

Abstract
Human Th17 cells express high levels of CD161, a member of the killer cell lectin-like receptor (KLR) family (also referred to as NK receptor-P1A (NKRP1A) or KLRB1), as a representative marker. CD161 is also expressed on natural killer (NK) cells and NKT cells. Lectin-like transcript 1 (LLT1), another KLR family member, was recently identified as a ligand for CD161. This interaction may play pivotal roles in the immunomodulatory functions of Th17 cells as well as those of NK and NKT cells. However, the molecular basis for the interaction is poorly understood. Here we show that the extracellular domain of CD161 bound directly to LLT1 with a Kd of 48 μm and with the fast kinetics typical of cell-cell recognition receptors. Mutagenesis revealed that the similar membrane-distal β-sheet and loop regions of both CD161 and LLT1 were utilized for the binding, and notably, these regions correspond to the ligand-binding sites for major histocompatibility complex (MHC)-recognizing KLRs. Furthermore, we found a pair of detrimental mutations for both molecules that restored the binding. These results reveal a new template model for the recognition mode between the KLR family members and provide insights into the molecular mechanism underlying Th17/NK/NKT-mediated immune responses.
Keywords: Cell Surface Receptor, Immunosupressor, MHC, Protein-Protein Interactions, Surface Plasmon Resonance (SPR), CD161, NKRP1, Th17 Cell, C-type Lectin-like Receptors, Natural Killer Cells
Introduction
Human CD161 (also called NK receptor-P1A (NKRP1A), killer cell lectin-like receptor subfamily B member 1 (KLRB1), or C-type lectin domain family 5 member B (CLEC5B)) is expressed on Th17 cells, natural killer (NK)3 cells, and NKT cells. CD161 belongs to the killer cell lectin-like receptor (KLR) family, and its members contain one C-type lectin-like domain in the extracellular region responsible for the ligand recognition. Although mice have several CD161 molecules, which are both activating and inhibitory receptors and known as the NKRP1 family, humans have only one CD161 molecule, which is an inhibitory receptor (1, 2).
The CD161/NKRP1 family molecules are type II transmembrane glycoproteins, which form disulfide-linked homodimers (2). Ligands for the murine NKRP1 family molecules were identified as C-type lectin-related (Clr)-g molecules, which also belong to the KLR family (3, 4). On the other hand, Aldemir et al. (5) and Rosen et al. (6) identified one of the KLR family members, the human lectin-like transcript-1 molecule (LLT1) (also referred to as CLEC2D) as a ligand for the human CD161, and this recognition inhibited the CD161+ NK cell cytotoxicity. Another study reported that proliferation-induced lymphocyte-associated receptor (PILAR) (also named KACL/CLEC2A) is a second ligand of human CD161 (7); however, recent reports using a tetramer staining technique revealed that PILAR cannot bind to CD161 but instead binds to NKp65, which also belongs to the KLR family (8, 9).
Recently, in addition to the CD161-LLT1 recognition, various interactions among KLR family members were reported, such as those between NKp80 and AICL and between NKp65 and PILAR (KACL) (8, 10). NKp80 binds to AICL with low affinity (Kd ∼4 μm), whereas NKp65 binds to PILAR (KACL) with high affinity (Kd ∼0.01 μm). However, their molecular recognition modes have remained elusive. Here, we have determined the biophysical characteristics of the CD161-LLT1 interaction, including the kinetic and thermodynamic properties, by a surface plasmon resonance analysis. Based on a broad mutagenesis study, we identified the binding sites for both LLT1 and CD161 as the membrane distal areas, which are also utilized for ligand binding by other KLR members (CD94-NKG2-MHCI and KLRG1-E-cadherin (11)), and revealed the first binding topology template for interactions among KLR family members.
EXPERIMENTAL PROCEDURES
Construction of Expression Plasmids
The DNA fragment encoding the extracellular domain (amino acid residues Leu71–Val191) of human LLT1 was amplified from the cDNA (code number EHS1001-437530, Open Biosystems). The amplified fragment was cloned into the expression vector pET-22b(+) (Novagen), to create the plasmid pET22bLLT1.
The DNA fragment encoding the extracellular domain (amino acid residues Gly90–Ser225) of human CD161 was amplified from human lymphocyte-derived cDNA. The fragment was cloned into a derivative of the expression vector pCA7 (12), to create the plasmid pCA7CD161. This vector contains the signal sequence (upstream of the protein-coding sequence) derived from the pHLsec vector (13). The biotin ligase (BirA) recognition sequence (GSLHHILDAQKMVWNHR) was inserted between the signal sequence and Gly90 of the CD161 sequence for the SPR analysis.
