

Abstract
Lipoxygenases have been implicated in cardiovascular disease. A rare single-nucleotide polymorphism causing T560M exchange has recently been described, and this mutation leads to a near null variant of the enzyme encoded for by the ALOX15 gene. When we inspected the three-dimensional structure of the rabbit ortholog, we localized Thr-560 outside the active site and identified a hydrogen bridge between its side chain and Gln-294. This interaction is part of a complex hydrogen bond network that appears to be conserved in other mammalian lipoxygenases. Gln-294 and Asn-287 are key amino acids in this network, and we hypothesized that disturbance of this hydrogen bond system causes the low activity of the T560M mutant. To test this hypothesis, we first mutated Thr-560 to amino acids not capable of forming side chain hydrogen bridges (T560M and T560A) and obtained enzyme variants with strongly reduced catalytic activity. In contrast, enzymatic activity was retained after T560S exchange. Enzyme variants with strongly reduced activity were also obtained when we mutated Gln-294 (binding partner of Thr-560) and Asn-287 (binding partner of Gln-294 and Met-418) to Leu. Basic kinetic characterization of the T560M mutant indicated that the enzyme lacks a kinetic lag phase but is rapidly inactivated. These data suggest that the low catalytic efficiency of the naturally occurring T560M mutant is caused by alterations of a hydrogen bond network interconnecting this residue with active site constituents. Disturbance of this bonding network increases the susceptibility of the enzyme for suicidal inactivation.
Keywords: Atherosclerosis, Eicosanoid-specific Enzymes, Enzyme Structure, Fatty Acid Metabolism, Lipid Oxidation, Lipoxygenase Pathway, Molecular Epidemiology, Single-Nucleotide Polymorphism
Introduction
Lipoxygenases (LOXs)2 catalyze the peroxidation of free and/or esterified polyunsaturated fatty acids to corresponding hydroperoxy compounds (1, 2). They constitute a heterogeneous family of lipid metabolizing enzymes and have been implicated in the pathogenesis of inflammatory (3, 4), degenerative (5, 6), and hyperproliferative (7, 8) diseases. The possible role of ALOX15 in cardiovascular diseases has been a matter of discussion for many years (9, 10) and pro- and anti-atherogenic (11–13) activities have been reported in various animal atherosclerosis models. In humans, expression of ALOX15 has been detected in atherosclerotic lesions (14), and structural analysis of deposited lipids suggested its catalytic activity in young human plaques (15). On the other hand, more recent studies failed to detect significant amounts of ALOX15 mRNA in advanced human lesions (16) but suggested significantly higher levels of ALOX5 transcripts. This finding and the pro-inflammatory character of ALOX5 are consistent with the hypothesis that atherosclerosis is an inflammatory disease of the vessel wall (17, 18).
In a population-based case control study (19) involving some 1800 subjects with clinically significant coronary artery disease and some 1700 healthy controls, a novel single-nucleotide polymorphism (SNP) in the coding region of the ALOX15 gene was reported. This nonsynonymous SNP exchanges Thr-560 to Met (T560M), and in vitro mutagenesis studies on the recombinant ALOX15 indicated a strong reduction of the catalytic activity of the T560M mutant (19). Heterozygous allele carriers had a significantly increased risk for coronary artery disease (adjusted odds ratio of 1.62; p = 0.02). When this SNP was genotyped in the patient cohort of the Atherosclerosis Risk in Communities study, heterozygote carriers also showed an increased risk for coronary artery disease (19), which was borderline significant (adjusted hazards ratio, 1.31; p = 0.06). In both studies, homozygote carriers were too rare to draw conclusions. In an independent large scale (some 2600 participants) case control study (20), a similar trend toward an increased risk for myocardial infarction was observed for heterozygote allele carriers of the T560M mutation (odd ratio, 1.7; p = 0.06).
The molecular basis for the strongly reduced catalytic activity of the T560M mutant has not been explored in detail. Structural modeling on the basis of the x-ray coordinates of the rabbit ortholog (21, 22) indicated that Thr-560 is not an immediate constituent of the active site. Instead, it is localized in a more flexible loop region that has no direct contact to the catalytic center. This study was aimed at exploring the mechanistic basis for the low catalytic efficiency of the naturally occurring T560M mutant of ALOX15. Our data suggest that the loss in catalytic activity is caused by a disturbance of a hydrogen bond network that surrounds the bottom of the substrate-binding pocket and that these alterations induce an increased susceptibility of the enzyme for catalytic inactivation.
MATERIALS AND METHODS
Chemicals
The chemicals used were obtained from the following sources: arachidonic acid (5Z,8Z,11Z,14Z-eicosatetraenoic acid) from Serva (Heidelberg, Germany); HPLC standards of 12S-HETE, 12(±)-HETE, 15S-HETE, and 15(±)-HETE from Cayman Chemical; sodium borohydride and ampicillin from Invitrogen; isopropyl-β-thiogalactopyranoside from Carl Roth GmbH (Karlsruhe, Germany). HPLC solvents were purchased from Baker (Deventer, The Netherlands). Restriction enzymes were obtained from Fermentas (St. Leon-Rot, Germany). Oligonucleotide synthesis was performed at BioTez (Berlin, Germany), and nucleic acid sequencing was carried out at Eurofins MWG Operon (Ebersberg, Germany). The Escherichia coli strain XL-1 blue was purchased from Stratagene (La Jolla, CA).
