


Abstract
Defects in the XPG DNA repair endonuclease gene can result in the cancer-prone disorders xeroderma pigmentosum (XP) or the XP–Cockayne syndrome complex. While the XPG cDNA sequence was known, determination of the genomic sequence was required to understand its different functions. In cells from normal donors, we found that the genomic sequence of the human XPG gene spans 30 kb, contains 15 exons that range from 61 to 1074 bp and 14 introns that range from 250 to 5763 bp. Analysis of the splice donor and acceptor sites using an information theory-based approach revealed three splice sites with low information content, which are components of the minor (U12) spliceosome. We identified six alternatively spliced XPG mRNA isoforms in cells from normal donors and from XPG patients: partial deletion of exon 8, partial retention of intron 8, two with alternative exons (in introns 1 and 6) and two that retained complete introns (introns 3 and 9). The amount of alternatively spliced XPG mRNA isoforms varied in different tissues. Most alternative splice donor and acceptor sites had a relatively high information content, but one has the U12 spliceosome sequence. A single nucleotide polymorphism has allele frequencies of 0.74 for 3507G and 0.26 for 3507C in 91 donors. The human XPG gene contains multiple splice sites with low information content in association with multiple alternatively spliced isoforms of XPG mRNA.
INTRODUCTION
Three rare, autosomal recessive inherited human disorders are associated with impaired nucleotide excision repair (NER) activity: xeroderma pigmentosum (XP), Cockayne Syndrome (CS) and trichothiodystrophy (reviewed in 1). XP has been studied most extensively. XP patients exhibit extreme sensitivity to sunlight, resulting in a high incidence of skin cancers (∼1000 times that of the general population) (2,3). About 20% of XP patients also develop neurologic abnormalities in addition to their skin problems. These clinical findings are associated with cellular defects, including hypersensitivity to killing and mutagenic effects of UV, and the inability of XP cells to repair UV-induced DNA damage (4). Seven different DNA NER genes, which correct seven distinct genetic XP complementation groups (XPA–XPG) have been identified (1). In addition, another entity, XP variant (XPV), exists. Patients suffering from XPV are defective in DNA polymerase η, which is responsible for error-free bypass of UV-induced DNA damage (5,6).
The human gene responsible for XP group G was identified as ERCC5 (7–9). The XPG gene maps to chromosome 13q32-33 (10), encodes a protein with a predicted molecular mass of 133 kDa (11) and is a founding member of the RAD2/XPG family (12–15), which comprises two related groups of nucleases (reviewed in 16,17). The XPG gene codes for a structure-specific endonuclease that cleaves damaged DNA ∼5 nt 3′ to the site of the lesion and is also required non-enzymatically for subsequent 5′ incision by the XPF/ERCC1 heterodimer during the NER process (18–20). While the XPG cDNA sequence was known (GenBank accession no. NM_000123), determining the genomic sequence is required for understanding its different functions. The lack of complete sequence information and the related functions delays the understanding of the role of XPG in NER. Recent evidence suggests that XPG is also involved in transcription-coupled repair of oxidative DNA lesions (21).
Mutations in the XPG gene not only result in the XP phenotype but also in a phenotype that combines features of XP and CS (XP–CS complex). XP–CS complex has been proposed as a distinct clinical entity (22,23) and patients suffering from XP–CS complex exhibit developmental retardation, dwarfism and severe neurologic abnormalities plus sun sensitivity and other abnormalities of XP, including skin cancer (24,25).
Here we determined the genomic sequence of the human XP group G gene. We identified the location of all 14 intron/exon borders, sizes of the introns and the sequence of the exon flanking splice donor and acceptor sites. We also found an unusually large number of alternatively spliced mRNA isoforms that occurred in normal human tissues. An information theory-based approach incorporating information weight matrices that reflect features of nearly 2000 published donor and acceptor sites (26) enabled us to analyze the contributions of the nucleotide sequences that flank the wild-type and alternative splice sites. We also measured the frequencies of two new single nucleotide polymorphisms, as well as the frequency of one known single nucleotide polymorphism in exon 15 of XPG.
MATERIALS AND METHODS
Cell lines, culture conditions and DNA/RNA extraction
GM0637, a normal SV40-immortalized fibroblast cell line, and F1-AG05247E and F2-AG05410 normal primary fibroblasts were obtained from the Human Genetic Cell Repositories (Camden, NJ). XPG fibroblast cell lines XP65BE (GM16398), XP82DC (GM16181) and XP96TA (GM16180) were kindly provided by Dr D.Busch (The Armed-Forces Institute, Washington, DC) and Dr H.Slor (Tel Aviv University, Tel Aviv, Israel). Cells were grown in DMEM supplemented with 2% glutamine and 10% FCS (Gibco BRL) in an 8% CO2 humidified incubator at 37°C. Total RNA and DNA were extracted from cells using the RNAqueous-Midi Kit (Ambion, TX) and DNAzol reagent (Gibco BRL), respectively. Multiple Choice first strand cDNA from various human tissues was obtained from Origene Technologies, Inc. (Rockville, MD).
Identification of alternatively spliced XPG mRNA isoforms
RNA (2 µg) was reverse transcribed using the SUPERSCRIPT preamplification System and Oligo(dT)12–18 primers for first strand cDNA synthesis according to the manufacturer’s protocol (Gibco BRL). The entire 3.8 kb coding region of the XPG gene was then amplified with two primers: UTR5′ (forward) and UTR3′ (reverse) (27) (Table 1) using the Advantage cDNA PCR Kit (Clontech, CA) at 71°C annealing/extension for 35 cycles and subsequently subcloned into pCR 2.1-TOPO vector (TOPO TA Cloning Kit; Invitrogen, CA). Because UTR5′ and UTR3′ each contain unique restriction sites at their ends (EagI and NsiI, respectively), the entire XPG cDNA could be released from the vector after vector amplification and checked for size differences compared to wild-type 3.8 kb XPG cDNA on an agarose gel. Single clones were then picked and subjected to sequencing of the whole XPG cDNA by cycle sequencing employing dideoxy termination chemistry and an ABI 373A automated DNA sequencer (PE Applied Biosystems, CA). A total of 13 different forward primers were used for this overlapping sequencing (Table 1): UTR5′ (27), 312–333 (9), T10 (28), 966–985 (9), 594R4 (28), T22 (28), 594R8b (28), 594R10 (28), 2472–2492 (9), T4 (28), XPG100 (29), T2 (28) and UTR3′ (27).