Preparation of Soluble LLT1 Proteins
The expression plasmid pET22bLLT1 was transformed into the Escherichia coli strain BL21(DE3)pLysS. The cells were cultured in LB medium with 100 mg/liter ampicillin (Nacalai Tesque, Kyoto, Japan) at 310 K. When the A600 reached 0.6, isopropyl β-d-thiogalactopyranoside (Nacalai Tesque) was added for induction, at a final concentration of 1 mm. The recombinant LLT1 protein was expressed as insoluble inclusion bodies. After 4 h of induction, the cells were harvested by centrifugation. The inclusion bodies were isolated from the cell pellet by sonication and were washed repeatedly with Triton wash buffer (50 mm Tris-HCl, pH 8.0, 100 mm NaCl, 0.5% Triton X-100). The purified LLT1 inclusion bodies were solubilized in denaturant buffer (50 mm MES, pH 6.5, 100 mm NaCl, 10 mm EDTA, 6 m guanidine HCl). The solubilized protein solution was slowly diluted by the addition of ice-cold refolding buffer (100 mm Tris-HCl, pH 8.0, 2 mm EDTA, 1 m l-arginine, 3.73 mm cystamine, 6.73 mm cysteamine) to a final protein concentration of 2 μm. After 72 h at 277 K, the refolded protein solution was concentrated with a VIVAFLOW50 system (Sartorius). The LLT1 protein was purified by size exclusion chromatography, using a Superdex-75 column (GE Healthcare).
Preparation of Soluble, Biotinylated CD161 Protein
The expression plasmid pCA7CD161 was transiently transfected, using polyethyleneimine, into 90% confluent HEK293T cells. The cells were cultured in DMEM (Sigma), supplemented with 10% FCS (Nichirei Bioscience), l-glutamine, and nonessential amino acids (Invitrogen). The DMEM medium was substituted with serum-free Opti-MEM (Invitrogen) medium immediately after transfection. The culture supernatant was harvested 72 h after transfection. The supernatant was concentrated and replaced with Tris buffer (20 mm Tris-HCl, pH 8.0), using an AmiconUltra filter (nominal molecular weight limit of 10,000; Millipore Corp.). The CD161 was biotinylated with the BirA enzyme. After biotinylation, the buffer of the reaction mixture was replaced with HBS-EP (10 mm HEPES, pH 7.4, 150 mm NaCl, 3 mm EDTA, 0.005% Surfactant P20).
Mutant Preparation of LLT1 and CD161
The mutagenesis of C163S and H176C of LLT1 was performed using a KOD Plus mutagenesis kit (Toyobo) with pET22bLLT1 as the template, to produce pET22bLLT1C163S and pET22bLLT1H176C, respectively. In addition, H176C-based mutagenesis (Y165A, N167A, K169E, R175E, R180E, and K181E) were performed, using pET22bLLT1H176C as the template. All LLT1 mutants were prepared in the same manner as the wild type.
Similarly, CD161 mutageneses (E162R, E179R, E179A, R181E, D183R, K185E, K185A, E186R, Y198A, E200R, E200A, Y201A, and E205R) were performed using pCA7CD161. All CD161 mutants were prepared in the same manner as the wild type.
Binding Analysis Using Surface Plasmon Resonance (SPR)
SPR experiments were performed using a BIAcore3000 (GE Healthcare). The biotinylated CD161 and control protein (biotinylated BSA) were immobilized on research grade CM5 chips (GE Healthcare), covalently coupled with streptavidin. The purified LLT1 proteins were buffer-substituted with HBS-EP and injected over the immobilized CD161, at a flow rate of 10 μl/min. The binding response at each concentration was calculated by subtracting the equilibrium response, measured in the control flow cell, from the response in each sample flow cell. The data were analyzed using BIAevaluation software, version 4.1, and ORIGIN software, version 7. Affinity constants (Kd) were derived by nonlinear curve fitting of the standard Langmuir binding isotherm.
For kinetics, the LLT1 was injected at a flow rate of 30 μl/min. The global fitting analysis using the 1:1 Langmuir binding model was simultaneously performed with the raw data for the association and dissociation phases at different concentrations of the LLT1 protein. Fitting was performed using the BIA evaluation software, version 4.1.
Equilibrium analyses of LLT1 were performed at five temperature points (283.15, 288.15, 293.15, 298.15, and 303.15 K). The standard state Gibbs energy change upon binding was obtained from Equation 1,
where Kd is the dissociation constant, expressed in units of mol/liter, and R is the gas constant. The ΔG values of each data set were plotted against the temperatures and were fitted with the nonlinear van't Hoff equation (Equation 2),
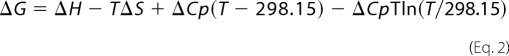 |
where ΔH and ΔS are the binding enthalpy and entropy at 298.15 K, respectively, and ΔCp is the heat capacity, which is assumed to be temperature-independent.