Bacterial Expression and Site-directed Mutagenesis of ALOX15
Wild-type human ALOX15 and its mutants were expressed as N-terminal His tag fusion proteins in E. coli as described before (23). For this purpose, the cDNA was cloned into the pQE-9 prokaryotic expression plasmid in such a way that the starting methionine of the LOX coding sequence was deleted. Because of technical reasons, the N terminus was elongated by additional amino acids including six consecutive His. Site-directed mutagenesis was performed using the QuikChangeTM site-directed mutagenesis kit (Stratagene, Amsterdam, The Netherlands). For each mutant, 5–10 clones were selected and screened for LOX expression, and one clone was completely sequenced to confirm mutagenesis.
Purification of Recombinant ALOX15
Wild-type human ALOX15 and selected mutants were affinity-purified on a Ni-TED matrix open bed column. For purification, LOX-active clones were picked with a sterilized toothpick, and 20 ml of LB medium containing ampicillin (0.1 mg/liter) were inoculated. After 8 h at 37 °C, 15 ml were added to 3 liters of LB medium containing ampicillin (0.1 mg/liter), and bacteria were grown at 37 °C overnight. The cultures were cooled to room temperature, and after expression of recombinant LOX was initiated by the addition of 1 mm isopropyl-β-thiogalactopyranoside further incubated for 4 h at 25 °C. Then cells were spun down, reconstituted in PBS, and lysed with an EmulsiFlex-C5 high pressure homogenizer (Avestin, Ottawa, Canada). Cell debris was spun down, the supernatant was added to 0.5 ml of Ni-TED (Machery-Nagel, Düren, Germany), and the suspension was incubated for 1 h at 4 °C. The Ni-TED was transferred to an open bed column and washed with washing buffer (50 mm NaH2PO4, 300 mm NaCl, pH 8.0), and the bound proteins were eluted with elution buffer (50 mm NaH2PO4, 300 mm NaCl, 200 mm imidazole, pH 8.0). The LOX activity of the elution fractions was tested employing a spectrophotometric assay. The pooled LOX containing Ni-TED fractions were supplemented with 10% glycerol and shock frozen in liquid nitrogen. An electrophoretic purity of ∼90% usually was obtained.
To determine the amount of LOX protein, Western blot analyses of wild-type and mutant enzymes were quantified (supplemental Fig. S1). Aliquots of the LOX containing Ni-TED fractions were separated by SDS gel electrophoresis and blotted on a nitrocellulose membrane, and His-tagged proteins were detected using an RGSHis antibody (Qiagen) at 1:5000 dilution. As secondary antibody, an anti-mouse IgG at a dilution of 1:5000 was used (Sigma-Aldrich).
In different batches of enzyme preparations, we found that the specific activity of purified wild-type human ALOX15 varied between 20 and 40 s−1 (under Vmax conditions), and these data are consistent with corresponding values obtained under comparable conditions for native and recombinant rabbit ALOX15 enzyme.
Cloning, Expression, and Activity Assay of Mouse 5-LOX (alox5)
500 μl of mouse blood were treated with fMet-Phe-Leu for 30 min at 37 °C, and total RNA was prepared using the QIAamp RNA blood mini kit from Qiagen. After reverse transcription (RevertAid H Minus reverse transcriptase; Fermentas, St. Leon-Rot, Germany), 100 ng of the resulting cDNA were used as template to amplify mouse alox5 by PCR. The PCR fragment was cloned into the pRSET A expression vector (Invitrogen), which contains an N-terminal His tag sequence, between unique XhoI and HindIII restriction sites. After sequencing, recombinant alox5 was expressed in the E. coli strain BL21(DE3)pLysS (Invitrogen) as described for ALOX15. The recombinant protein was purified on a Co-Sepharose open bed column (BD Talon; BD Biosciences, Heidelberg, Germany), and elution of the protein was achieved by 200 mm imidazole. The fractions containing 5-LOX activity were pooled and used for activity assays.
As for ALOX15, catalytic activity of alox5 was assayed by HPLC quantification of arachidonic acid oxygenation products. For this purpose, aliquots of the purified enzyme preparation were incubated with arachidonic acid (100 μm) for 10 min in PBS containing 0.4 mm Ca2+, 0.1 mm ATP, and 40 μg/ml phosphatidyl choline (final concentrations). Product preparation and HPLC analysis were carried out as described for ALOX15.
Expression and Activity Assay of Human Platelet 12-LOX (ALOX12)
Human platelet 12-LOX (ALOX12) was expressed as His tag fusion protein as described before (24). Enzyme purification and activity assays of wild-type and mutant enzyme species were carried out as described above for ALOX15.
Determination of Iron Content of Wild-type ALOX15 and Selected Mutants
To determine the iron content, LOX containing Ni-TED fractions were further purified by FPLC using a Resource Q column (GE Healthcare). The electrophoretically homogeneous FPLC fractions were concentrated and desalted by gel filtration, and the iron content was measured by atom absorption spectroscopy on a PerkinElmer Life Sciences AA800 instrument equipped with an AS800 autosampler. The iron content was related to LOX protein that was quantified spectrophotometrically (1 mg/ml pure ALOX15 has an absorbance of 1.78 at 280 nm).
HPLC-based Activity Assays
For routine activity assays, one sequenced clone was replated, five well separated colonies were picked, and the bacteria were cultured overnight at 37 °C in 5 ml of LB medium containing 0.1 mg/ml ampicillin. LOX expression was induced by the addition of isopropyl-β-thiogalactopyranoside (final concentration, 1 mm). After 2 h at 30 °C, the bacteria were spun down, washed, and reconstituted in 0.5 ml of PBS. Arachidonic acid was added on ice (final concentration, 100 μm), and the cells were lysed by sonication with a Labsonic U-tip sonifier (Braun, Melsungen, Germany). The mixture was incubated for 10 min at 25 °C, the hydroperoxy compounds formed were reduced with sodium borohydride, and after acidification to pH 3 (acetic acid), 0.5 ml of methanol were added. The protein precipitate was spun down, and aliquots of the clear supernatant were injected directly for quantification of the LOX products to reverse phase HPLC. For most activity assays, instead of the 5-ml overnight cultures, purified LOX fractions were used applying the same protocol.