Table 1. Human XPG PCR and sequencing primers.
 |
*Location of primer sequence in XPG cDNA GenBank accession no. NM_000123, where 1 is the beginning of the 5′UTR.
Characterization of genomic XPG DNA and alternatively spliced isoforms
A total of nine primer pairs (all but one in the XPG coding region; GenBank accession no. NM_000123; base pair 1 represents the beginning of the 5′ UTR) were developed that allowed us to PCR amplify the entire genomic XPG DNA, which spans 30 kb (Table 1). All PCR reactions were performed using the Advantage cDNA PCR Kit (Clontech, CA) as per the manufacturer’s instructions and annealing/extension temperatures were optimized for each primer pair. The PCR steps were conducted as follows: 94°C for 3 min, then 35 cycles of amplification (94°C for 20 s and optimized annealing/extension temperature for 3 min), ending with final optimized annealing/extension temperature for 3 min.
The primer pair 156–174 and 432–411 results in an ∼6 kb fragment (67°C annealing/extension); 302–323 and Intron 4R result in an ∼2.3 kb fragment (66°C); T10 (28) and 853–832 result in an ∼4.1 kb fragment (67°C); 826–844 and E7-3R (28) result in an ∼3.2 kb fragment (66°C); 966–985 (9) and T1 (28) result in an ∼1.1 kb fragment (67°C); 594R8b (28) and 593F10 (28) result in an ∼2.8 kb fragment (69°C); 2305–2324 and 2550–2531 result in an ∼0.9 kb fragment (69°C); 2472–2492 (9) and XPG101 (29) result in an ∼5.9 kb fragment (69°C); finally, XPG100 (29) and UTR3′ (27) result in an ∼3.7 kb fragment (69°C).
The intronic sequence of the exon flanking splice donor and acceptor sites was determined by sequencing the nine agarose gel-purified genomic XPG PCR fragments, as described above, using the following additional primers: 517–496, 503–522, 470–450, 696–672, 669–693, 991–1010, 1220–1201, 593F7 (28), 594R6 (28), 593F9 (28), 1977–1998, 2233–2215, 2529–2550, 594R9 (28), 2730–2747, 2780–2759, XPG102 (29), 3096–3117 and 3290–3269.
To locate the alternatively spliced exons that are positioned within intron 1 and intron 6, two primer pairs for each alternatively spliced exon were designed to amplify overlapping fragments reaching from the 5′ exon to the alternatively spliced exon, and from there to the 3′ exon. Primer pairs 266–285 and Intron 1R, and Intron 1F and 381–360, located the alternatively spliced exon in the middle of intron 1 (35 cycles, 69°C annealing/extension; Advantage cDNA PCR Kit, Clontech, CA). Primer pairs 826–844 and Intron 6R, and Intron 6F and E7-3R (28), located the alternatively spliced exon in the first third of intron 6 (35 cycles, 66°C annealing/extension; Advantage cDNA PCR Kit, Clontech, CA). These primers were also used to sequence the region flanking the alternatively spliced sites.
The primers for PCR amplification used to assess the presence of six different XPG splice variants in different human tissues were designed on the basis of the XPG cDNA sequence (GenBank accession no. NM_000123): pair I, 156–174 and 432–411; pair II, 503–522 and 696–672; pair III, 826–844 and E7-3R (28); pair IV, 991–1010 and 1220–1201; pair V 594R8b (28) and 593F10 (28); pair VI, 2305–2324 and 2550–2531. For primer pairs I–IV, the annealing/extension was performed at 66°C, while for primer pairs V and VI, annealing/extension was conducted at 69°C.
Splice donor and acceptor site sequence analysis with an information theory-based model
Sequences were scanned with the donor and acceptor individual information weight matrices and the identified sites were displayed and interpreted as described previously (26,30–32). These analyses can be performed on a Web server: http://www.lecb.ncifcrf.gov/~toms/delilaserver.html.
Determination of exon 15 polymorphism frequencies
In order to examine two possibly new polymorphisms in exon 15 of the XPG gene, we screened DNA from 91 anonymous donors (men and women employees and unrelated children, age range 1–76 years, 62% men) (33). A sample of buccal swabs was obtained from each individual and genomic DNA extracted as described (34). A 455 bp region within exon 15 of the XPG cDNA (GenBank accession no. NM_000123), which contains the new C3354G and C3435G as well as the previously reported G3507C (9) sites, was PCR-amplified using T2 forward (28) and 3624–3607 reverse (9) primers. The Advantage cDNA PCR Kit (Clontech, CA) was utilized following the manufacturer’s protocol at 60°C annealing/extension for 35 cycles. After agarose gel purification, the PCR product was sequenced using primer 3330–3349 (9), as described above.
RESULTS
Exon–intron organization of the human XPG gene
The human XPG gene is essential for DNA nucleotide excision repair and individuals suffering from a defect in the XPG gene are sun sensitive and at high risk of developing sunlight-induced skin cancers. In order to identify the genotype of such individuals, detailed knowledge about the genomic architecture of XPG is crucial. We developed primer pairs and PCR conditions to amplify the entire 30 kb genomic XPG DNA (see Materials and Methods). The whole XPG gene can be amplified with nine overlapping PCR fragments, spanning the UTR5′ region to exon 2 (6 kb fragment), from exon 2 to the beginning of intron 4 (2.3 kb fragment), from exon 4 to exon 6 (4.1 kb fragment), from exon 6 to exon 7 (3.2 kb fragment), from exon 7 to exon 8 (1.1 kb fragment), from exon 8 to exon 9 (2.8 kb fragment), from exon 9 to exon 11 (0.9 kb fragment), from exon 10 to exon 14 (5.9 kb fragment) and from exon 13 to the UTR3′ region (3.7 kb fragment). The human XPG gene is organized into 15 exons, which range from 61 to 1074 bp in size (Fig. 1). The 14 introns vary from 250 (intron 10) to 5763 bp (intron 1). We determined the nucleotide sequence of these fragments: GenBank accession nos AF255431–AF255442 (Fig. 1). Comparative nucleotide sequence analysis revealed two unannotated clones from high-throughput sequencing (AL137246 and AL157769), each containing part of the genomic XPG sequence (Fig. 1). These clones complete the intronic gaps of our sequencing efforts and appear to contain XPG sequence up to 2 kb 5′ of exon 1 (AL137246).
Figure 1.