Circular Dichroism (CD) Spectrum
CD spectra were recorded on a JASCO model J-805 CD spectrometer. Far-UV CD measurements were performed with a 20 μm concentration of each protein in HBS-EP buffer, using a 1-mm cell and a bandwidth of 1 nm. Spectra were accumulated four times.
RESULTS AND DISCUSSION
Expression and Purification of Soluble LLT1 and CD161 Proteins
The extracellular domain of human LLT1 receptor was expressed in E. coli as inclusion bodies and was refolded in vitro by a dilution method. The refolded LLT1 migrated as a monomer and was purified by size exclusion chromatography, and the final product exhibited high purity (Fig. 1A). The wild type LLT1 tended to aggregate. We compared the amino acid sequence of the extracellular region of LLT1 with those of other C-type lectin family members, by a Basic Local Alignment Search Tool (BLAST) search. Human CD69 (hCD69), whose crystal structure was determined previously, shares 40% identity with the extracellular region of LLT1 (Fig. 2, A and C). The crystal structure of hCD69 revealed that its C-type lectin-like domain has three disulfide bonds. The Cys163 and His176 residues of LLT1 are equivalent to the Cys173 and Cys186 residues of hCD69, respectively. His176 cannot form the disulfide bond with Cys163, and thus Cys163 is a free cysteine. We expected that this might destabilize the protein and thus introduced the H176C mutation, to allow disulfide bond formation with Cys163. As another option, we substituted Cys163 with serine, to eliminate the free cysteine residue. The C163S and H176C mutants were both expressed and refolded by the same method as that used for the wild type. The size exclusion chromatography profiles and the CD spectra of these mutants were essentially the same as those of the wild type (supplemental Fig. S1). The stability of the C163S mutant was not improved, and the protein still tended to precipitate; however, the H176C mutant was stable enough for use in further biophysical experiments.
FIGURE 1.

Binding analyses of CD161-LLT1 interaction. A, size exclusion chromatography analysis of LLT1. Chromatogram of wild type LLT1 on HiLoad 26/60 Superdex 75 pg size exclusion column. The bars indicate the elution positions of the molecular mass markers. Inset, SDS-PAGE analysis of the peak fractions from the size exclusion chromatography. The LLT1 protein is indicated by the arrow on the right side of the gel. B, measuring the affinity of the CD161-LLT1 interaction. The affinity of the CD161-LLT1 interaction was determined by equilibrium binding experiments. Ten 2-fold dilutions of LLT1 (H176C) (174–0.34 μm) were injected over the CD161, and the responses were plotted against the concentrations of injected LLT1 (H176C) protein. Inset, Scatchard plot of the same data is shown. The solid line is linear fit. C, kinetic analysis of the CD161-LLT1 interaction. LLT1 (H176C) at the indicated concentrations was injected (solid bar) over CD161 (430 response units (RU)). Rate equations derived from the 1:1 binding model were fitted to the association and dissociation phases of all four injections (global fitting). Residual errors from the fits are shown in the bottom panel. D, thermodynamic analysis of the CD161-LLT1 interaction. The dissociation constant (Kd) for the CD161-LLT1 interaction was measured at five temperatures (283.15–303.15 K) and was converted into the standard free energy of binding (ΔG). Values for the enthalpic (ΔH) and standard entropic (−TΔS) changes (at 298.15 K) and the specific heat capacity (ΔCp) were determined by fitting the non-linear van't Hoff equation to these data. E, comparison of the thermodynamic properties of several protein-protein interactions (at 298.15 K). The values for protein-protein interactions (excluding antibody-antigen interactions) are the mean ± S.E. (error bars) of 30 distinct interactions taken from Stites (20).
FIGURE 2.

Amino acid sequence alignment and structure of CTLDs. Shown are amino acid sequence alignments of the CTLDs of LLT1 with PILAR and AICL (A) and CD161 with NKp65 and NKp80 (B). Secondary structure elements (yellow arrows indicate β-strands, red boxes indicate α-helices, and purple boxes indicate 310 helices) of hCD69 and mDectin-1 are displayed above the alignments. The asterisks indicate residues mutated in this study, with detrimental effect in red and modest effect in orange. The magenta arrows indicate the pair of residues that showed detrimental effects when mutated independently but restored the binding when mutated simultaneously. C, ribbon diagrams of hCD69 (left) and mDectin-1 (right). Each diagram shows side and top views. Secondary structure elements are labeled as follows: α-helices (red), β-strands (yellow), and 310 helices (purple).