For time dependence measurements, the activity assays were incubated for the scheduled time periods at 25 °C, and then the reaction was stopped by the addition of 500 μl of ice-cold methanol. Hydroperoxides were reduced with sodium borohydride, samples were acidified with acetic acid, and protein precipitation was spun down. Aliquots of the supernatant were used for reverse phase HPLC quantification.
HPLC analysis was performed on a Shimadzu instrument equipped with a Hewlett-Packard diode array detector 1040 A by recording the absorbance at 235 nm. Reverse phase HPLC was carried out on a Nucleodur C18 gravity column (Machery-Nagel; 250 × 4 mm, 5-μm particle size) coupled with a guard column (8 × 4 mm, 5-μm particle size). A solvent system of methanol/water/acetic acid (85/15/0.05, v/v/v) was used at a flow rate of 1 ml/min. 15-HETE enantiomers were separated by chiral phase HPLC as free fatty acids on a Chiralcel OD column (Daicel Chem. Ind., Ltd.) using a solvent system consisting of hexane/2-propanol/acetic acid (100/5/0.1, v/v/v) and a flow rate of 1 ml/min. 12-HETE enantiomers were separated as methyl esters after incubating with diazomethane for 15 min at room temperature on the Chiralcel OD column using a solvent system of hexane/2-propanol/acetic acid (100/2/0.1, v/v/v) and a flow rate of 1 ml/min.
Structural Modeling and Amino Acid Sequence Alignments
Human and rabbit ALOX15 proteins share 81% sequence identity. For the rabbit enzyme, the crystal structure was solved, and the Protein Data Bank entry 2P0M was used to model the structure of the human ortholog. The recently solved crystal structure of human 5-LOX (25) was employed to construct a model of mouse 5-LOX (alox5). The VMD software package (26) was employed for structural analysis and figure preparation. Hydrogen bonds were identified with the Swiss PdbViewer v4.0.1. Multiple sequence alignments were performed applying clustalW2 program provided by the European Bioinformatics Institute.
Statistics
The experimental raw data were statistically evaluated employing the Microsoft Excel® software package (version 12.0). The means ± standard deviations were calculated. For significance calculations, the two-sided (type 2) Student's t test was employed.
RESULTS
Thr-560 Is Not an Active Site Residue but Forms a Hydrogen Bridge with Gln-294
Human ALOX15 shares a high degree (81%) of sequence identity with the ortholog rabbit enzyme, and thus, the x-ray coordinates of the rabbit enzyme were employed to draw structural conclusions on the human enzyme. For clarity reasons, we use amino acid numbering of human ALOX15 throughout this paper.
When we inspected the structure of the rabbit ALOX15, we found that Thr-560 is not an active site residue. Although it is localized in proximity to Ile-417 (Fig. 1A), which contributes to form the bottom of the substrate-binding pocket (27), the closest distance between the two residues is 4.12 Å. This is beyond the binding distance of noncovalent interactions. Thr-560 is localized in a loop region that lacks stable secondary structural elements. Its OH group (hydrogen donor) bridges with the side chain oxygen of the amide group (hydrogen acceptor) of Gln-294 (Fig. 1B), which is located in a neighboring loop. This hydrogen bridge might stabilize this structural microenvironment.
FIGURE 1.

Localization of Thr-560 in the three-dimensional structure of ALOX15 and its involvement in a network of hydrogen bonds. A, Thr-560 is not an immediate active site residue. Although it is located in proximity to Ile-417, there is no direct interaction to any of the triade constituents (Phe-352, Ile-417, Met-418, and Ile-592; numbering according to human ALOX15). The shortest distance (4.12 Å) was measured between the backbone carbonyl of Ile-417 and the backbone nitrogen of Thr-560, which is beyond the binding distance of noncovalent interactions. B, the side chain OH group of Thr-560 hydrogen bridges with side chain oxygen of Gln-294. This hydrogen bridge might contribute to stabilize the less well structured loop regions indicated in green and pink. C, the loop regions surrounding Thr-560 and Gln-294 do not undergo major structural rearrangement upon ligand binding at the active site. In fact, the bonding distance of the Thr-560–Gln-294 hydrogen bridge, which is 2.62 Å in the ligand-free structure, only increases to 2.72 Å in the ligand-bound form. As indicated before, there is a pronounced relocation of helix 2 during ligand binding. Gray, ligand-free conformer; light blue, ligand-bound conformer; orange, arachidonic acid. D, Gln-294 is a key residue in a hydrogen bond network, which connects the side chain of Thr-560 with Met-418, a member of the amino acid triade that determines the positional specificity of mammalian 12/15-LOXs. The images were prepared with the VMD software package (26). Hydrogen bridges were determined with Swiss-PdbViewer, v4.0.1.
The rabbit ALOX15 undergoes structural rearrangement when a ligand is bound at the active site, and two major conformers (ligand-free and ligand-bound) have been described (22). The conformational changes include dislocation of surface helix 2 and, to make room for ligand binding, retreat of helix 18 from the active site. To explore whether the three-dimensional structure of the Thr-560 region is altered upon ligand binding, we compared the two conformers but did not find major structural differences (Fig. 1C). In fact, the bonding distance of the Thr-560–Gln-294 hydrogen bridge was nearly unchanged (2.62 Å for the ligand-free conformer versus 2.73 Å for the ligand-bound conformer). More detailed structural investigations indicated that the Thr-560–Gln-294 hydrogen bridge is part of a more complex hydrogen bond network (Fig. 1D). Here, Gln-294 and Asn-287 appear to play major roles because each of these residues interacts with four different binding partners. Interestingly, this hydrogen bond network interconnects Thr-560 with Met-418, which has previously been shown to directly interact with substrate fatty acids (24, 27).
