

Abstract
Activation of acid-sensing ion channels (ASICs) contributes to neuronal death during stroke, to axonal degeneration during neuroinflammation, and to pain during inflammation. Although understanding ASIC gating may help to modulate ASIC activity during these pathologic situations, at present it is poorly understood. The ligand, H+, probably binds to several sites, among them amino acids within the large extracellular domain. The extracellular domain is linked to the two transmembrane domains by the wrist region that is connected to two anti-parallel β-strands, β1 and β12. Thus, the wrist region together with those β-strands may have a crucial role in transmitting ligand binding to pore opening and closing. Here we show that amino acids in the β1-β2 linker determine constitutive opening of ASIC1b from shark. The most crucial residue within the β1-β2 linker (Asp110), when mutated from aspartate to cysteine, can be altered by cysteine-modifying reagents much more readily when channels are closed than when they are desensitized. Finally, engineering of a cysteine at position 110 and at an adjacent position in the β11-β12 linker leads to spontaneous formation of a disulfide bond that traps the channel in the desensitized conformation. Collectively, our results suggest that the β1-β2 and β11-β12 linkers are dynamic during gating and tightly appose to each other during desensitization gating. Hindrance of this tight apposition leads to reopening of the channel. It follows that the β1-β2 and β11-β12 linkers modulate gating movements of ASIC1 and may thus be drug targets to modulate ASIC activity.
Keywords: Acid-sensing Ion Channels (ASIC), Disulfide, Ion Channels, Neurotransmitter Receptors, Oocyte, Receptor Desensitization, Receptor Structure-Function, Site-directed Mutagenesis, Sodium Channels
Introduction
Acid-sensing ion channels (ASICs)2 are H+-gated Na+ channels and are abundantly expressed throughout the central and the peripheral nervous system (1). They probably contribute to the excitatory postsynaptic current in many neurons (2–6) and to detection of painful acidosis in peripheral tissues (7, 8). ASIC1a is the most abundant ASIC subunit in the mammalian CNS, and most ASICs in central neurons are homomeric ASIC1a or heteromeric ASIC1a/2a (2, 3, 9). During prolonged acidosis that accompanies brain ischemia and autoimmune inflammation, ASIC1a gets activated and enhances brain injury and axonal damage, respectively (4, 10). Thus, a better understanding of ASIC1 gating is desirable and may lead to pharmacological interventions aimed at modulating ASIC1 activity during diverse neuropathological states (11).
After a rapid drop in pH, ASICs open within milliseconds (12). During prolonged acidification they desensitize; the kinetics of desensitization varies over a 100-fold range from 10 ms (13, 14) to several seconds (15). For most ASICs, desensitization is complete, but some ASICs have small, sustained currents that do not desensitize in the continuous presence of protons (16–18). Such sustained currents could have a major contribution to the harmful effects of ASIC activity during prolonged acidosis. However, despite the importance of desensitization gating and sustained opening of ASICs, our molecular understanding of these processes is incomplete.
Fig. 1 illustrates the gating kinetics of rat ASIC1a (rASIC1a) to mild and strong acidification (pH 6.4 and 5.0). Application of pH 6.4 or 5.0 elicits transient currents that completely desensitize with a time constant τ = ∼2 s. Recently we characterized ASIC1b from shark (sASIC1b) that shares 70% amino acid identity with rASIC1a but desensitizes strikingly differently (18). As illustrated in Fig. 1, application of pH 6.4 elicits transient sASIC1b currents that decline at least 40-fold faster than that of rASIC1a (τ < 50 ms). More importantly, desensitization is incomplete, and a sustained current remains as along as pH is acidic. The level of the sustained current is ∼5% of the peak current amplitude (Fig. 1). pH 5.0 also elicits transient currents, and in addition, shortly after the initial peak, a second current component develops with variable amplitude ∼50% of the amplitude of the transient current (Fig. 1). This second current component desensitizes much more slowly than the initial transient current. We referred to the typical transient ASIC current as the “transient current” and to the second slow current component at pH 5.0 as the “slow current” (18). Thus, both the kinetics and the extent of desensitization are different between sASIC1b and rASIC1a.
FIGURE 1.