On the other hand, unlike LLT1, the CD161 protein could not be properly refolded by the dilution method. However, using HEK293T cells, the extracellular region of CD161 with the C-terminal biotinylation tag could be expressed transiently, as a soluble protein (for details, see “Experimental Procedures”). The secreted CD161 was biotinylated with the BirA enzyme and was specifically immobilized on the streptavidin-coupled sensor chip for the SPR analysis.
Surface Plasmon Resonance Analysis of the CD161-LLT1 Interaction
To analyze the molecular interaction between CD161 and LLT1, we performed an SPR analysis, using the above mentioned recombinant proteins. Soluble LLT1 proteins were injected over sensor surfaces to which biotinylated CD161 or biotinylated BSA (as a negative control) had been immobilized. The affinity constants of LLT1 binding to CD161 were determined by an equilibrium binding analysis. The response derived from the negative control was subtracted from each response derived from the CD161 protein. Fig. 1B shows the conventional plots of these binding data. CD161 bound the wild type, C163S, and H176C LLT1 proteins with similar affinities, and the respective dissociation constants (Kd) were 53, 52, and 48 μm at 298.15 K. We confirmed that the CD161 binding to the immobilized LLT1 in the inverted orientation also shows similar binding characteristics with weak affinity and fast kinetics (data not shown); however, the soluble CD161 was prone to aggregation, and thus the precise binding parameters could not be determined.
Binding Kinetics of the CD161-LLT1 Interaction
The equilibrium binding data for CD161 binding with the wild type and C163S and H176C mutants of LLT1 indicated fast kinetics (supplemental Fig. S2). Both the wild type and C163S LLT1 proteins were unstable and tended to aggregate, making it difficult to obtain precise association and dissociation rates. On the other hand, the H176C mutant exhibited much higher solubility and stability than the others. Therefore, the H176C LLT1 mutant was used as the standard template for further analyses, including kinetics, thermodynamics, and mutagenesis studies (hereafter, the H176C LLT1 protein is referred to simply as the “LLT1 protein”). The detailed kinetic parameters of the CD161-LLT1 interaction were determined by global fitting of the monoexponential rate equations, derived from the simple 1:1 Langmuir binding model, to the binding responses. Fig. 1C illustrates the reasonable fitting with small residual errors (bottom panel) to the data for LLT1 binding, which yielded typical association (kon = 1.1 ± 0.1 × 105 m−1 s−1) and fast dissociation (koff = 5.3 ± 0.55 s−1) kinetics similar to those of other cell-cell recognition receptors (Table 1 and supplemental Table SI). The kinetically derived Kd and that determined by equilibrium binding were essentially the same, providing further evidence that these kinetic parameters are correct.
TABLE 1.
Kinetic and thermodynamic parameters of the interaction between LLT1 and CD161 at 25 °C
| Immobilized | Analyte | Kineticsa |
|||
|---|---|---|---|---|---|
| Kd, eqb | kon (× 105) | koff | Kd, kinc | ||
| μm | m−1s−1 | s−1 | μm | ||
| CD161 | Wild type LLT1 | 52.6 | |||
| C163S LLT1 | 51.8 | ||||
| H176C LLT1 | 48.2 | 1.1 ± 0.1 | 5.3 ± 0.55 | 48.5 ± 7.5 | |
| Thermodynamicsd |
|||||
|---|---|---|---|---|---|
| ΔG | ΔH | −TΔS | ΔCp | ||
| kcal/mol | kcal/mol/K | ||||
| CD161 | H176C LLT1 | −5.9 ± 0.02 | −3.2 ± 0.13 | −2.7 ± 0.11 | −0.41 ± 0.01 |
a The values are means ± range of two experiments.
b Kd, eq values were obtained from the equilibrium analysis.
c Kd, kin values were obtained from global fitting.
d The values are means ± S.E. of three experiments.
Thermodynamic Properties of the CD161-LLT1 Interaction
The non-linear van't Hoff analysis (Equation 2) was performed to determine the thermodynamic parameters. The binding affinities of the CD161-LLT1 interaction were determined at five temperatures, from 283.15 to 303.15 K. Reasonable fitting of the non-linear van't Hoff equation to the data produced the proper thermodynamic parameters (Fig. 1D). The binding of LLT1 to CD161 was characterized by favorable entropic and enthalpic changes at 298.15 K (Table 1 and Fig. 1E), in contrast to the TCR-MHC interactions (14–19), which exhibited a large, unfavorable entropic change and a large heat capacity change (ΔCp ∼1 kcal mol−1 K−1) (supplemental Table SII). In addition, the TCR-MHC interaction exhibited slow kinetics. This characteristic is believed to be caused by the adjustment of the flexible ligand binding site (“induced fit”) and/or the trapping of water molecules at the binding interface. The CD161-LLT1 interaction exhibited a moderate ΔCp of ∼0.41 kcal mol−1 K−1. Taken together, the binding properties of the CD161-LLT1 interaction are characterized by entropically and enthalpically driven binding, a small ΔCp, and fast kinetics (20), indicating that only small conformational changes may be required for binding. This is similar to other low affinity interactions of cell-cell recognition molecules, including the KIR2DL3-HLA-Cw7 (21), LILRB2-HLA-G (22), and NKG2D-ligand (MIC-A, ULBP3, and RAE-1β) (23) interactions.
