherited thrombophilia was not associated with a statistically significant increased risk of VTE in pregnancy or in puerperium. In overall carriers of FVL, recurrence in puerperium was reported 14.2% and 30% in carriers with pregnancy related first VTE, with a risk 6.8 times higher than in women without thrombophilia and with a non pregnancy related first VTE. Thrombophilic tests were more often positive in women who had VTE during the first trimester.
Combined oral hormone replacement therapy and (or) oral contraceptives are recognized risk factors for VTE in thrombophilic women. In female carriers of FVL and PT 20210A, the risk of VTE increased exponentially (35, and 16-fold respectively).70,71 Recently Wu et al72 conducted metaanalysis of clinical trials of women with inherited thrombophilia taking oral contraceptives or hormone replacement therapy. In the oral contraceptive group, significant associations of the risk of VTE were found in women with FVL, PT 20210A, and deficiencies of AT III, PC, PS, and elevated levels of factor VIII. For hormone replacement therapy, a significant association was found in women with FVL.72
There is a lack of information about genetic testing of inherited thrombophilic factors as PE prevention. Inherited thrombophilic factors that have been tested include FVL, PT 20210A, methylenetetrahydrofolate reductase C677T and plasminogen activator inhibitor-1.73,74 Conflicting results hence its clinical utility demand further study to clearly stratify the clinical risk of major inherited thrombophilic factors and its genetic testing to prevent PE.
Authors’ Contributions
FK is the sole author of the “Work”. “Work” is original and does not infringe upon any copyright, proprietary, or personal right of any third party, and neither part of it nor any “Work” based on substantially similar data has been submitted to another journal for publication.
Footnotes
Conflict of interest: Authors have no conflict of interest
References
- 1.Rossi E, Za T, Ciminello A, Leone G, De SV. The risk of symptomatic pulmonary embolism due to proximal deep venous thrombosis differs in patients with different types of inherited thrombophilia. Thromb Haemost. 2008;99(6):1030–4. doi: 10.1160/TH08-02-0069. [DOI] [PubMed] [Google Scholar]
- 2.Egeberg O. Inherited antithrombin deficiency causing thrombophilia. Thromb Diath Haemorrh. 1965;13:516–30. [PubMed] [Google Scholar]
- 3.Picard V, Nowak-Gottl U, Biron-Andreani C, Fouassier M, Frere C, Goualt-Heilman M, et al. Molecular bases of antithrombin deficiency: twenty-two novel mutations in the antithrombin gene. Hum Mutat. 2006;27(6):600. doi: 10.1002/humu.9425. [DOI] [PubMed] [Google Scholar]
- 4.Griffin JH, Evatt B, Zimmerman TS, Kleiss AJ, Wideman C. Deficiency of protein C in congenital thrombotic disease. J Clin Invest. 1981;68(5):1370–3. doi: 10.1172/JCI110385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Comp PC, Nixon RR, Cooper MR, Esmon CT. Familial protein S deficiency is associated with recurrent thrombosis. J Clin Invest. 1984;74(6):2082–8. doi: 10.1172/JCI111632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tollefsen DM, Majerus DW, Blank MK. Heparin cofactor II: purification and properties of a heparindependent inhibitor of thrombin in human plasma. J Biol Chem. 1982;257(5):2162–9. [PubMed] [Google Scholar]