Structural map of the XPG gene. The 15 exons and 14 introns are numbered and their size in base pairs is indicated below. The parts of the genomic XPG sequence that we determined are indicated and were submitted to GenBank (accession nos AF255431–AF255442). AL137246 and AL157769 are unannotated sequences that were subsequently found in GenBank.
Analysis of the exon–intron boundaries
The human intronic splice donor and acceptor site sequences and their location in the coding XPG sequence are listed in Table 2 and compared to the mouse and Drosophila sequences. We analyzed the effects of the splice sequences on RNA processing using an information theory-based approach incorporating information weight matrices that reflect features of nearly 2000 published donor and acceptor sites (26). Information is the only measure of sequence conservation which is additive, and it describes the degree to which a member contributes to the conservation of an entire sequence family rather than looking at only the consensus sequence (35). Information content is defined as the number of choices needed to describe a sequence pattern, using a logarithmic scale in bits. The magnitude of the information content indicates how strongly conserved a base is in natural splice junction binding sites with ∼2.4 bits being the apparent minimal functional value (31). The conserved splice sites exhibit information contents that range from 3.3 bits (5′ intron 9) to 12.7 bits (3′ intron 2), consistent with functional activity but of considerable variation in strength (Table 2).
Table 2. Human XPG gene: sequence and information content of splice sites.
 |
The three non-conserved splice sites (5′ intron 1, 5′ and 3′ intron 13) exhibit very low information content (0, –14.6 and –4.8 bits, respectively) (Table 2). These sites appear to represent a variant class of splice junctions that might be spliced via a spliceosome mechanism employing factors distinct from those used for the usual splice junctions. The 5′ intron splice donors of introns 1 and 13 are a perfect match to the rare U12 5′ splice site for nucleotides 2–7, TATCCT (36,37). These splice sites are also conserved in the mouse XPG gene but not in the Drosophlia XPG gene (Table 2). The human 3′ splice acceptor site of intron 13 reads ‘cat’ instead of ‘cag’. However, the mouse intron 13 3′ splice acceptor site ends with ‘cac’ rather than ‘cat’, which matches the U12 3′ splice site sequence. Interestingly, the 3′ intron 9 splice site has an unusually low information content (–2.1 bits) but does not follow the U12 spliceosome sequence. This might point towards a rare third spliceosome mechanism utilized for this site.
Identification of alternatively spliced isoforms among human XPG transcripts
Using RT–PCR, we amplified the entire coding region of XPG from total RNA isolated from fibroblasts from one normal and four XPG patients (see Materials and Methods). Using special primer pairs and PCR conditions, we identified all six splice variants (I–VI) separately in all these fibroblasts (Fig. 2 and Table 3). Retention of the alternatively spliced exon in intron 1 (isoform I) leads to the insertion of 37 codons and a frameshift after the insert that results in a TAG stop codon two amino acids downstream. Isoform II (complete intron 3 retention) and isoform III (alternatively spliced exon in intron 6) might be of special functional interest as they comprise inframe insertions. The first inserts 138 new codons, including seven stop codons. However, a new methionine is also inserted 18 bases downstream from the last stop codon. Similarly, the alternatively spliced exon in intron 6 inserts 46 codons, including one TAG stop codon (9th codon) followed by a new methionine 201 bases downstream in exon 7. The partial skipping of exon 8 (isoform IV) was previously reported and results in a TGA stop codon nine amino acids later (9). Partial retention of the beginning of intron 8 (isoform V) leads to the addition of 22 codons including a TAG stop codon (12th codon) and a frameshift after the insert that results in a TGA stop codon nine amino acids later. Isoform VI (complete retention of intron 9; 117 new codons including seven stop codons) also leads to a frameshift after the insert that results in a TAG stop codon 11 amino acids downstream. These sequences have been deposited in GenBank, accession no. AH009656.
Figure 2.

Structural map of alternatively spliced XPG mRNA isoforms. To identify the isoforms, total RNA was reverse transcribed, RT–PCR amplified and subsequently subcloned into a cloning vector for sequencing (see Materials and Methods). The splice isoforms (I–VI, bottom) are compared to the wild-type XPG mRNA (middle) and the wild-type genomic XPG DNA (top). The genomic location and size (in base pairs) of insertions/deletions due to alternative splicing are indicated.
Table 3. Human XPG mRNA: sequence of alternatively spliced isoforms and information content of cryptic splice sites.
 |
*GenBank sequence AH009656.
We analyzed the effects of the alternatively spliced sequences on RNA processing using the information theory-based approach (Table 3). All splice sites except the splice donor of isoform I carry an information content greater than or equal to the minimal functional value (see above). The information content (measured in bits) of all novel alternatively spliced donor and acceptor sites in splice isoforms where the predominant splice sites were skipped (isoforms II, IV, V, VI) exceeded or were similar to those of the corresponding predominant sites. The splice donor site of isoform I exhibits a very low information content (–16 bits) and is a perfect match to the minor U12 5′ splice site for nucleotides 2–7, TATCCT (36,37). This is in agreement with our findings for the predominant splice donors in intron 1 and intron 13 (Table 2).
Several combinations of these alternatively spliced isoforms were observed in 16 subcloned XPG cDNA samples from the fibroblast lines. Isoforms I, II and IV were detected alone. Isoform II was also found in combination with isoform IV or VI. Isoform VI was only found together with isoform II. Isoforms III and V were only found together.
Using semi-quantitative RT–PCR, we looked for the presence of normal and alternatively spliced isoforms of XPG mRNA in cultured normal human skin fibroblasts from two normal donors (F1–AG05247E and F2–AG05410), and from normal human brain, liver, lung, kidney, spleen and prostate tissue (Fig. 3). The normally spliced isoform was present in all samples (black arrows). Isoforms II and VI (and to a lesser extent isoform IV) were readily detected in the normal human tissues. There appeared to be variations in the amount of these isoforms between different donors (for example, skin fibroblasts F1 and F2, isoforms II and VI; Fig. 3, top panel) and between different tissues (for example, reduced level of isoform VI in kidney versus other tissues; Fig. 3, bottom panel). The possible functional importance of these differences is not known.
Figure 3.