Representative current traces for rASIC1a (top panels) and sASIC1b (bottom panels) illustrating the different desensitization kinetics at mild and strong acidification (pH 6.4 and 5.0). The time constant of desensitization of transient rASIC1a currents was τ = 2.1 ± 0.2 s at pH 6.4 (n = 13) and τ = 1.8 ± 0.2 s at pH 5.0 (n = 13; p = 0.1). Desensitization was complete, and no sustained current remained. Desensitization of the transient sASIC1b currents was much faster than for rASIC1a (τ < 50 ms, n = 13; p < 0.001) but incomplete. Desensitization of the second current component at pH 5.0 was best described by two time constants (τ1 = 7 ± 0.4 s and τ2 = 2.1 ± 0.4 s; n = 6). Note that current amplitudes at pH 5.0 were larger than at pH 6.4.
The sustained and slow sASIC1b currents are both unselective, whereas the transient current is Na+-selective (18). Moreover, the slow current cross-desensitizes the sustained but not the transient current (18). These characteristics suggest that the sustained and slow sASIC1b currents share a conformation that is different from the typical transient open conformation. Thus, there are clearly two different open states for sASIC1b that can be separated macroscopically. The slight delay of onset of the slow current at pH 5.0 suggests that the second unselective open state is reached from the desensitized state, which can be described by the following kinetic scheme.
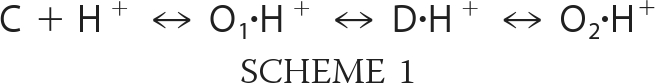 |
This scheme is solely intended to illustrate the basic idea of two macroscopically separable open states, O1 and O2, and for the sake of simplicity, it does not incorporate further closed and open states. Moreover, states could also be cyclically connected, with the unselective open state O2 directly connected to the closed state C but still connected to the desensitized state D, not affecting the major implications of this scheme. The scheme implies that sASIC1b opens to a Na+-selective open state O1 from which it quickly reaches the desensitized state D, from which it reopens to an unselective open state O2. It follows that the desensitized state of sASIC1b is energetically unstable, whereas for most other ASICs, including rASICa, it is stable in the continued presence of H+, and channels do not reopen. Because the sustained sASIC1b current is comparatively small, at equilibrium most (>90%) of the channels are probably in the desensitized state, and few channels (<10%) are in the open state O2.
The crystal structure of chicken ASIC1 (cASIC1) has been solved at acidic pH (19, 20), probably representing the desensitized conformation of the channel. It provides a structural framework to understand ASIC gating (21), in particular desensitization gating. The cASIC1 structure is characterized by the symmetric arrangement of three subunits. Each subunit has two transmembrane domains (TMDs) that are linked to the large extracellular domain (ECD) by an apparently flexible wrist. The ECD resembles a clenched hand and consists of five subdomains, namely the palm, thumb, finger, knuckle, and β-ball domains (19).
In this study, we identified amino acids in the β1-β2 and β11-β12 linkers of the palm domain that determine the presence of a sustained current in sASIC1b. Moreover, our results indicate that the β1-β2 and β11-β12 linkers are dynamic during gating and come in close apposition in the desensitized state. Hindrance of this tight apposition destabilizes the desensitized state, inducing sustained reopening of the channel. Covalently linking the two linkers traps the channel in the desensitized state. These linkers have a similar role in rASIC1a, suggesting that they have a conserved role for ASIC1 gating. The crucial role of these two linkers makes them interesting targets for drugs that modulate ASIC gating.
EXPERIMENTAL PROCEDURES
Molecular Biology
Chimeras of rat ASIC1a and shark ASIC1b were obtained by recombinant PCR. Amino acids substitutions were generated by site-directed mutagenesis using standard protocols. KAPPA HiFi polymerase (peqlab; Erlangen) was used for all PCRs and PCR-derived fragments were controlled by sequencing. All of the constructs were cloned in the oocyte expression vector pRSSP, which is optimized for functional expression in Xenopus oocytes (12). Using the mMessage mMachine kit (Ambion, Austin, TX), capped cRNA was generated by SP6 RNA polymerase from linearized plasmids.
Electrophysiology
Surgical removal of oocytes was done as described elsewhere (18). Between 0.016 and 8 ng of cRNA were injected into stage V or VI oocytes of Xenopus laevis, and the oocytes were kept in OR-2 medium (82.5 mm NaCl, 2.5 mm KCl, 1.0 mm Na2HPO4, 5.0 mm HEPES, 1.0 mm MgCl2, 1.0 mm CaCl2, and 0.5 g/liter polyvinylpyrrolidone) at 19 °C and studied 24–72 h after injection. Whole cell currents were recorded with a TurboTec 03X amplifier (npi electronic, Tamm, Germany) using an automated, pump-driven solution exchange system together with the oocyte testing carousel controlled by the interface OTC-20 (npi electronic) (22). With this system, 80% of the bath solution (10–90%) is exchanged within 300 ms (23). Data acquisition and solution exchange were managed using CellWorks version 5.1.1 (npi electronic). The data were filtered at 20 Hz and acquired at 1 kHz. Holding potential was −70 mV, except when otherwise indicated. All of the experiments were performed at room temperature (20–25 °C). The bath solution for two-electrode voltage clamp contained 140 mm NaCl, 1.8 mm CaCl2, 1.0 mm MgCl2, and 10 mm HEPES. For solutions with a pH ≤ 6.6, HEPES was replaced by MES.