- 7.Tollefsen DM. Heparin cofactor II deficiency. Arch Pathol Lab Med. 2002;126(11):1394–400. doi: 10.5858/2002-126-1394-HCID. [DOI] [PubMed] [Google Scholar]
- 8.Dahlback B, Carlsson M, Svensson PJ. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: prediction of a cofactor to activated protein C. Proc Natl Acad Sci U S A. 1993;90(3):1004–8. doi: 10.1073/pnas.90.3.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koster T, Rosendaal FR, de Ronde H, Briet E, Vandenbroucke JP, Bertina RM. Venous thrombosis due to poor anticoagulant response to activated protein C: Leiden Thrombophilia Study. Lancet. 1993;342(8886, 8887):1503–6. doi: 10.1016/s0140-6736(05)80081-9. [DOI] [PubMed] [Google Scholar]
- 10.Poort SR, Rosendaal FR, Reitsma PH, Bertina RM. A common genetic variation in the 3-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood. 1996;88(10):3698–703. [PubMed] [Google Scholar]
- 11.Miyao N, Suzuki Y, Takeshita K, Kudo H, Ishii M, Hiraoka R, et al. Various adhesion molecules impair microvascular leukocyte kinetics in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2006;290(6):L1059–68. doi: 10.1152/ajplung.00365.2005. [DOI] [PubMed] [Google Scholar]
- 12.Ding BS, Gottstein C, Grunow A, Kuo A, Ganguly K, Albelda SM, et al. Endothelial targeting of a recombinant construct fusing a PECAM-1 singlechain variable antibody fragment (scFv) with prourokinase facilitates prophy-lactic thrombolysis in the pulmonary vasculature. Blood. 2005;106(13):4191–8. doi: 10.1182/blood-2005-05-2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murciano JC, Muro S, Koniaris L, Christofidou-Solomidou M, Harshaw DW, Albelda SM, et al. ICAM-directed vascular immunotargeting of antithrombotic agents to the endothelial luminal surface. Blood. 2003;101(10):3977–84. doi: 10.1182/blood-2002-09-2853. [DOI] [PubMed] [Google Scholar]
- 14.Remillard CV, Yuan JX. PGE2 and PAR-1 in pulmonary fibrosis: a case of biting the hand that feeds you? Am J Physiol Lung Cell Mol Physiol. 2005;288(5):L789–92. doi: 10.1152/ajplung.00016.2005. [DOI] [PubMed] [Google Scholar]
- 15.Carbajal JM, Gratrix ML, Yu CH, Schaeffer RC., Jr ROCK mediates thrombin's endothelial barrier dysfunction. Am J Physiol Cell Physiol. 2000;279(1):C195–C204. doi: 10.1152/ajpcell.2000.279.1.C195. [DOI] [PubMed] [Google Scholar]
- 16.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91(4):1487–500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 17.Garcia JG, Verin AD, Schaphorst K, Siddiqui R, Patterson CE, Csortos C, et al. Regulation of endothelial cell myosin light chain kinase by Rho, cortactin, and p60(src) Am J Physiol. 1999;276(6 Pt 1):L989–98. doi: 10.1152/ajplung.1999.276.6.L989. [DOI] [PubMed] [Google Scholar]
- 18.Reuner KH, Ruf A, Litfin F, Patscheke H. The mutation G20210 --> A in the prothrombin gene is a strong risk factor for pulmonary embolism. Clin Chem. 1998;44(6 Pt 1):1365–6. [PubMed] [Google Scholar]
- 19.Deitcher SR, Caiola E, Jaffer A. Demystifying two common genetic predispositions to venous thrombosis. Cleve Clin J Med. 2000;67(11):825–6. doi: 10.3949/ccjm.67.11.825. [DOI] [PubMed] [Google Scholar]
- 20.Rosendorff A, Dorfman DM. Activated protein C resistance and factor V Leiden: a review. Arch Pathol Lab Med. 2007;131(6):866–71. doi: 10.5858/2007-131-866-APCRAF. [DOI] [PubMed] [Google Scholar]