Mutation of Thr-560 to Residues Not Capable of Forming Side Chain Hydrogen Bridges Impairs Catalytic Activity
The naturally occurring T560M mutant in the human enzyme lacks the hydrogen donating side chain OH group, and the purified recombinant protein exhibits a strongly impaired catalytic activity (Fig. 2A and Table 1). This corresponds well with the previously reported 5% residual activity for this mutant when expressed in E. coli (19). Similar results were obtained when wild-type human ALOX15 and its T560M mutant were expressed in human embryonic kidney cells (19), and we confirmed this data in the current study (not shown). The T560A mutant, which also lacks the hydrogen donating OH group, exhibited a residual catalytic activity of only 17% (Table 1). In contrast, when Thr-560 was mutated to Ser, an amino acid that carries a hydrogen donating OH group, the catalytic activity was largely retained. In fact, we measured a residual activity of more than 70% (Table 1). These data confirm the catalytic importance of the Thr-560–Gln-294 hydrogen bridge. Moreover, we found that these mutants exhibited reaction specificities similar to those of the wild-type enzyme (Table 1), suggesting that no major alterations in enzyme-substrate interaction were induced.
FIGURE 2.

T560M and Q294L exchanges inactivate human ALOX15. Wild-type and mutant enzymes were expressed and purified as described under “Materials and Methods.” LOX concentrations of each preparation were quantified by immunoblotting, and equal amounts of enzyme were used in the activity assays. Representative example chromatograms are shown. The y axis was scaled for the product amount formed by the wild-type enzyme. In A, the mobile phase of the HPLC system consisted of 80% methanol, 20% water, and 0.05% acetic acid, resulting in longer retention times. For B, the solvent composition was 85% methanol, 15% water, and 0.05% acetic acid as described under “Materials and Methods.”
TABLE 1.
Catalytic activity and reaction specificity of human ALOX15 Thr-560 mutants
Recombinant expression of human ALOX15 variants (n = 5 for each mutant), product preparation, and HPLC analysis were performed as described under “Materials and Methods” with the crude cell lysate of 5-ml cultures. The ALOX15 content of the different samples was quantified by immunoblotting, and comparable amounts of LOX protein were used for activity assays. The amounts of HETE-isomers (15-HETE + 12-HETE) were quantified for each sample, and HETE formation of the wild-type ALOX15 was set at 100%.
| LOX species | Relative catalytic activity | Share of 15-HETE |
|---|---|---|
| % | % | |
| Wild-type | 100 ± 9.9 | 83.6 ± 3.5 |
| T560M | 3.8 ± 1.0 | 86.5 ± 1.2 |
| T560A | 16.6 ± 5.7 | 85.0 ± 1.4 |
| T560S | 70.7 ± 24.6 | 84.5 ± 0.7 |
Q294L Exchange Also Leads to a Loss in Catalytic Activity
As suggested in Fig. 1B, the hydroxy group of Thr-560 hydrogen bridges with the side chain of Gln-294. If this hydrogen bridge is important for the catalytic activity, mutation of Gln-294 to a residue lacking a hydrogen acceptor should also lead to an inactive enzyme. Indeed, when we tested the purified Q294L mutant in our HPLC-based activity assay, we observed a strongly reduced catalytic activity (Fig. 2B). Further activity assays using higher (5-fold) enzyme concentration of the Q294L mutant indicated a product pattern that was very similar to that of the wild-type enzyme (84% 15-HETE, 16% 12-HETE; see supplemental Fig. S2). Taken together, the strongly impaired catalytic activity of the T560M mutant and the even lower activity of the Q294L variant suggest the functional importance of the hydrogen bridge between Thr-560 and Gln-294.
Asn-287 is a Key Residue in the Hydrogen Bonding Network and Its Mutation Also Inactivated the ALOX15
A second key element in this hydrogen bond network is Asn-287 (Fig. 1D). Its side chain amide forms hydrogen bridges with the backbone of Val-288, with Asp-415, and with Met-418. When we mutated this residue to Leu (N287L), an amino acid that cannot form any side chain hydrogen bridges, we obtained a mutant enzyme that exhibited less than 1% residual catalytic activity (Table 2). We next replaced Asn-287 with Asp. The side chain of aspartate might act as hydrogen acceptor, and thus the hydrogen bridge with the backbone amide of Val-288 should have been retained. However, because there is no amide nitrogen in the side chain, the hydrogen bridges to Asp-415 and Met-418 (Fig. 1D) should have been lost. Although the N287D mutant exhibits a strongly reduced catalytic activity when compared with the wild-type enzyme (Table 2), its reaction rate was significantly (p < 0.016) higher than that of the N287L mutant. Finally, we created the N287Q mutant. Asparagine and glutamine carry identical functional side chain residues but differ with respect to their hydrocarbon chain length. In principle, the side chain hydrogen bridges formed by Asn-287 might be retained in the N287Q mutant despite the additional CH2 group in the side chain. When we compared the catalytic activity of the purified N287Q mutant with that of the wild-type enzyme, we found that the mutant enzyme exhibited only ∼4% residual activity (Table 2). However, when compared with the N287D and N287L mutants, the catalytic activity of the N287Q variant was significantly higher (Fig. 3). The product pattern of the N287Q mutant was similar to that of the wild-type enzyme.
TABLE 2.