2-Aminoethyl methanethiosulfonate (MTSEA) (Toronto Research Chemicals, North York, Canada) was dissolved in bath solution and kept on ice when not in use. Fresh solutions were prepared every 20 min to ensure desired concentrations. Wild-type channels showed no detectable changes upon exposure to methanethiosulfonate compounds, DTT or H2O2. Before application, pH was adjusted for bath solutions containing MTS compounds, DTT or H2O2.
Data Analysis
Data were collected and pooled from at least two preparations of oocytes isolated on different days from different animals. The data were analyzed with the software IgorPro (Wavemetrics, Lake Oswego, OR). Concentration response curves were fit to the Hill-Function,
where EC50 is the pH at which half-maximal activation/desensitization of the transient current component was achieved, and n is the Hill coefficient. Time constants of desensitization were determined by fitting the decay phase of current traces to a mono-exponential function. The amplitude of the sustained current was determined when it had reached steady state and was normalized to the amplitude of the transient peak current.
Reversal potential of the transient current of the rASIC1a-MDS mutant was determined by activation of the channel with pH 5.0 at different holding potentials between −70 and +50 mV. Reversal potential of the sustained current at pH 5.0 was determined by stepping voltage in 20 mV steps between −70 and +70 mV. Currents after identical voltage steps at pH 7.4 (before activation of channels) were subtracted from currents at pH 5.0 to yield reversal potentials of pH 5.0 induced currents. The results are reported as the means ± S.E. They represent the mean of n individual measurements on different oocytes. Statistical analysis was done with Student's unpaired t test.
RESULTS
The Proximal Ectodomain Controls Sustained Opening of ASIC1
To identify the amino acids that determine sustained opening of sASIC1b, we generated a series of chimeras in which we exchanged different parts of the ECD of sASC1b by corresponding sequences from rASIC1a. For these chimeras, we then measured currents at pH 6.4 and 5, estimated the time constant of desensitization of the transient current, and determined the presence and amplitude of a sustained current; we did not investigate the desensitization of the second slow current, which will be briefly discussed at the end of this manuscript. First, we exchanged the whole ECD of sASIC1b. The resulting chimera (srs6) showed transient inward currents upon application of low pH (Fig. 2). At pH 5, currents desensitized completely with a time constant τ = 1.5 ± 0.2 s (n = 14), not significantly different from rASIC1a (p = 0.44). Moreover, the desensitization rate at pH 6.4 was similar to that at pH 5 (Fig. 2). Thus, desensitization of this chimera was indistinguishable from rASIC1a, showing that the ECD determines the kinetics of desensitization and the presence of a sustained current in ASIC1. Therefore, we substituted gradually smaller parts of the ECD of sASIC1b. Substitution of either the first two-thirds or the first one-third of the ECD of sASIC1b (chimeras srs5 and srs4) yielded channels that desensitized approximately three times more rapidly than rASIC1a currents (at pH 5: τ = 0.7 ± 0.1 s, n = 14, and τ = 0.5 ± 0.04 s, n = 29, respectively; p < 0.001) and more than ten times more slowly than sASIC1b currents (p < 0.001). For both chimeras, the desensitization rate at pH 6.4 was similar to that at pH 5 (Fig. 2). Most importantly, both chimeras desensitized completely to pH 6.4 and 5, and there was no sustained or slow current. Thus, the first one-third of the ECD of sASIC1b is necessary for the fast desensitization and the presence of the slow sustained current of this channel.
FIGURE 2.

A small region shortly after TMD1 determines sustained opening of sASIC1b. Left panel, schematic drawings of rASIC1a and sASIC1b and chimeras. Middle panel, representative traces of currents at pH 6.4 and 5.0. The scale bars correspond to 2 μA, except when otherwise indicated. Right panel, time constants of desensitization of the transient current at pH 6.4 (open circles) and 5 (filled circles). n > 6. ***, p < 0.001 (compared with sASIC1b).