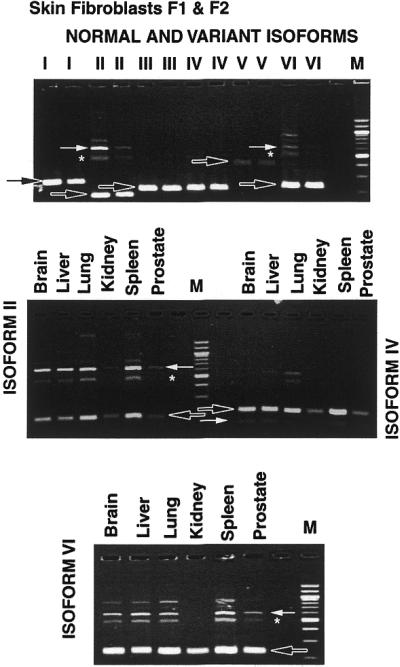
XPG mRNA: normal and variant splice isoforms in human tissues. Top, cultured normal primary skin fibroblasts (F1-AG05247E and F2-AG05410). RNA was isolated from each cell line, cDNA was prepared by RT–PCR and amplified separately by use of primer pairs that cover both normal splice isoforms and each of the variant splice isoforms (I–VI). The PCR products were separated by agarose gel electrophoresis (see Materials and Methods). Each pair of lanes present results from F1 and F2, respectively, for each of the primer pairs. The black arrows indicate the location of the normally spliced isoforms. Detectable alternatively spliced isoforms are indicated by white arrows. The asterisk indicates a heterodimer of the normal and the abnormally spliced isoforms (II and VI). The normal isoform is present in each lane. Alternatively spliced isoforms II and VI are seen on the gel indicating their relative abundance. Middle and bottom, cDNA obtained from human brain, liver, lung, kidney, spleen and prostate tissue was amplified by use of primer pairs for XPG mRNA isoform II (middle, left), isoform IV (middle, right) and isoform VI (bottom) and separated by agarose gel electrophoresis. The black arrows indicate the normally spliced isoforms. The alternatively spliced isoforms (white arrows) and heterodimers between the normal and alternatively spliced forms (asterisk) are detectable in the different tissues. There appears to be a relative reduction in alternatively spliced isoforms II, IV and VI in the kidney compared to the other human organs.
Single nucleotide polymorphisms in exon 15
When we compared the XPG nucleotide sequence from our individuals with the two unannotated GenBank clones (accession nos AL137246 and AL157769), we found an exact exonic sequence match, except two base changes: XPG cDNA (GenBank accession no. NM_000123) positions 3354 (C) and 3435 (C) in exon 15. In the AL137246 clone, these bases read G instead of C. However, a C was found in all our clones including DNA from normal cells. These base changes lead to single amino acid changes Arg1053Gly and Arg1080Gly. Comparative computer analysis of the mouse exon 15 also revealed a G at position 3354. However, the area around position 3435 in the mouse exon 15 is absent.
We tested DNA from 91 individuals, randomly selected from NIH (33), to determine the frequencies of these possibly new single nucleotide polymorphisms, which are in the vicinity of another single nucleotide polymorphism, His1104Asp (G3507C) (9). Due to a lack of suitable restriction sites, we sequenced the part of exon 15 spanning those sites after PCR amplification from genomic DNA. In all 91 samples we found a C at positions 3354 and 3435. This indicates that the two base changes C3354G and C3435G represent either two quite rare single nucleotide polymorphisms or sequencing errors in the unannotated clone AL137246. In contrast, the single nucleotide polymorphism C3507G is quite common. As shown in Table 4 the overall allele frequencies for 3507G and 3507C are 74 and 26%, respectively. The observed genotype distribution also matched the expected genotype distribution as predicted by the Hardy–Weinberg theory (Table 4). Thus, this common single nucleotide polymorphism may be useful for further genetic studies.
Table 4. XPG exon 15 polymorphism (G3507C; Asp1104His) allele frequencies and genotype distribution.
| NIH donors | Alleles | Genotype distribution observed [expected]a | |||||
| total | 3507G | 3507C | G/G | G/C | C/C | ||
| |
|
|
p |
q |
p2 |
2pq |
q2 |
| Number | 91 | 182 | 135 | 47 | 51 | 33 | 7 |
| Frequency | 100% | 100% | 74.2% | 25.8% | 56% [55%] | 36.3% [38.4%] | 7.7% [6.6%] |
aHardy–Weinberg frequencies.
DISCUSSION
XPG gene functions and associated clinical symptoms
In this study we characterized the whole human XPG gene at the genomic level, as well as the mRNA expression level (alternative splicing). All XP genes (XPA–XPG) are involved in the NER process. NER eliminates a wide variety of DNA damage including UV photoproducts (38–41). The sequence of the NER process consists of two broad steps: (i) lesion recognition, strand incision and damaged nucleotide displacement; and (ii) gap filling by DNA polymerization and ligation (42,43). Two NER subpathways have been discerned: ‘global genome repair’ (GGR) and ‘transcription-coupled repair’ (TCR) (44). GGR operates genome-wide and is able to remove DNA lesions from all locations in the genome at any moment in the cell cycle. TCR specifically acts on the transcribed strand of active genes, where it rapidly removes elongation-blocking lesions (45). The XPG gene, with its 3′ endonuclease activity, is involved in both NER subpathways (4). Clinically, however, patients with defects in the XPG gene may present mild XP symptoms, XP symptoms together with neurologic abnormalities, or combined features of XP and Cockayne syndrome (XP–CS complex) (1).
Based on the mutational analysis of two XPG patients suffering from only mild XP symptoms (9) and of four patients suffering from XPG–CS complex (27,28), a common mutational pattern for XPG–CS was proposed that implies a second XPG function (21,27). XPG mutations that confer XPG–CS complex were those that severely truncated the protein, whereas conservative single amino acid substitutions that eliminate NER but produce full-length protein resulted in the XP phenotype only (27).
There is evidence that defective TCR of endogenously generated oxidative DNA damage may underlie the clinical appearance of CS, as suggested by the fact that this process is defective in cells from patients with XPG–CS as well as CSB (CS complementation group B), but not in patients with mild XP symptoms only (21,46,47). In addition, comparative analysis of the Drosophila melanogaster XPG primary amino acid sequence led to the identification of a new conserved domain in the C-terminus of the protein, downstream of the previously identified nuclease domain I (48). A short stretch of amino acids in the N-terminal region of the XPG polypeptide, which is highly conserved in the human, mouse, Xenopus and Drosophila sequences, but not in the yeasts Schizosaccharomyces pombe and Saccharomyces cerevisiae, was also identified. This region includes the core amino acid sequence HEILTD, which is completely conserved in all four higher eukaryotes. This might also support the notion of a second unique function for XPG in higher eukaryotes (48). In addition, XPG protein may be involved in immunoglobulin class switching and DNA recombination (49).