- 21.Martinelli I, Mannucci PM, De S V, Taioli E, Rossi V, Crosti F, et al. Different risks of thrombosis in four coagulation defects associated with inherited thrombophilia: a study of 150 families. Blood. 1998;92(7):2353–8. [PubMed] [Google Scholar]
- 22.Mateo J, Oliver A, Borrell M, Sala N, Fontcuberta J. Laboratory evaluation and clinical characteristics of 2,132 con-secutive unselected patients with venous thromboembolism--results of the Spanish Multicentric Study on Thrombophilia (EMET-Study) Thromb Haemost. 1997;77(3):444–51. [PubMed] [Google Scholar]
- 23.Bauduer F, Lacombe D. Factor V Leiden, prothrombin 20210A, methylenetetrahydrofolate reductase 677T, and population genetics. Mol Genet Metab. 2005;86(1-2):91–9. doi: 10.1016/j.ymgme.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 24.Williamson D, Brown K, Luddington R, Baglin C, Baglin T. Factor V Cambridge: a new mutation (Arg306 --> Thr) associated with resistance to activated protein C. Blood. 1998;91(4):1140–4. [PubMed] [Google Scholar]
- 25.Koeleman BP, Reitsma PH, Allaart CF, Bertina RM. Activated protein C resistance as an additional risk factor for thrombosis in protein C-deficient families. Blood. 1994;84(4):1031–5. [PubMed] [Google Scholar]
- 26.Reitsma PH, Bernardi F, Doig RG, Gandrille S, Greengard JS, Ireland H, et al. Protein C deficiency: a database of mutations, 1995 update. On behalf of the Subcommittee on Plasma Coagulation Inhibitors of the Scientific and Standardization Committee of the ISTH. Thromb Haemost. 1995;73(5):876–89. [PubMed] [Google Scholar]
- 27.Stamatoyannopoulos G, Majerus PW, Perlmutter RM, Varmus H. 3rd ed. Philadelphia: Saunders; 2000. The molecular basis of blood disease. [Google Scholar]
- 28.Heeb MJ, Mesters RM, Tans G, Rosing J, Griffin JH. Binding of protein S to factor Va associated with inhibition of prothrombinase that is independent of activated protein C. J Biol Chem. 1993;268(4):2872–7. [PubMed] [Google Scholar]
- 29.Van Wijnen M, Stam JG, van’t Veer C, Meijers JC, Reitsma PH, Bertina RM, et al. The interaction of protein S with the phospholipid surface is essential for the activated protein C-independent activity of protein S. Thromb Haemost. 1996;76(3):397–403. [PubMed] [Google Scholar]
- 30.Engesser L, Broekmans AW, Briet E, Brommer EJ, Bertina RM. Hereditary protein S deficiency: clinical manifesta-tions. Ann Intern Med. 1987;106(5):677–82. doi: 10.7326/0003-4819-106-5-677. [DOI] [PubMed] [Google Scholar]
- 31.Ohlin AK, Marlar RA. The first mutation identified in the thrombomodulin gene in a 45-year-old man presenting with thromboembolic disease. Blood. 1995;85(2):330–6. [PubMed] [Google Scholar]
- 32.Bertina RM, Leiden Factor V, et al. Factor mutations affecting thrombotic risk. Clin Chem. 1997;43(9):1678–83. [PubMed] [Google Scholar]
- 33.Anderson FA, Jr, Spencer FA. Risk factors for venous thromboembolism. Circulation. 2003;107(23 Suppl 1):I-9–I-16. doi: 10.1161/01.CIR.0000078469.07362.E6. [DOI] [PubMed] [Google Scholar]
- 34.Ishiguro K, Kojima T, Kadomatsu K, Nakayama Y, Takagi A, Suzuki M, et al. Complete antithrombin deficiency in mice results in embryonic lethality. J Clin Invest. 2000;106(7):873–8. doi: 10.1172/JCI10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tollefsen DM, Pestka CA. Heparin cofactor II activity in patients with disseminated intravascular coagulation and hepatic failure. Blood. 1985;66(4):769–74. [PubMed] [Google Scholar]