Mapping of Binding Sites
To identify the CD161 binding site on LLT1, a wide range of LLT1 mutants were prepared (see “Experimental Procedures”) and used for the SPR analysis. LLT1 and CD161 are quite similar to the other KLRs (whereas LLT1 displays 40% amino acid identity to hCD69, CD161 displays 30% to murine Dectin-1 (mDectin-1)) (Fig. 2). The typical structure of a KLR, as depicted by KLRG1 in Fig. 3A, reveals a conventional C-type lectin-like domain (CTLD) fold, comprising two α-helices (α1 and α2) and two antiparallel β-sheets (β0-β1-β5-β1′ and β2′-β2-β3-β4-β2″). Although the ligands for the KLRs are different (for MHC, Ly49 (24) and CD94/NKG2 (25); for E-cadherin, KLRG1 (11)), they utilize their membrane-distal head region for ligand binding (Fig. 3A and supplemental Fig. S3). Based on the crystal structures of hCD69 (for LLT1) and mDectin-1 (for CD161), we designed mutations in the putative ligand binding regions to identify the interaction sites (Figs. 2 and 3B). Six LLT1 mutants (Y165A, N167A, K169E, R175E, R180E, and K181E) were properly refolded (supplemental Fig. S1), and the SPR analysis showed that all of the mutants exhibited reduced or undetectable CD161 binding (Fig. 3C). Notably, the K169E, R175E, R180E, and K181E mutations completely disrupted the CD161 binding. The Y165A and N167A mutations of LLT1 had moderate effects on the interaction, resulting in a 4–5-fold decrease in the interaction affinity.
FIGURE 3.

Structure of E-cadherin-KLRG1 complex and mutagenesis studies. A, structure of the E-cadherin-KLRG1 complex structure. KLRG1 is depicted by a yellow ribbon diagram. Ligand binding region of KLRG1 is colored red. E-cadherin is shown as a dark gray surface representation. Two views of KLRG1 are presented (side and top views). Secondary structure elements are labeled. B, binding affinities of LLT1 mutants for immobilized CD161 mutants at 298.15 K. C, ribbon diagrams of model structures of LLT1 (top, blue) and CD161 (bottom, green). Each diagram shows side and top views. Residues mutated in this study are shown as spheres with detrimental effect in red and modest effect in orange. The magenta spheres indicate the pair of residues that showed detrimental effects when mutated independently but restored the binding when mutated simultaneously.
In another orientation, to clarify the LLT1 binding site on CD161, 13 CD161 mutants (E162R, E179R, E179A, R181E, D183R, K185E, K185A, E186R, Y198A, E200R, E200A, Y201A, and E205R) were expressed and biotinylated, using the same method as for the wild type (Figs. 2 and 3B). We examined the conformations of the CD161 proteins using a mouse anti-human CD161 monoclonal antibody (mAb) B199.2 (Abcam), which only recognizes the conformational epitope of CD161 (26). Wild type CD161 and all of the mutants were recognized by mAb B199.2 (supplemental Fig. S4), indicating that they are all properly folded. The SPR analysis revealed that, although the E179R, K185E, and E200R mutations had no substantial impact on LLT1 binding, the alanine mutations of these three residues disrupted the LLT1 binding. The R181E and E186R mutations of CD161 had modest effects on binding, resulting in a 2–3-fold decrease in the affinity. The E162R, D183R, Y198A, Y201A, and E205R mutations of CD161 had detrimental effects on binding (Fig. 3C).
We performed SPR binding analyses of combinations between the LLT1 and CD161 mutants to identify the mutation combination(s) restoring the binding activity and thus to provide the putative binding pair(s). Although almost all of the LLT1 mutations were unable to bind to the CD161 mutants, the K169E mutation restored the binding to the CD161 E205R mutant (Fig. 4A and Table 2). This strongly suggests that Lys169 of LLT1 directly interacts with Glu205 of CD161. Y198A and Y201A CD161 mutants abolished binding to all LLT1 mutants, and E162R and D183R CD161 mutants displayed little affinity to the wild type LLT1 and no binding to the other mutants. On the other hand, R175E, R180E, and K181E mutants rendered LLT1 unable to bind to all of the CD161 mutants. These residues showing the most significant effects are located close together and a bit far from the Glu205(CD161)-Lys169(LLT1) binding partners. The same strategy has been employed to determine binding orientations, by identifying the effects of charge swap mutations on the typical low affinity binding often observed in cell-cell recognition events (e.g. CD2-CD48 (27) and TCR-MHC complexes (28)). Together with the present study, the results suggest that amino acid substitutions in these low affinity interactions are relatively tolerated. Therefore, the charge swap mutation strategy would be useful for determining the binding orientation in cell-cell recognition receptors.