Genomic XPG sequence and exon/intron splice sites
We determined the genomic sequence of the human XPG gene and the organization of its coding sequence (Fig. 1). The human XPG gene is comprised of 15 exons that range from 61 to 1074 bp in size and 14 introns that range from 250 to 5763 bp in size, which spans 30 kb in total. There is an overall 66% identity to the mouse XPG gene at the amino acid level. At the conserved regions of the RAD2 family the identity is >80% (50). Compared to the protein sequence of the Drosophila ortholog of the human XPG gene, there is only 28% identity overall. However, looking at the N and I domains, there are identities of 60 and 62%, respectively (48).
We also analyzed the exon/intron boundaries of the human XPG gene (Table 2). An information theory-based approach incorporating information weight matrices that reflect features of nearly 2000 published human donor and acceptor sites (26) was used to study the effects of the splice sequences on RNA processing. All conserved human XPG splice sites exhibit information content between 3.3 bits (5′ intron 9) and 12.7 bits (3′ intron 2). Evidence from analysis of many other human splice junction sequences indicates that sites with this information content are fully functional with 2.4 bits being the minimal functional value. Information content below 2.4 bits often results in skipping of the preceding exon (31).
We determined that the human XPG gene also contains three non-conserved sites for RNA splicing (Table 2). These sites comprise the splice donors 5′ of intron 1 and 5′ of intron 13, as well as the splice acceptor 3′ of intron 13. The corresponding information content was 0 bits, –14.6 bits and –4.8 bits, respectively. These non-conserved splice sites seem to be components of the minor (U12) spliceosome (36,37) and are also strongly conserved in the mouse XPG gene but not in the Drosophila XPG gene (50). Interestingly, an alternatively spliced XPG mRNA isoform, previously described (28), skipped exons 2–13 and appears to utilize the minor U12 spliceosome. Unfortunately, analysis of the effects of mouse or Drosophila splice sequences on RNA processing using an information theory-based approach was not feasible due to a lack of information weight matrices that reflect features of mouse or Drosophila donor and acceptor sites.
Alternatively spliced XPG isoforms
Alternative splicing of human gene transcripts that occurs normally is well documented. For example, alternatively spliced isoforms for the human polymerase β gene were reported with deletion of exon II, inclusion of intron 9 or deletion of exon XI (51). Other alternatively spliced genes include the RecQ5 gene (52) or the xeroderma pigmentosum group C gene, where low levels of alternatively spliced isoforms of the XPC mRNA containing exon 9a were detected in normal donors (33).
We identified six alternatively spliced XPG mRNA isoforms (I–VI) that occurred normally (Fig. 2). The alternatively spliced XPG isoforms showed retained alternatively spliced exons (I, III), full intron retentions (II, VI), partial intron retention (V) and partial exon skipping (IV). Interestingly, isoforms II and III comprise inframe insertions. Although the retained sequences introduce new termination signals, these signals are followed by a new methionine. Under some circumstances reinitiation of translation can occur in eukaryotes (reviewed in 53). XPG mRNA isoform IV was previously reported by Nouspikel and Clarkson (9). All the alternative transcripts are listed in GenBank (accession no. AF255442). We did not observe the previously reported intronic dinucleotide repeat polymorphism (54). Another alternatively spliced XPG mRNA isoform involving deletion of exons 2–13 was also previously reported (28).
Analysis of the alternatively spliced sequences with the information theory-based approach revealed that all splice sites carry an information content greater than or equal to the minimal functional information content which exceeded or was similar to the information content value of the corresponding natural splice site (Table 3). Interestingly, the splice donor site of isoform I (–16 bits) also seems to be part of the minor U12 spliceosome (36,37). In addition, combinations of several alternatively spliced isoforms were detected on the same cloned message.
Using semi-quantitative RT–PCR techniques, three of the six alternatively spliced XPG mRNA isoforms (II, IV and VI) were readily detectable in human fibroblasts (Fig. 3, top panel). There was inter-individual variation in the relative abundance of these alternatively spliced isoforms in fibroblasts from different individuals. We found a ubiquitous expression pattern of normal XPG mRNA and of the alternatively spliced isoforms II, IV and VI in different adult human tissues except kidney tissue (Fig. 3, middle and lower panels).
The functional consequences of the observations, especially the functional role(s) of the potential protein products generated by these splicing events, remain to be elucidated in further studies. One possibility is that the alternatively spliced isoforms are functionally compromised but compete with normally spliced XPG mRNA. In this case the relative abundance of alternatively spliced XPG transcripts would lead to a decrease in one or more XPG functions, possibly including a reduced nucleotide excision repair capacity. Recently, it was reported that lung cancer patients had significantly reduced expression levels of normal XPG mRNA compared to healthy controls (55). Previously, it was also demonstrated that reduced DNA repair capacity as measured by the host cell reactivation assay is associated with an increased risk of lung cancers (56). Inter-individual variation of DNA repair capacities is well documented (1,57,58). Thus, higher relative expression levels of alternatively spliced XPG mRNA might lead to altered cancer susceptibility.
In addition, the relative reduction in alternatively spliced isoforms II, IV and VI in the kidney might point towards a new, still unknown XPG function. For example, Shannon et al. (59) found significantly elevated levels of XPF transcripts and protein in adult mouse testis compared to other mouse tissues, which is consistent with a role for the XPF gene in male germ cell development. Clearly, in the case of XPG, further studies are indicated, including more sensitive techniques like real-time PCR for quantitative gene expression (60).
Single nucleotide polymorphisms in XPG
There is evidence in the literature that normal individuals who carry specific polymorphic single nucleotide base changes in DNA repair genes, which lead to amino acid substitutions, may have an increased risk of certain cancers (61). Dybdahl et al. (62) found that individuals who carry a certain single nucleotide polymorphism (SNP) in the coding region of the XPD gene (Lys751) had a higher risk of developing basal cell carcinomas compared to individuals who did not carry this SNP (Glu751). Another study demonstrated that rare microsatellite polymorphisms in the DNA repair genes XRCC1 and XRCC3 were associated with breast and internal cancers (63). Loss of heterozygosity of the XPG gene was found in primary prostate cancers and metastases (64). We compared our XPG nucleotide sequence with the unannotated GenBank clone, accession no. AL137246, and found two new non-conserved SNPs in the coding sequence of the XPG gene (Arg1053Gly and Arg1080Gly) in addition to the already reported SNP His1104Asp (9). We found the latter to be a relatively common SNP (25.8% 1104His) in the XPG gene (Table 4). Thus, this SNP might also be useful in further population studies to investigate cancer susceptibility in the normal population.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr S.Clarkson for his support and information about some of the XPG primers, and Tala Shahlavi for technical help. S.E. was supported in part by a grant from the Deutsche Forschungsgemeinschaft (DFG).