- 36.Parker KA, Tollefsen DM. The protease specificity of heparin cofactor II: inhibition of thrombin generated during coagulation. J Biol Chem. 1985;260(6):3501–5. [PubMed] [Google Scholar]
- 37.Tollefsen DM, Pestka CA, Monafo WJ. Activation of heparin cofactor II by dermatan sulfate. J Biol Chem. 1983;258(11):6713–6. [PubMed] [Google Scholar]
- 38.Kraaijenhagen RA, In’t Anker PS, Koopman MM, Reitsma PH, Prins MH, Van den EA, et al. High plasma concentration of factor VIIIc is a major risk factor for venous thromboembolism. Thromb Haemost. 2000;83(1):5–9. [PubMed] [Google Scholar]
- 39.Wise RJ, Dorner AJ, Krane M, Pittman DD, Kaufman RJ. The role of von Willebrand factor multimers and propeptide cleavage in binding and stabilization of factor VIII. J Biol Chem. 1991;266(32):21948–55. [PubMed] [Google Scholar]
- 40.Koster T, Blann AD, Briet E, Vandenbroucke JP, Rosendaal FR. Role of clotting factor VIII in effect of von Wille-brand factor on occurrence of deepvein thrombosis. Lancet. 1995;345(8943):152–5. doi: 10.1016/s0140-6736(95)90166-3. [DOI] [PubMed] [Google Scholar]
- 41.Kyrle PA, Minar E, Hirschl M, Bialonczyk C, Stain M, Schneider B, et al. High plasma levels of factor VIII and the risk of recurrent venous thromboembolism. N Engl J Med. 2000;343(7):457–62. doi: 10.1056/NEJM200008173430702. [DOI] [PubMed] [Google Scholar]
- 42.Ten Wolde M, Duffels MGJ, Bank I, Bakhtiari K, Meijers JCM, Hutten BA. High levels of coagulation factors IX or XI: risk factors for pulmonary embolism? J.Thromb.Haemost. 2003;1(suppl 1) Abstract number: P1455. [Google Scholar]
- 43.Lammle B, Wuillemin WA, Huber I, Krauskopf M, Zurcher C, Pflugshaupt R, et al. Thromboembolism and bleeding tendency in congenital factor XII deficiency--a study on 74 subjects from 14 Swiss families. Thromb Haemost. 1991;65(2):117–21. [PubMed] [Google Scholar]
- 44.Girolami A, Randi ML, Gavasso S, Lombardi AM, Spiezia F. The occasional venous thromboses seen in patients with severe (homozygous) FXII deficiency are probably due to associated risk factors: a study of prevalence in 21 patients and review of the literature. J Thromb Thrombolysis. 2004;17(2):139–43. doi: 10.1023/B:THRO.0000037670.42776.cd. [DOI] [PubMed] [Google Scholar]
- 45.Lindahl AK, Sandset PM, Abildgaard U. The present status of tissue factor pathway inhibitor. Blood Coagul Fibri-nolysis. 1992;3(4):439–49. [PubMed] [Google Scholar]
- 46.Aoki N, Moroi M, Sakata Y, Yoshida N, Matsuda M. Abnormal plasminogen: a hereditary molecular abnormality found in a patient with recurrent thrombosis. J Clin Invest. 1978;61(5):1186–95. doi: 10.1172/JCI109034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Francis CW. Factor XIII polymorphisms and venous thromboembolism. Arch Pathol Lab Med. 2002;126(11):1391–3. doi: 10.5858/2002-126-1391-FXPAVT. [DOI] [PubMed] [Google Scholar]
- 48.Mudd SH, Finkelstein JD, Irreverre F, Laster L. Homocystinuria: an enzymatic defect. Science. 1964;143:1443–5. doi: 10.1126/science.143.3613.1443. [DOI] [PubMed] [Google Scholar]
- 49.Undas A, Brozek J, Szczeklik A. Homocysteine and thrombosis: from basic science to clinical evidence. Thromb Haemost. 2005;94(5):907–15. doi: 10.1160/TH05-05-0313. [DOI] [PubMed] [Google Scholar]