FIGURE 4.

Model structure of CD161-LLT1 complex. A, SPR binding analyses of combinations between the LLT1 and CD161 mutants. Filled squares and solid line, E205R CD161/wild type LLT1; filled circles and dashed line, wild type CD161/K169E LLT1; filled triangles and solid line, E205R CD161/K169E LLT1. B, schematic model of LLT1 recognition by CD161. Ribbon diagrams and spheres are the same as in Fig. 3C. C, amino acid sequence alignments of the putative receptor/ligand binding regions of LLT1 with PILAR and AICL (top) and CD161 with NKp65 and NKp80 (bottom). Secondary structure elements of hCD69 and mDectin-1 are displayed above the alignments. The asterisks indicate residues mutated in this study, with color representations corresponding to those in B and Fig. 3C.
TABLE 2.
Binding affinities of LLT1 mutants for immobilized CD161 mutants at 25 °C
| CD161 |
Kd |
||||||
|---|---|---|---|---|---|---|---|
| WT LLT1 | Y165A | N167A | K169E | R175E | R180E | K181E | |
| μm | |||||||
| Wild type | 48 | 240 | 180 | NBa | NB | NB | NB |
| E162R | 310 | NB | NB | NB | NB | NB | NB |
| E179R | 34 | 150 | 120 | 300 | NB | NB | NB |
| E179A | 393 | NB | NB | NB | NB | NB | NB |
| R181E | 100 | 210 | 140 | NB | NB | NB | NB |
| D183R | 390 | NB | NB | NB | NB | NB | NB |
| K185E | 55 | 170 | 140 | 310 | NB | NB | NB |
| K185A | 236 | 180 | 149 | 318 | NB | NB | NB |
| E186R | 150 | NB | NB | NB | NB | NB | NB |
| Y198A | NB | NB | NB | NB | NB | NB | NB |
| E200R | 45 | NB | NB | NB | NB | NB | NB |
| E200A | NB | NB | NB | NB | NB | NB | NB |
| Y201A | NB | NB | NB | NB | NB | NB | NB |
| E205R | 370 | NB | NB | 26 | NB | NB | NB |
a NB, no detectable binding.
Considering the above results together with the electrostatic and hydrophilic/hydrophobic complementation, we constructed a feasible model of the CD161-LLT1 complex, using the hCD69 and mDectin-1 structures as templates. Both CD161 and LLT1 utilize similar membrane-distal head regions, from the β3 strand (Tyr165) to the β5 strand (Lys181) of LLT1 and from the β2-β3 loop (Glu162) to the β4-β5 loop (Glu205) of CD161, for binding, and thus the complex model resembles a symmetrical dimer (Fig. 4B). This complex structure supports the previous report that PILAR (KACL) cannot bind to CD161 (8, 9) because Asp152 of PILAR (KACL), which corresponds to Lys169 of LLT1, probably disrupts the interaction with Glu205 of CD161 (Fig. 4, B and C). We further investigated the binding modes of other KLR-KLR interactions, such as NKp80-AICL and NKp65-PILAR. Notably, the putative binding sites (especially the β3 to β4-β5 loop regions) of the CD161-LLT1 complex model were relatively conserved, in either the CD161 relatives (CD161/NKp65/NKP80) or the LLT1 relatives (LLT1/PILAR/AICL) (Figs. 2, A and B, and 4C). This strongly suggests that the KLR-KLR recognition involves a common binding mode. On the other hand, the amino acid differences, including the binding pair residues, Val224(NKp80)-Asp129(AICL) and Thr188(NKp65)-Asp152(PILAR), corresponding to Glu205(CD161)-Lys169(LLT1) (Fig. 4, B and C), would contribute to discrete ligand specificities.
We constructed the CD161 dimer-LLT1 dimer complex model structure without any disruption of the proposed interaction topology, as shown in supplemental Fig. S5. If the dimer-dimer interaction occurred on the cell-cell interface, then this would probably show an avidity effect to enhance the binding activity.
CONCLUSION
In this report, we showed that CD161 binds LLT1 with low affinity, fast kinetics, and entropically and enthalpically driven thermodynamics with a small heat capacity, which are typical features for interactions mediating cell-cell recognition. Furthermore, we revealed a new recognition mode between KLR family members. As in the other KLRs, CD161 and LLT1 both utilize similar membrane-distal head β-sheet and loop regions for binding. The binding pair residues, revealed by the mutation combinations, suggested a plausible CD161-LLT1 complex model, which is the first template model for the interactions between KLR family members, such as NKp65-PILAR and NKp80-AICL, that explains their binding specificities.