Catalytic activity and reaction specificity of human ALOX15 Q294L and Asn-287 mutants
Recombinant expression of human ALOX15, enzyme purification, product preparation, and HPLC analysis were performed as described under “Materials and Methods.” Equal amounts of enzyme were used, products (15-HETE + 12-HETE) were quantified for each sample, and the wild-type ALOX15 was set at 100%. For each mutant, three independent activity assays were carried out, and the means ± S.D. are given in the Table. ND, not determined because of lack of sufficient material. NS, not significant versus wild-type enzyme.
| LOX species | Relative catalytic activity | Share of 15-HETE |
|---|---|---|
| % | % | |
| Wild-type | 100 ± 5.8 | 89.0 ± 0.1 |
| Q294L | 0.6 ± 0.05 | ND |
| N287L | 0.5 ± 0.07a | ND |
| N287D | 1.1 ± 0.3b | 88.8 ± 0.2 (NS) |
| N287Q | 3.9 ± 0.1 | 88.1 ± 1.2 (NS) |
a p = 0.007 (versus N287Q).
b p = 0.016 (versus N287Q).
FIGURE 3.

Catalytic activity of various ALOX15 Q287 mutants. Wild-type and mutant enzymes were expressed and purified as described under “Materials and Methods.” LOX concentrations of each preparation were quantified by immunoblotting, and equal amounts of enzyme were used in the activity assays. The y axis was normalized to the product amount formed by the N287Q mutant. Note that in comparison with Table 2, the amount of enzyme used was increased 3-fold.
The Thr-560–Gln-294 Hydrogen Bridge May Be Conserved in a Large Number of Animal LOXs
To explore whether the Thr-560–Gln-294 hydrogen bridge is peculiar for human ALOX15, we searched the publically available sequence databases and found that Thr-560 of the human enzyme is strongly conserved in a large number of animal LOX isoforms (supplemental Table S1). In some cases Thr is exchanged for a Ser, which also acts as a hydrogen donor. In fact, our mutagenesis studies indicated only a minor loss in catalytic activity for a Thr-to-Ser exchange (Table 1). The binding partner of Thr-560 in human ALOX15 (Gln-294) is conserved in almost every LOX isoform that contains a Thr or an Ser at the position aligning with Thr-560 of ALOX15 (supplemental Table S1). Except for the epidermal LOXs, the third key residue in this hydrogen bond network (Asn-287) is also conserved. Taken together, these data suggest that similar hydrogen bridges may also exist in other LOX isoforms.
Recently, the crystal structure of human 5-LOX has been solved (25), and we inspected this ALOX5 structure for the existence of a similar hydrogen bond. Indeed, a hydrogen bridge was present between Thr-570 (corresponds to Thr-560 in ALOX15) and Gln-303 (corresponds to Gln-294 in ALOX15) with a binding distance of 2.21 Å. On the other hand, Thr-560, Gln-294, and Asn-287 of ALOX15 are not conserved in plant or prokaryotic LOXs. For these LOX isoforms, the hydrogen bond network may not be of functional importance.
Functional Importance of Thr-560 and Gln-294 in Other Mammalian LOX Isoforms
If the hydrogen bond network identified for human ALOX15 is conserved, as suggested by the alignment data, one would expect impaired catalytic activities when our mutagenesis scheme is applied to other LOX isoforms. To test this hypothesis, we created corresponding mutants for human ALOX12 (platelet type 12-LOX) and mouse alox5 (5-LOX). Sequence alignments identified the target amino acids (Thr-560 and Gln-294 for ALOX12 and T570 and Gln-303 for alox5, respectively), and we introduced a Met and a Leu at these positions. The results summarized in Table 3 indicate that the mutant enzyme species exhibited strongly impaired catalytic activities and that the drop in activity was more pronounced for the Q294L/Q303L exchanges as compared with T560M/T570M mutation. These data confirm the findings made for human ALOX15 for two additional mammalian LOXs and suggest the functional importance of the identified hydrogen bond network for these enzyme species.
TABLE 3.
Catalytic activity and reaction specificity of human ALOX12 (platelet-type 12-LOX) and alox5 (mouse 5-LOX) mutants
Recombinant expression of human ALOX12, product preparation, and HPLC analysis were performed as described under “Materials and Methods” with the crude cell lysate of 5-ml cultures (n = 5 for each mutant). For alox5 wild-type and mutant enzyme species, aliquots of co-Sepharose purified enzyme preparations were used containing equal amounts of LOX protein as determined by Western blot. The amounts of HETE isomers were quantified for each sample by HPLC and used as measure for the catalytic activity. HETE formation by the wild type was set 100% for each LOX isoform.
| LOX species | Relative catalytic activity |
|---|---|
| % | |
| ALOX12 wild-type | 100.0 ± 22.1 |
| T560M | 25.3 ± 7.4 |
| Q294L | 16.1 ± 8.0 |
| alox5 wild-type | 100.0 ± 1.3 |
| T570M | 8.2 ± 1.5 |
| Q303L | 3.2 ± 0.2 |
The T560M Mutant Lacks a Kinetic Lag Phase and Is More Susceptible for Suicidal Inactivation
To further explore the molecular basis for the low catalytic efficiency of the T560M mutant, we compared the progress curves of arachidonic acid oxygenation (Fig. 4). We found that the wild-type enzyme shows a pronounced kinetic lag phase but then the reaction rate increases continuously prior to enzyme inactivation. In contrast, no kinetic lag phase was observed for the T560M mutant. Instead, a continuous decline of the oxygenation rate was observed from the very beginning of the reaction, and after 2 min the reaction has virtually ceased. The addition of fresh substrate did not restart the reaction (Fig. 5) as indicated by HPLC analysis of the reaction products, and these data exclude a shortage of fatty acid substrate as molecular basis for the impaired catalytic activity. In contrast, the addition of fresh enzyme induced a further increase in product formation, indicating suicidal inactivation of the enzyme during fatty acid oxygenation as a molecular basis for the loss in catalytic activity. These data suggested that the T560M mutant does not exhibit a kinetic lag phase but appears to be more susceptible for suicidal inactivation.