DDBJ/EMBL/GenBank accession nos: AF255431–AF255442 and AH009656
References
- 1.Bootsma D., Kraemer,K.H., Cleaver,J.E. and Hoeijmakers,J.H. (1998) Nucleotide excision repair syndromes: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. In Vogelstein,B. and Kinzler,K.W. (eds), The Genetic Basis of Human Cancer. McGraw-Hill, New York, NY, pp. 245–274.
- 2.Kraemer K.H., Lee,M.M. and Scotto,J. (1987) Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch. Dermatol., 123, 241–250. [DOI] [PubMed] [Google Scholar]
- 3.Kraemer K.H., Lee,M.M., Andrews,A.D. and Lambert,W.C. (1994) The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch. Dermatol., 130, 1018–1021. [PubMed] [Google Scholar]
- 4.van Steeg H. and Kraemer,K.H. (1999) Xeroderma pigmentosum and the role of UV-induced DNA damage in skin cancer. Mol. Med. Today, 5, 86–94. [DOI] [PubMed] [Google Scholar]
- 5.Masutani C., Kusumoto,R., Yamada,A., Dohmae,N., Yokoi,M., Yuasa,M., Araki,M., Iwai,S., Takio,K. and Hanaoka,F. (1999) The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature, 399, 700–704. [DOI] [PubMed] [Google Scholar]
- 6.Johnson R.E., Kondratick,C.M., Prakash,S. and Prakash,L. (1999) hRAD30 mutations in the variant form of xeroderma pigmentosum. Science, 285, 263–265. [DOI] [PubMed] [Google Scholar]
- 7.Mudgett J.S. and MacInnes,M.A. (1990) Isolation of the functional human excision repair gene ERCC5 by intercosmid recombination. Genomics, 8, 623–633. [DOI] [PubMed] [Google Scholar]
- 8.O’Donovan A. and Wood,R.D. (1993) Identical defects in DNA repair in xeroderma pigmentosum group G and rodent ERCC group 5. Nature, 363, 185–188. [DOI] [PubMed] [Google Scholar]
- 9.Nouspikel T. and Clarkson,S.G. (1994) Mutations that disable the DNA repair gene XPG in a xeroderma pigmentosum group G patient. Hum. Mol. Genet., 3, 963–967. [DOI] [PubMed] [Google Scholar]
- 10.Takahashi E., Shiomi,N. and Shiomi,T. (1992) Precise localization of the excision repair gene, ERCC5, to human chromosome 13q32.3-q33.1 by direct R-banding fluorescence in situ hybridization. Jpn J. Cancer Res., 83, 1117–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Constantinou A., Gunz,D., Evans,E., Lalle,P., Bates,P.A., Wood,R.D. and Clarkson,S.G. (1999) Conserved residues of human XPG protein important for nuclease activity and function in nucleotide excision repair. J. Biol. Chem., 274, 5637–5648. [DOI] [PubMed] [Google Scholar]
- 12.Scherly D., Nouspikel,T., Corlet,J., Ucla,C., Bairoch,A. and Clarkson,S.G. (1993) Complementation of the DNA repair defect in xeroderma pigmentosum group G cells by a human cDNA related to yeast RAD2. Nature, 363, 182–185. [DOI] [PubMed] [Google Scholar]
- 13.MacInnes M.A., Dickson,J.A., Hernandez,R.R., Learmonth,D., Lin,G.Y., Mudgett,J.S., Park,M.S., Schauer,S., Reynolds,R.J. and Strniste,G.F. (1993) Human ERCC5 cDNA-cosmid complementation for excision repair and bipartite amino acid domains conserved with RAD proteins of Saccharomyces cerevisiae and Schizosaccharomyces pombe. Mol. Cell Biol., 13, 6393–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shiomi T., Harada,Y., Saito,T., Shiomi,N., Okuno,Y. and Yamaizumi,M. (1994) An ERCC5 gene with homology to yeast RAD2 is involved in group G xeroderma pigmentosum. Mutat. Res., 314, 167–175. [DOI] [PubMed] [Google Scholar]
- 15.Murray J.M., Tavassoli,M., al Harithy,R., Sheldrick,K.S., Lehmann,A.R., Carr,A.M. and Watts,F.Z. (1994) Structural and functional conservation of the human homolog of the Schizosaccharomyces pombe rad2 gene, which is required for chromosome segregation and recovery from DNA damage. Mol. Cell Biol., 14, 4878–4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harrington J.J. and Lieber,M.R. (1994) Functional domains within FEN-1 and RAD2 define a family of structure-specific endonucleases: implications for nucleotide excision repair. Genes Dev., 8, 1344–1355. [DOI] [PubMed] [Google Scholar]
- 17.Robins P., Pappin,D.J., Wood,R.D. and Lindahl,T. (1994) Structural and functional homology between mammalian DNase IV and the 5′-nuclease domain of Escherichia coli DNA polymerase I. J. Biol. Chem., 269, 28535–28538. [PubMed] [Google Scholar]
- 18.Aboussekhra A., Biggerstaff,M., Shivji,M.K., Vilpo,J.A., Moncollin,V., Podust,V.N., Protic,M., Hubscher,U., Egly,J.M. and Wood,R.D. (1995) Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell, 80, 859–868. [DOI] [PubMed] [Google Scholar]
- 19.Mu D., Hsu,D.S. and Sancar,A. (1996) Reaction mechanism of human DNA repair excision nuclease. J. Biol. Chem., 271, 8285–8294. [DOI] [PubMed] [Google Scholar]
- 20.Wakasugi M., Reardon,J.T. and Sancar,A. (1997) The non-catalytic function of XPG protein during dual incision in human nucleotide excision repair. J. Biol. Chem., 272, 16030–16034. [DOI] [PubMed] [Google Scholar]
- 21.Le Page F., Kwoh,E.E., Avrutskaya,A., Gentil,A., Leadon,S.A., Sarasin,A. and Cooper,P.K. (2000) Transcription-coupled repair of 8-oxoguanine: requirement for XPG, TFIIH, and CSB and implications for Cockayne syndrome. Cell, 101, 159–171. [DOI] [PubMed] [Google Scholar]