CD161 is predominantly expressed on all human Th17 cells. Th17 cells have important roles in autoimmune and chronic inflammatory disorders, such as rheumatoid arthritis (31–33). The control of the Th17 responses has already yielded successful results in a clinical trial with rheumatoid arthritis patients (29). Although CD161 has neither activating nor inhibitory signaling motifs, CD161 acts as both an activating and inhibitory receptor, depending on the cell type. The CD161-LLT1 interaction leads to the inhibition of NK cell cytotoxicity (6). However, the interaction in combination with TCR signaling activates T cells to secrete interferon (IFN)-γ (5). CD161 on Th17 cells also acts as a co-activating receptor and promotes T cell expansion. Furthermore, the expression level of CD161 on Th17 cells is largely up-regulated in inflammatory disorders (30). Although the detailed mechanisms of CD161 regulation of immune cells are not well understood, our results provided here show that the membrane-distal head region of CD161 is a new target to inhibit the CD161-LLT1 interaction, leading to the regulation of autoimmune and chronic inflammatory disorders.
Supplementary Material
Acknowledgments
We thank K. Furuno, N. Sugihara, T. Ose, Y. Tanaka, I. Tanaka, and K. Akasaki for helpful discussions.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5 and Tables SI and SII.
- NK
- natural killer
- KLR
- killer cell lectin-like receptor
- SPR
- surface plasmon resonance
- CTLD
- C-type lectin-like domain.
REFERENCES
- 1. Lanier L. L., Chang C., Phillips J. H. (1994) J. Immunol. 153, 2417–2428 [PubMed] [Google Scholar]
- 2. Mesci A., Ljutic B., Makrigiannis A. P., Carlyle J. R. (2006) Immunol. Res. 35, 13–26 [DOI] [PubMed] [Google Scholar]
- 3. Iizuka K., Naidenko O. V., Plougastel B. F., Fremont D. H., Yokoyama W. M. (2003) Nat. Immunol. 4, 801–807 [DOI] [PubMed] [Google Scholar]
- 4. Carlyle J. R., Jamieson A. M., Gasser S., Clingan C. S., Arase H., Raulet D. H. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 3527–3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aldemir H., Prod'homme V., Dumaurier M. J., Retiere C., Poupon G., Cazareth J., Bihl F., Braud V. M. (2005) J. Immunol. 175, 7791–7795 [DOI] [PubMed] [Google Scholar]
- 6. Rosen D. B., Bettadapura J., Alsharifi M., Mathew P. A., Warren H. S., Lanier L. L. (2005) J. Immunol. 175, 7796–7799 [DOI] [PubMed] [Google Scholar]
- 7. Huarte E., Cubillos-Ruiz J. R., Nesbeth Y. C., Scarlett U. K., Martinez D. G., Engle X. A., Rigby W. F., Pioli P. A., Guyre P. M., Conejo-Garcia J. R. (2008) Blood 112, 1259–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Spreu J., Kuttruff S., Stejfova V., Dennehy K. M., Schittek B., Steinle A. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 5100–5105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Germain C., Bihl F., Zahn S., Poupon G., Dumaurier M. J., Rampanarivo H. H., Padkjær S. B., Spee P., Braud V. M. (2010) J. Biol. Chem. 285, 36207–36215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Welte S., Kuttruff S., Waldhauer I., Steinle A. (2006) Nat. Immunol. 7, 1334–1342 [DOI] [PubMed] [Google Scholar]
- 11. Li Y., Hofmann M., Wang Q., Teng L., Chlewicki L. K., Pircher H., Mariuzza R. A. (2009) Immunity. 31, 35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takeda M., Ohno S., Seki F., Nakatsu Y., Tahara M., Yanagi Y. (2005) J. Virol. 79, 14346–14354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aricescu A. R., Assenberg R., Bill R. M., Busso D., Chang V. T., Davis S. J., Dubrovsky A., Gustafsson L., Hedfalk K., Heinemann U., Jones I. M., Ksiazek D., Lang C., Maskos K., Messerschmidt A., Macieira S., Peleg Y., Perrakis A., Poterszman A., Schneider G., Sixma T. K., Sussman J. L., Sutton G., Tarboureich N., Zeev-Ben-Mordehai T., Jones E. Y. (2006) Acta. Crystallogr. D Biol. Crystallogr. 62, 1114–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boniface J. J., Reich Z., Lyons D. S., Davis M. M. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 11446–11451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Willcox B. E., Gao G. F., Wyer J. R., Ladbury J. E., Bell J. I., Jakobsen B. K., van der Merwe P. A. (1999) Immunity 10, 357–365 [DOI] [PubMed] [Google Scholar]