FIGURE 4.

Time dependence of arachidonic acid oxygenation by purified wild-type human ALOX15 and its T560M mutant. Wild-type and T560M mutant were expressed and purified as described under “Materials and Methods.” LOX concentrations of each preparation were quantified by immunoblotting, and in this experiment, the amount of T560M used was 5-fold higher than the wild type. Activity assays were carried out as described, and the reaction was stopped by the addition of 500 μl of ice-cold methanol. After reduction of hydroperoxides and acidification, the samples were centrifuged, and aliquots of the supernatant were used for HPLC analysis.
FIGURE 5.
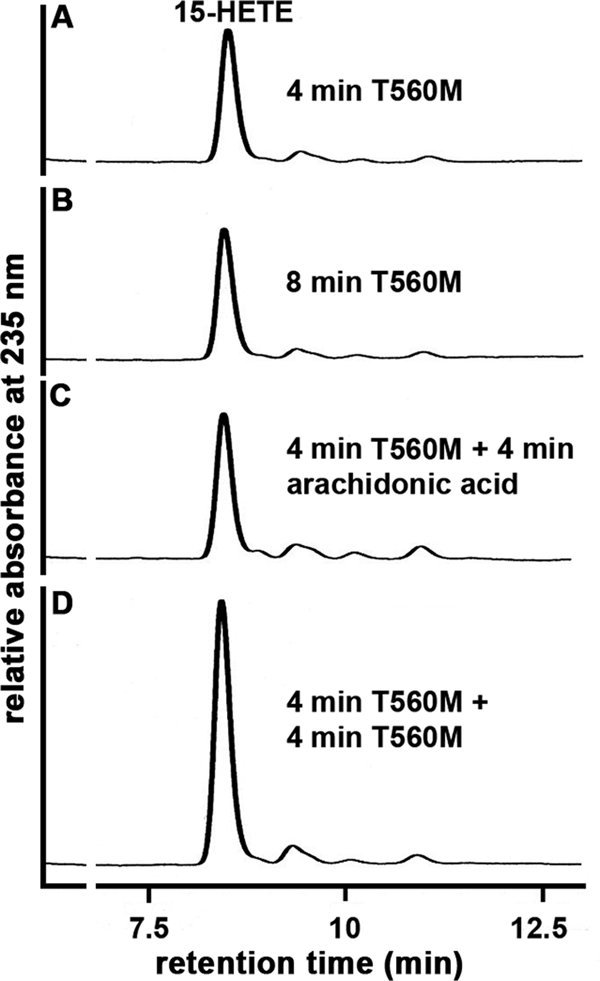
Suicidal inactivation of the T560M mutant of ALOX15 during arachidonic acid oxygenation. The T560M mutant was expressed and purified as described under “Materials and Methods.” Activity assays were carried out as described, and the reaction was stopped after 4 min of incubation by the addition of 500 μl of ice-cold methanol (A). Additional samples were further incubated for 4 min with no additives (B), with fresh arachidonic acid (C), or with fresh enzyme (D). After reduction of the hydroperoxides and acidification, samples were centrifuged, and aliquots of the supernatant were analyzed by HPLC. Representative chromatograms for one of three independent experiments are shown.
Lack of Iron Incorporation Is Not Responsible for the Reduced Catalytic Activity
LOXs contain equimolar amounts of non-heme iron, and iron incorporation into recombinant proteins might be a critical step when iron-containing enzymes are expressed in E. coli. To exclude that a difference in iron incorporation between wild-type and mutant enzyme species is responsible for the reduced catalytic activity of the enzyme species, we compared the iron content of wild-type ALOX15, its T560M and N287D mutant. We quantified an iron load of 80.9 ± 0.04, 76.8 ± 0.09, and 80.4 ± 0.03% for wild-type ALOX15, T560M, and N287D, respectively. These data indicate an 80% iron load of the recombinant enzyme and strongly suggest that the loss in catalytic activity may not be related to impaired iron incorporation.
DISCUSSION
The role of ALOX15 in atherogenesis has been a matter of discussion for many years, and pro- and anti-atherogenic effects have been reported (9–16). Recent population-based case control studies yielded evidence for an anti-atherogenic effect of the enzyme (19). The molecular basis for this effect remains unclear, but involvement of ALOX15 in vasodilatation was discussed as a possible reason in one of these studies (20). However, because the T560M SNP is very rare in Caucasians (less than 1% heterozygous allele carriers), both studies were underpowered to draw definite conclusions on the pathophysiological relevance of this SNP in myocardial infarction and/or coronary artery disease.
Nevertheless, the T560M exchange occurs in vivo, and its lacking catalytic activity was quite surprising because the Thr-to-Met exchange should not directly affect the active site. Inspecting the three-dimensional structure of the rabbit ALOX15, which shares 81% identity with the human enzyme, we found that the side chain hydroxy group of Thr-560 hydrogen bridges with the side chain oxygen of Gln-294. This hydrogen bridge is part of a more complex hydrogen bond network, which apparently stabilizes the structure of this region of the protein, which lacks stable secondary structural elements such as helices or β-barrels. Moreover, via this hydrogen bond network, Thr-560 is interconnected with Met-418, which, according to the triade concept, interacts with the methyl end of the substrate fatty acid (24, 27). Thus, our data suggest that exchange of a peripheral amino acid might induce via the hydrogen bond network conformational alterations at the catalytic center, which are mirrored by a strongly impaired catalytic activity. More generally spoken, structural alterations outside the active site might impact the enzyme activity if these alterations are translated to the catalytic center.