- 50.Rees MM, Rodgers GM. Homocysteinemia: association of a metabolic disorder with vascular disease and thrombosis. Thromb Res. 1993;71(5):337–59. doi: 10.1016/0049-3848(93)90160-p. [DOI] [PubMed] [Google Scholar]
- 51.Fowler B, Kraus J, Packman S, Rosenberg LE. Homocystinuria. Evidence for three distinct classes of cystathionine betasynthase mutants in cultured fibroblasts. J Clin Invest. 1978;61(3):645–53. doi: 10.1172/JCI108976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yap S, Boers GH, Wilcken B, Wilcken DE, Brenton DP, Lee PJ, et al. Vascular outcome in patients with homocystinuria due to cystathionine betasynthase deficiency treated chronically: a multicenter observational study. Arterioscler Thromb Vasc Biol. 2001;21(12):2080–5. doi: 10.1161/hq1201.100225. [DOI] [PubMed] [Google Scholar]
- 53.Ignatescu M, Kostner K, Zorn G, Kneussl M, Maurer G, Lang IM, et al. Plasma Lp(a) levels are increased in patients with chronic thromboembolic pulmonary hypertension. Thromb Haemost. 1998;80(2):231–2. [PubMed] [Google Scholar]
- 54.Von Depka M, Nowak-Gottl U, Eisert R, Dieterich C, Barthels M, Scharrer I, et al. Increased lipoprotein (a) levels as an independent risk factor for venous thromboembolism. Blood. 2000;96(10):3364–8. [PubMed] [Google Scholar]
- 55.Tracy PB, Giles AR, Mann KG, Eide LL, Hoogendoorn H, Rivard GE. Factor V (Quebec): a bleeding diathesis associated with a qualitative platelet Factor V deficiency. J Clin Invest. 1984;74(4):1221–8. doi: 10.1172/JCI111531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kufrin D, Eslin DE, Bdeir K, Murciano JC, Kuo A, Kowalska MA, et al. Antithrombotic thrombocytes: ectopic expression of urokinase-type plasminogen activator in platelets. Blood. 2003;102(3):926–33. doi: 10.1182/blood-2003-01-0054. [DOI] [PubMed] [Google Scholar]
- 57.McKay H, Derome F, Haq MA, Whittaker S, Arnold E, Adam F, et al. Bleeding risks associated with inheritance of the Quebec platelet disorder. Blood. 2004;104(1):159–65. doi: 10.1182/blood-2003-11-4077. [DOI] [PubMed] [Google Scholar]
- 58.Mammen EF. Sticky platelet syndrome. Semin Thromb Hemost. 1999;25(4):361–5. doi: 10.1055/s-2007-994939. [DOI] [PubMed] [Google Scholar]
- 59.Reeves WC, Demers LM, Wood MA, Skarlatos S, Copenhaver G, Whitesell L, et al. The release of thromboxane A2 and prostacyclin following experimental acute pulmonary embolism. Prostaglandins Leukot Med. 1983;11(1):1–10. doi: 10.1016/0262-1746(83)90104-x. [DOI] [PubMed] [Google Scholar]
- 60.Ghuysen A, Lambermont B, Dogne JM, Kolh P, Tchana-Sato V, Morimont P, et al. Effect of BM-573 [N-terbutyl-N’-[2-(4’-methylphenylamino)-5-nitro-benzenesulfonyl]urea], a dual thromboxane synthase inhibitor and thromboxane receptor antagonist, in a porcine model of acute pulmonary embolism. J Pharmacol Exp Ther. 2004;310(3):964–72. doi: 10.1124/jpet.104.066852. [DOI] [PubMed] [Google Scholar]
- 61.Eldor A, Rachmilewitz EA. The hypercoagulable state in thalassemia. Blood. 2002;99(1):36–43. doi: 10.1182/blood.v99.1.36. [DOI] [PubMed] [Google Scholar]
- 62.Stein PD, Beemath A, Meyers FA, Skaf E, Olson RE. Deep venous thrombosis and pulmonary embolism in hospitalized patients with sickle cell disease. Am J Med. 2006;119(10):897.e7–11. doi: 10.1016/j.amjmed.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 63.Cappellini MD, Robbiolo L, Bottasso BM, Coppola R, Fiorelli G, Mannucci AP. Venous thromboembolism and hypercoagulability in splenectomized patients with thalassaemia intermedia. Br J Haematol. 2000;111(2):467–73. doi: 10.1046/j.1365-2141.2000.02376.x. [DOI] [PubMed] [Google Scholar]