- 16. Garcia K. C., Radu C. G., Ho J., Ober R. J., Ward E. S. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 6818–6823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Anikeeva N., Lebedeva T., Krogsgaard M., Tetin S. Y., Martinez-Hackert E., Kalams S. A., Davis M. M., Sykulev Y. (2003) Biochemistry 42, 4709–4716 [DOI] [PubMed] [Google Scholar]
- 18. Lee J. K., Stewart-Jones G., Dong T., Harlos K., Di Gleria K., Dorrell L., Douek D. C., van der Merwe P. A., Jones E. Y., McMichael A. J. (2004) J. Exp. Med. 200, 1455–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Davis-Harrison R. L., Armstrong K. M., Baker B. M. (2005) J. Mol. Biol. 346, 533–550 [DOI] [PubMed] [Google Scholar]
- 20. Stites W. E. (1997) Chem. Rev. 97, 1233–1250 [DOI] [PubMed] [Google Scholar]
- 21. Maenaka K., Juji T., Nakayama T., Wyer J. R., Gao G. F., Maenaka T., Zaccai N. R., Kikuchi A., Yabe T., Tokunaga K., Tadokoro K., Stuart D. I., Jones E. Y., van der Merwe P. A. (1999) J. Biol. Chem. 274, 28329–28334 [DOI] [PubMed] [Google Scholar]
- 22. Shiroishi M., Kuroki K., Tsumoto K., Yokota A., Sasaki T., Amano K., Shimojima T., Shirakihara Y., Rasubala L., van der Merwe P. A., Kumagai I., Kohda D., Maenaka K. (2006) J. Mol. Biol. 355, 237–248 [DOI] [PubMed] [Google Scholar]
- 23. McFarland B. J., Strong R. K. (2003) Immunity. 19, 803–812 [DOI] [PubMed] [Google Scholar]
- 24. Dam J., Guan R., Natarajan K., Dimasi N., Chlewicki L. K., Kranz D. M., Schuck P., Margulies D. H., Mariuzza R. A. (2003) Nat. Immunol. 4, 1213–1222 [DOI] [PubMed] [Google Scholar]
- 25. Petrie E. J., Clements C. S., Lin J., Sullivan L. C., Johnson D., Huyton T., Heroux A., Hoare H. L., Beddoe T., Reid H. H., Wilce M. C., Brooks A. G., Rossjohn J. (2008) J. Exp. Med. 205, 725–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bennett I. M., Zatsepina O., Zamai L., Azzoni L., Mikheeva T., Perussia B. (1996) J. Exp. Med. 184, 1845–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van der Merwe P. A., McNamee P. N., Davies E. A., Barclay A. N., Davis S. J. (1995) Curr. Biol. 5, 74–84 [DOI] [PubMed] [Google Scholar]
- 28. Jorgensen J. L., Esser U., Fazekas de St Groth B., Reay P. A., Davis M. M. (1992) Nature 355, 224–230 [DOI] [PubMed] [Google Scholar]
- 29. Genovese M. C., Van den Bosch F., Roberson S. A., Bojin S., Biagini I. M., Ryan P., Sloan-Lancaster J. (2010) Arthritis Rheum. 62, 929–939 [DOI] [PubMed] [Google Scholar]
- 30. Kleinschek M. A., Boniface K., Sadekova S., Grein J., Murphy E. E., Turner S. P., Raskin L., Desai B., Faubion W. A., de Waal Malefyt R., Pierce R. H., McClanahan T., Kastelein R. A. (2009) J. Exp. Med. 206, 525–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cosmi L., Cimaz R., Maggi L., Santarlasci V., Capone M., Borriello F., Frosali F., Querci V., Simonini G., Barra G., Piccinni M. P., Liotta F., Palma R. D., Maggi E., Romagnani S., Annunziato F. (2011) Arthritis Rheum., in press [DOI] [PubMed] [Google Scholar]
- 32. Maggi L., Santarlasci V., Capone M., Peired A., Frosali F., Crome S. Q., Querci V., Fambrini M., Liotta F., Levings M. K., Maggi E., Cosmi L., Romagnani S., Annunziato F. (2010) Eur. J. Immunol. 40, 2174–2181 [DOI] [PubMed] [Google Scholar]
- 33. Cosmi L., De Palma R., Santarlasci V., Maggi L., Capone M., Frosali F., Rodolico G., Querci V., Abbate G., Angeli R., Berrino L., Fambrini M., Caproni M., Tonelli F., Lazzeri E., Parronchi P., Liotta F., Maggi E., Romagnani S., Annunziato F. (2008) J. Exp. Med. 205, 1903–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.