The question of why the T560M exchange does not alter the reaction specificity of the mutant enzyme was not addressed experimentally. However, for alterations in the reaction specificity, the spatial properties of the amino acid side chain are important because introduction of less space-filling residues at this position provides more space for deeper penetration of the substrate fatty acid into the substrate binding pocket (24, 27). In contrast, in the wild-type enzyme, the hydrogen bond network interconnects Thr-560 with the peptide backbone of Met-418, which does not alter the spatial properties of this amino acid. Thus, the structural alterations induced by T560M and M418V(A) exchange are different, and alterations in the positional specificity cannot necessarily be expected as functional consequences of T560M exchange. We hypothesize that T560M exchange might impact the alignment of fatty acid substrate at the active site in such a way that the rate of hydrogen abstraction from C13 is impaired but that the stereochemistry of oxygen insertion is hardly altered. It may be of interest in this context that the M418A mutant of the rabbit 12/15-LOX only exhibits 30% residual catalytic activity (28).
Detailed sequence comparison (supplemental Table S1) suggests that this hydrogen bond network might be conserved in animal LOXs, and our findings indicate that disruption of this network (mutations at Thr-560 or Gln-294) induces a loss in enzymatic activity of ALOX15, ALOX12, and alox5. Thus, our data suggest that this structural element is of functional importance for these and probably for other animal LOX isoforms.
It is interesting to note that T560M exchange induced less severe functional consequences (partial inactivation) than mutations at Gln-294, and this was the case for all three LOX isoforms tested. These differences may become plausible if one considers the fact that Thr-560 only forms a single hydrogen bond within the hydrogen bridge network. In contrast, Gln-294 is involved in multiple hydrogen bonds (Fig. 1D), and thus, the structural alterations induced by mutations of Gln-294 are expected to be more severe.
When we characterized the catalytic activity of the T560M mutant in more detail, we noticed that the difference in the reaction rate between wild-type and mutant enzyme strongly depended on the duration of the incubation period (Fig. 4), suggesting that the two enzyme species exhibited different reaction kinetics. In general, kinetics of the LOX reaction are characterized by two peculiarities: (i) kinetic lag phase (29, 30) and (ii) suicidal enzyme inactivation (31). When we recorded the kinetic progress curves for human wild-type ALOX15 and its T560M mutant (Fig. 4), we found that the wild-type enzyme follows the usual reaction kinetics but that the T560M mutant lacks the kinetic lag phase. In fact, the reaction starts with the maximal rate, but the enzyme quickly undergoes inactivation. These kinetic peculiarities appear to be related to the hydrogen bond network involving Thr-560, Gln-294, and Asn-287, which thus might play a role in the activation/inactivation processes. The detailed molecular basis, however, remains to be explored.
The kinetic lag phase has previously been related to oxidation of the non-heme iron from its silent ferrous state to its catalytically active ferric form (29, 30). However, although oxidation of the non-heme iron is involved in the activation process, more recent kinetic studies on the rabbit ALOX15 suggested an oxygen dependence of the activation process (32). Although the detailed mechanism of enzyme activation still remains elusive, oxygen dependence has recently been confirmed experimentally for another mammalian LOX (33, 34).
Some LOX isoforms undergo suicidal inactivation during fatty acid oxygenation (31, 35, 36), and we confirmed this kinetic peculiarity for both recombinant wild-type human ALOX15 and its T560M mutant. However, the inactivation rate of the mutant enzyme appears to be much faster (Fig. 4) because the enzyme was completely inactivated after 1–2 min. More detailed studies on the molecular basis for this difference are difficult, because the principle mechanism of suicidal inactivation of LOXs or other enzymes of the arachidonic acid cascade such as cyclooxygenase isoforms has not been clarified. Originally, it has been suggested for LOXs that peroxide-induced oxidation of a single methionine residue at the active site is the critical step in suicidal inactivation (35). However, when this methionine was exchanged by site-directed mutagenesis to a residue that cannot be oxidized, the enzyme still underwent suicidal inactivation. These data indicate that methionine oxidation may not be a crucial process in suicidal inactivation (36). Although the molecular basis of suicidal inactivation still remains unclear, the T560M mutant of the human ALOX15 might constitute a suitable model for further investigation into this mechanism.
Supplementary Material
This work was supported by Deutsche Forschungsgemeinschaft Grant GRK1673.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Figs. S1 and S2.
- LOX
- lipoxygenase
- SNP
- single-nucleotide polymorphism.