- 22.Robbins J.H. (1988) Xeroderma pigmentosum. Defective DNA repair causes skin cancer and neurodegeneration. J. Am. Med. Assoc., 260, 384–388. [DOI] [PubMed] [Google Scholar]
- 23.Robbins J.H., Kraemer,K.H., Lutzner,M.A., Festoff,B.W. and Coon,H.G. (1974) Xeroderma pigmentosum. An inherited disease with sun sensitivity, multiple cutaneous neoplasms, and abnormal DNA repair. Ann. Intern. Med., 80, 221–248. [DOI] [PubMed] [Google Scholar]
- 24.Moriwaki S., Stefanini,M., Lehmann,A.R., Hoeijmakers,J.H., Robbins,J.H., Rapin,I., Botta,E., Tanganelli,B., Vermeulen,W., Broughton,B.C. et al. (1996) DNA repair and ultraviolet mutagenesis in cells from a new patient with xeroderma pigmentosum group G and cockayne syndrome resemble xeroderma pigmentosum cells. J. Invest. Dermatol., 107, 647–653. [DOI] [PubMed] [Google Scholar]
- 25.Rapin I., Lindenbaum,Y., Dickson,D., Kraemer,K.H. and Robbins,J.H. (2000) Cockayne syndrome and xeroderma pigmentosum: DNA repair disorders with overlaps and paradoxes. Neurology, 55, 1442–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneider T.D. (1997) Sequence walkers: a graphical method to display how binding proteins interact with DNA or RNA sequences [published erratum appears in Nucleic Acids Res. (1998), 26, following 1134]. Nucleic Acids Res., 25, 4408–4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nouspikel T., Lalle,P., Leadon,S.A., Cooper,P.K. and Clarkson,S.G. (1997) A common mutational pattern in Cockayne syndrome patients from xeroderma pigmentosum group G: implications for a second XPG function. Proc. Natl Acad. Sci. USA, 94, 3116–3121. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Okinaka R.T., Perez-Castro,A.V., Sena,A., Laubscher,K., Strniste,G.F., Park,M.S., Hernandez,R., MacInnes,M.A. and Kraemer,K.H. (1997) Heritable genetic alterations in a xeroderma pigmentosum group G/Cockayne syndrome pedigree. Mutat. Res., 385, 107–114. [DOI] [PubMed] [Google Scholar]
- 29.Ellison A.R., Nouspikel,T., Jaspers,N.G., Clarkson,S.G. and Gruenert,D.C. (1998) Complementation of transformed fibroblasts from patients with combined xeroderma pigmentosum–Cockayne syndrome. Exp. Cell Res., 243, 22–28. [DOI] [PubMed] [Google Scholar]
- 30.Schneider T.D. (1997) Information content of individual genetic sequences. J. Theor. Biol., 189, 427–441. [DOI] [PubMed] [Google Scholar]
- 31.Rogan P.K., Faux,B.M. and Schneider,T.D. (1998) Information analysis of human splice site mutations [published erratum appears in Hum. Mutat. (1999), 13, 82]. Hum. Mutat., 12, 153–171. [DOI] [PubMed] [Google Scholar]
- 32.Khan S.G., Levy,H.L., Legerski,R., Quackenbush,E., Reardon,J.T., Emmert,S., Sancar,A., Li,L., Schneider,T.D., Cleaver,J.E. et al. (1998) Xeroderma pigmentosum group C splice mutation associated with autism and hypoglycinemia [published erratum appears in J. Invest. Dermatol. (1999), 12, 402]. J. Invest. Dermatol., 111, 791–796. [DOI] [PubMed] [Google Scholar]
- 33.Khan S.G., Metter,E.J., Tarone,R.E., Bohr,V.A., Grossman,L., Hedayati,M., Bale,S.J., Emmert,S. and Kraemer,K.H. (2000) A new xeroderma pigmentosum group C poly(AT) insertion/deletion polymorphism. Carcinogenesis, 21, 1821–1825. [DOI] [PubMed] [Google Scholar]
- 34.Richards B., Skoletsky,J., Shuber,A.P., Balfour,R., Stern,R.C., Dorkin,H.L., Parad,R.B., Witt,D. and Klinger,K.W. (1993) Multiplex PCR amplification from the CFTR gene using DNA prepared from buccal brushes/swabs. Hum. Mol. Genet., 2, 159–163. [DOI] [PubMed] [Google Scholar]
- 35.Mount S.M. (1982) A catalogue of splice junction sequences. Nucleic Acids Res., 10, 459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hall S.L. and Padgett,R.A. (1996) Requirement of U12 snRNA for in vivo splicing of a minor class of eukaryotic nuclear pre-mRNA introns. Science, 271, 1716–1718. [DOI] [PubMed] [Google Scholar]
- 37.Hall S.L. and Padgett,R.A. (1994) Conserved sequences in a class of rare eukaryotic nuclear introns with non-consensus splice sites. J. Mol. Biol., 239, 357–365. [DOI] [PubMed] [Google Scholar]
- 38.Ma L., Hoeijmakers,J.H. and van der Eb,A.J. (1995) Mammalian nucleotide excision repair. Biochim. Biophys. Acta, 1242, 137–163. [DOI] [PubMed] [Google Scholar]
- 39.Sancar A. (1996) DNA excision repair [published erratum appears in Annu. Rev. Biochem. (1997), 66, VII]. Annu. Rev. Biochem., 65, 43–81. [DOI] [PubMed] [Google Scholar]
- 40.Wood R.D. (1996) DNA repair in eukaryotes. Annu. Rev. Biochem., 65, 135–167. [DOI] [PubMed] [Google Scholar]
- 41.de Laat W.L., Jaspers,N.G. and Hoeijmakers,J.H. (1999) Molecular mechanism of nucleotide excision repair. Genes Dev., 13, 768–785. [DOI] [PubMed] [Google Scholar]
- 42.Emmert S., Kobayashi,N., Khan,S.G. and Kraemer,K.H. (2000) The xeroderma pigmentosum group C gene leads to selective repair of cyclobutane pyrimidine dimers rather than 6-4 photoproducts. Proc. Natl Acad. Sci. USA, 97, 2151–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li R.Y., Calsou,P., Jones,C.J. and Salles,B. (1998) Interactions of the transcription/DNA repair factor TFIIH and XP repair proteins with DNA lesions in a cell-free repair assay. J. Mol. Biol., 281, 211–218. [DOI] [PubMed] [Google Scholar]