- 64.Shigekiyo T, Ohshima T, Oka H, Tomonari A, Azuma H, Saito S. Congenital histidinerich glycoprotein deficiency. Thromb Haemost. 1993;70(2):263–5. [PubMed] [Google Scholar]
- 65.Castaman G, Ruggeri M, Burei F, Rodeghiero F. High levels of histidinerich glycoprotein and thrombotic diathesis: report of two unrelated families. Thromb Res. 1993;69(3):297–305. doi: 10.1016/0049-3848(93)90027-l. [DOI] [PubMed] [Google Scholar]
- 66.Margaglione M, Brancaccio V, De Lucia D, Martinelli I, Ciampa A, Grandone E, et al. Inherited thrombophilic risk factors and venous thromboembolism: distinct role in peripheral deep venous thrombosis and pulmonary embolism. Chest. 2000;118(5):1405–11. doi: 10.1378/chest.118.5.1405. [DOI] [PubMed] [Google Scholar]
- 67.Okumus G, Kiyan E, Arseven O, Tabak L, Abaci N, Unaltuna NE, et al. Inherited thrombophilic risk factors in venousthromboembolism: factor V leiden and prothrombin 20210 A. Turkish Resp J. 2004;5(2):82–5. [Google Scholar]
- 68.Okumus G, Kiyan E, Arseven O, Tabak L, Diz-Kucukkaya R, Unlucerci Y, et al. Hereditary thrombophilic risk factors and venous thromboembolism in Istanbul, Turkey: the role in different clinical manifestations of venous thromboembolism. Clin Appl Thromb Hemost. 2008;14(2):168–73. doi: 10.1177/1076029607305620. [DOI] [PubMed] [Google Scholar]
- 69.De SV, Martinelli I, Rossi E, Battaglioli T, Za T, Mannuccio MP, et al. The risk of recurrent venous thromboembolism in pregnancy and puerperium without antithrombotic prophylaxis. Br J Haematol. 2006;135(3):386–91. doi: 10.1111/j.1365-2141.2006.06317.x. [DOI] [PubMed] [Google Scholar]
- 70.Vandenbroucke JP, Koster T, Briet E, Reitsma PH, Bertina RM, Rosendaal FR. Increased risk of venous thrombosis in oral-contraceptive users who are carriers of factor V Leiden mutation. Lancet. 1994;344(8935):1453–7. doi: 10.1016/s0140-6736(94)90286-0. [DOI] [PubMed] [Google Scholar]
- 71.Martinelli I, Taioli E, Bucciarelli P, Akhavan S, Mannucci PM. Interaction between the G20210A mutation of the prothrombin gene and oral contraceptive use in deep vein thrombosis. Arterioscler Thromb Vasc Biol. 1999;19(3):700–3. doi: 10.1161/01.atv.19.3.700. [DOI] [PubMed] [Google Scholar]
- 72.Wu O, Robertson L, Langhorne P, Twaddle S, Lowe GD, Clark P, et al. Oral contraceptives, hormone replacement therapy, thrombophilias and risk of venous thromboembolism: a systematic review. The Thrombosis: Risk and Economic Assessment of Thrombophilia Screening (TREATS) Study. Thromb Haemost. 2005;94(1):17–25. doi: 10.1160/TH04-11-0759. [DOI] [PubMed] [Google Scholar]
- 73.Westrich GH, Weksler BB, Glueck CJ, Blumenthal BF, Salvati EA. Correlation of thrombophilia and hypofibrinolysis with pulmonary embolism following total hip arthroplasty: an analysis of genetic factors. J Bone Joint Surg Am. 2002;84-A(12):2161–7. doi: 10.2106/00004623-200212000-00006. [DOI] [PubMed] [Google Scholar]
- 74.Kruse L, Mitchell AM, Camargo CA, Jr, Hernandez J, Kline JA. Frequency of thrombophilia-related genetic variations in patients with idiopathic pulmonary embolism in an urban emergency department. Clin Chem. 2006;52(6):1026–32. doi: 10.1373/clinchem.2005.061861. [DOI] [PubMed] [Google Scholar]