REFERENCES
- 1. Brash A. R. (1999) J. Biol. Chem. 274, 23679–23682 [DOI] [PubMed] [Google Scholar]
- 2. Andreou A., Feussner I. (2009) Phytochemistry 70, 1504–1510 [DOI] [PubMed] [Google Scholar]
- 3. Wymann M. P., Schneiter R. (2008) Nat. Rev. Mol. Cell Biol. 9, 162–176 [DOI] [PubMed] [Google Scholar]
- 4. Kühn H., O'Donnell V. B. (2006) Prog. Lipid. Res. 45, 334–356 [DOI] [PubMed] [Google Scholar]
- 5. Palacios-Pelaez R., Lukiw W. J., Bazan N. G. (2010) Mol. Neurobiol. 41, 367–374 [DOI] [PubMed] [Google Scholar]
- 6. Bishnoi M., Patil C. S., Kumar A., Kulkarni S. K. (2005) Methods Find Exp. Clin. Pharmacol. 27, 465–470 [DOI] [PubMed] [Google Scholar]
- 7. Moreno J. J. (2009) Biochem. Pharmacol. 77, 1–10 [DOI] [PubMed] [Google Scholar]
- 8. Liu S. H., Shen C. C., Yi Y. C., Tsai J. J., Wang C. C., Chueh J. T., Lin K. L., Lee T. C., Pan H. C., Sheu M. L. (2010) Br. J. Pharmacol. 160, 1963–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hersberger M. (2010) Clin. Chem. Lab. Med. 48, 1063–1073 [DOI] [PubMed] [Google Scholar]
- 10. Poeckel D., Funk C. D. (2010) Cardiovasc. Res. 86, 243–253 [DOI] [PubMed] [Google Scholar]
- 11. Zhao L., Funk C. D. (2004) Trends Cardiovasc. Med. 14, 191–195 [DOI] [PubMed] [Google Scholar]
- 12. Funk C. D. (2006) Arterioscler. Thromb. Vasc. Biol. 26, 1204–1206 [DOI] [PubMed] [Google Scholar]
- 13. Wittwer J., Hersberger M. (2007) Prostaglandins Leukot. Essent. Fatty Acids 77, 67–77 [DOI] [PubMed] [Google Scholar]
- 14. Ylä-Herttuala S., Rosenfeld M. E., Parthasarathy S., Glass C. K., Sigal E., Witztum J. L., Steinberg D. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 6959–6963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kühn H., Heydeck D., Hugou I., Gniwotta C. (1997) J. Clin. Invest. 99, 888–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Spanbroek R., Grabner R., Lotzer K., Hildner M., Urbach A., Ruhling K., Moos M. P., Kaiser B., Cohnert T. U., Wahlers T., Zieske A., Plenz G., Robenek H., Salbach P., Kuhn H., Radmark O., Samuelsson B., Habenicht A. J. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 1238–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Krishnaswamy G. (2010) Cardiovasc. Hematol Disord. Drug Targets 10, 234 [DOI] [PubMed] [Google Scholar]
- 18. Klingenberg R., Hansson G. K. (2009) Eur. Heart. J. 30, 2838–2844 [DOI] [PubMed] [Google Scholar]
- 19. Assimes T. L., Knowles J. W., Priest J. R., Basu A., Borchert A., Volcik K. A., Grove M. L., Tabor H. K., Southwick A., Tabibiazar R., Sidney S., Boerwinkle E., Go A. S., Iribarren C., Hlatky M. A., Fortmann S. P., Myers R. M., Kuhn H., Risch N., Quertermous T. (2008) Atherosclerosis 198, 136–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hersberger M., Müller M., Marti-Jaun J., Heid I. M., Coassin S., Young T. F., Waechter V., Hengstenberg C., Meisinger C., Peters A., König W., Holmer S., Schunkert H., Klopp N., Kronenberg F., Illig T. (2009) Atherosclerosis 205, 192–196 [DOI] [PubMed] [Google Scholar]
- 21. Gillmor S. A., Villaseñor A., Fletterick R., Sigal E., Browner M. F. (1997) Nat. Struct. Biol. 4, 1003–1009 [DOI] [PubMed] [Google Scholar]
- 22. Choi J., Chon J. K., Kim S., Shin W. (2008) Proteins 70, 1023–1032 [DOI] [PubMed] [Google Scholar]
- 23. Walther M., Anton M., Wiedmann M., Fletterick R., Kuhn H. (2002) J. Biol. Chem. 277, 27360–27366 [DOI] [PubMed] [Google Scholar]
- 24. Vogel R., Jansen C., Roffeis J., Reddanna P., Forsell P., Claesson H. E., Kuhn H., Walther M. (2010) J. Biol. Chem. 285, 5369–5376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gilbert N. C., Bartlett S. G., Waight M. T., Neau D. B., Boeglin W. E., Brash A. R., Newcomer M. E. (2011) Science 331, 217–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Humphrey W., Dalke A., Schulten K. (1996) J. Mol. Graphics 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 27. Sloane D. L., Leung R., Craik C. S., Sigal E. (1991) Nature 354, 149–152 [DOI] [PubMed] [Google Scholar]
- 28. Borngräber S., Browner M., Gillmor S., Gerth C., Anton M., Fletterick R., Kühn H. (1999) J. Biol. Chem. 274, 37345–37350 [DOI] [PubMed] [Google Scholar]
- 29. Schilstra M. J., Veldink G. A., Vliegenthart J. F. (1993) Biochemistry 32, 7686–7691 [DOI] [PubMed] [Google Scholar]
- 30. Schilstra M. J., Veldink G. A., Verhagen J., Vliegenthart J. F. (1992) Biochemistry 31, 7692–7699 [DOI] [PubMed] [Google Scholar]
- 31. Rapoport S. M., Schewe T., Wiesner R., Halangk W., Ludwig P., Janicke-Höhne M., Tannert C., Hiebsch C., Klatt D. (1979) Eur. J. Biochem. 96, 545–561 [DOI] [PubMed] [Google Scholar]
- 32. Ivanov I., Saam J., Kuhn H., Holzhütter H. G. (2005) FEBS J. 272, 2523–2535 [DOI] [PubMed] [Google Scholar]
- 33. Zheng Y., Brash A. R. (2010) J. Biol. Chem. 285, 39876–39887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zheng Y., Brash A. R. (2010) J. Biol. Chem. 285, 39866–39875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rapoport S., Härtel B., Hausdorf G. (1984) Eur. J. Biochem. 139, 573–576 [DOI] [PubMed] [Google Scholar]
- 36. Gan Q. F., Witkop G. L., Sloane D. L., Straub K. M., Sigal E. (1995) Biochemistry 34, 7069–7079 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.