- 44.Sugasawa K., Ng,J.M., Masutani,C., Iwai,S., van der Spek,P.J., Eker,A.P., Hanaoka,F., Bootsma,D. and Hoeijmakers,J.H. (1998) Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol. Cell, 2, 223–232. [DOI] [PubMed] [Google Scholar]
- 45.van Hoffen A., Venema,J., Meschini,R., van Zeeland,A.A. and Mullenders,L.H. (1995) Transcription-coupled repair removes both cyclobutane pyrimidine dimers and 6-4 photoproducts with equal efficiency and in a sequential way from transcribed DNA in xeroderma pigmentosum group C fibroblasts. EMBO J., 14, 360–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klungland A., Hoss,M., Gunz,D., Constantinou,A., Clarkson,S.G., Doetsch,P.W., Bolton,P.H., Wood,R.D. and Lindahl,T. (1999) Base excision repair of oxidative DNA damage activated by XPG protein. Mol. Cell, 3, 33–42. [DOI] [PubMed] [Google Scholar]
- 47.Cooper P.K., Nouspikel,T., Clarkson,S.G. and Leadon,S.A. (1997) Defective transcription-coupled repair of oxidative base damage in Cockayne syndrome patients from XP group G. Science, 275, 990–993. [DOI] [PubMed] [Google Scholar]
- 48.Houle J.F. and Friedberg,E.C. (1999) The Drosophila ortholog of the human XPG gene. Gene, 234, 353–360. [DOI] [PubMed] [Google Scholar]
- 49.Tian M. and Alt,F.W. (2000) Transcription-induced cleavage of immunoglobulin switch regions by nucleotide excision repair nucleases in vitro. J. Biol. Chem., 275, 24163–24172. [DOI] [PubMed] [Google Scholar]
- 50.Ludwig D.L., Mudgett,J.S., Park,M.S., Perez-Castro,A.V. and MacInnes,M.A. (1996) Molecular cloning and structural analysis of the functional mouse genomic XPG gene. Mamm. Genome, 7, 644–649. [DOI] [PubMed] [Google Scholar]
- 51.Chyan Y.J., Ackerman,S., Shepherd,N.S., McBride,O.W., Widen,S.G., Wilson,S.H. and Wood,T.G. (1994) The human DNA polymerase β gene structure. Evidence of alternative splicing in gene expression. Nucleic Acids Res., 22, 2719–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sekelsky J.J., Brodsky,M.H., Rubin,G.M. and Hawley,R.S. (1999) Drosophila and human RecQ5 exist in different isoforms generated by alternative splicing. Nucleic Acids Res., 27, 3762–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kozak M. (1999) Initiation of translation in prokaryotes and eukaryotes. Gene, 234, 187–208. [DOI] [PubMed] [Google Scholar]
- 54.Samec S., Clarkson,S.G., Blaschak,J., Chakravarti,A., Morris,M.A., Scherly,D. and Antonarakis,S.E. (1994) Dinucleotide repeat polymorphism within ERCC5 gene. Hum. Mol. Genet., 3, 214. [DOI] [PubMed] [Google Scholar]
- 55.Cheng L., Spitz,M.R., Hong,W.K. and Wei,Q. (2000) Reduced expression levels of nucleotide excision repair genes in lung cancer: a case-control analysis. Carcinogenesis, 21, 1527–1530. [PubMed] [Google Scholar]
- 56.Wei Q.Y., Cheng,L., Hong,W.K. and Spitz,M.R. (1996) Reduced DNA repair capacity in lung cancer patients. Cancer Res., 56, 4103–4107. [PubMed] [Google Scholar]
- 57.Cheng L., Eicher,S.A., Guo,Z.Z., Hong,W.K., Spitz,M.R. and Wei,Q.Y. (1998) Reduced DNA repair capacity in head and neck cancer patients. Cancer Epidemiol. Biomarkers Prev., 7, 465–468. [PubMed] [Google Scholar]
- 58.Oesch F., Aulmann,W., Platt,K.L. and Doerjer,G. (1987) Individual differences in DNA repair capacities in man. Arch. Toxicol. Suppl., 10, 172–179. [DOI] [PubMed] [Google Scholar]
- 59.Shannon M., Lamerdin,J.E., Richardson,L., McCutchen-Maloney,S.L., Hwang,M.H., Handel,M.A., Stubbs,L. and Thelen,M.P. (1999) Characterization of the mouse Xpf DNA repair gene and differential expression during spermatogenesis. Genomics, 62, 427–435. [DOI] [PubMed] [Google Scholar]
- 60.Bieche I., Nogues,C., Paradis,V., Olivi,M., Bedossa,P., Lidereau,R. and Vidaud,M. (2000) Quantitation of hTERT gene expression in sporadic breast tumors with a real-time reverse transcription–polymerase chain reaction assay. Clin. Cancer Res., 6, 452–459. [PubMed] [Google Scholar]
- 61.Mohrenweiser H.W. and Jones,I.M. (1998) Variation in DNA repair is a factor in cancer susceptibility: a paradigm for the promises and perils of individual and population risk estimation? Mutat. Res., 400, 15–24. [DOI] [PubMed] [Google Scholar]
- 62.Dybdahl M., Vogel,U., Frentz,G., Wallin,H. and Nexo,B.A. (1999) Polymorphisms in the DNA repair gene XPD: correlations with risk and age at onset of basal cell carcinoma. Cancer Epidemiol. Biomarkers Prev., 8, 77–81. [PubMed] [Google Scholar]
- 63.Price E.A., Bourne,S.L., Radbourne,R., Lawton,P.A., Lamerdin,J., Thompson,L.H. and Arrand,J.E. (1997) Rare microsatellite polymorphisms in the DNA repair genes XRCC1, XRCC3 and XRCC5 associated with cancer in patients of varying radiosensitivity. Somat. Cell Mol. Genet., 23, 237–247. [DOI] [PubMed] [Google Scholar]
- 64.Hyytinen E.R., Frierson,H.F.,Jr, Sipe,T.W., Li,C.L., Degeorges,A., Sikes,R.A., Chung,L.W. and Dong,J.T. (1999) Loss of heterozygosity and lack of mutations of the XPG/ERCC5 DNA repair gene at 13q33 in prostate cancer. Prostate, 41, 190–195. [DOI] [PubMed] [Google Scholar]