Substitution of either the first 123 or the first 24 amino acids of the sASIC1b ECD (chimeras srs3 and srs2) had essentially the same effect: the kinetics of desensitization was intermediate to rASIC1a and sASIC1b (at pH 5: τ = 0.3 ± 0.02 s, n = 15, and τ = 0.2 ± 0.02 s, n = 13, respectively; p < 0.001) and similar at pH 6.4 and 5. Moreover, there was no sustained or slow current. When we substituted only the first 14 amino acids of the ECD, we obtained a chimera (srs1) with a very different desensitization kinetics: desensitization of the transient current was very fast (τ < 50 ms) and incomplete (amplitude of the sustained current was 11 ± 2% of the peak current at pH 6.4, n = 3, and 13 ± 2% at pH 5, n = 5). Thus, this chimera desensitized basically as sASIC1b wild type. In summary, the most striking difference in desensitization was observed between chimeras srs1 and srs2, which differ in only 7 amino acids.
The results obtained with the chimeras between sASIC1b and rASIC1a revealed that a small region of the ECD (residues 109–115) shortly after TMD1 is necessary for the presence of a sustained current at pH 6.4 and a slow current at pH 5. Moreover, in agreement with a previous report (13), this same region largely determines the kinetics of desensitization of the transient current; other regions that determine the kinetics of desensitization to a lesser extent seem to be scattered over the ECD. As is shown in Fig. 3, the critical region lies in a linker that connects β-strand 1 of the palm domain to β-strand 2 of the β-ball. We next analyzed in more detail the effect on desensitization of these 7 amino acids, Met109–Tyr115, in the β1-β2 linker.
FIGURE 3.

Residues 109–115 of sASIC1b localize to the β1-β2 linker. A, ribbon representation of the desensitized cASIC1 structure. The different domains of the ECD are shown in different colors (TMDs in red, palm in yellow, thumb in green, knuckle in turquoise, finger in purple, and β-ball in orange). B, β-strands 1 and 12 from one subunit and the linkers that connect them to β-strands 2 and 11, respectively, are shown. C, schematic representation of B. D, amino acid sequences of the β1-β2 and the β11-β12 linkers of sASIC1b, rASIC1a, and cASIC1, respectively. The carboxyl-carboxylate pair between β-strands 1 and 12 is illustrated by a red line.
Amino Acids 109–111 Control Sustained Opening of ASIC1; Amino Acid 110 Is Especially Important
A previous study identified an amino acid triplet in the proximal ECD as a determinant of the slow desensitization kinetics of rASIC1a (13). This triplet is 83SQL in rASIC1a and 112PFM in sASIC1b and falls into the critical region identified by our chimeras (Fig. 3). Therefore, we next substituted the PFM triplet of sASIC1b with the SQL triplet of rASIC1a. sASIC1b-SQL desensitized rapidly with a time constant τ < 50 ms (n = 10), and it still had a sustained current at pH 6.4 and a slow current at pH 5 (5 ± 2% of the peak current amplitude at pH 6.4 and 7 ± 1% at pH 5, n = 8; Fig. 4A), clearly showing that the PFM triplet is not necessary for sustained sASIC1b opening. Unexpectedly, although because of the fast desensitization we cannot exclude some slowing of the desensitization of sASIC1b by the SQL substitution, the SQL triplet did not determine slow desensitization kinetics of the transient sASIC1b current (Fig. 4A).
FIGURE 4.

Aspartate 110 is most crucial for sustained opening of sASIC1b. A and B, left panels, representative current traces at pH 6.4 and 5.0 for sASIC1b (A), rASIC1a (B), and amino acid substitutions in the β1-β2 linker. The scale bars correspond to 2 μA, except when otherwise indicated. The insets show the current decline after washout of acidic pH on a 4-fold expanded scale. Right panels, time constants of desensitization of the transient current at pH 6.4 (open circles) and 5 (filled circles). n > 8. *, p < 0.05; ***, p < 0.001 (compared with wild type). C, left panel, current-voltage relationship for the transient and the sustained current of rASIC1a-MDS at pH 5.0. Channels had been repeatedly activated at different holding potentials and currents normalized to the current at −70 mV. The absolute values of the current amplitudes at −70 mV were 7.2 ± 1.7 μA (transient current; n = 11), and 0.77 ± 0.13 μA (sustained current; n = 10), respectively. The current-voltage relationship for voltages between −30 and +50 mV for rASIC1a-wt is shown for comparison as dotted squares; currents were normalized to the current at −30 mV (n = 3). Right panel, representative current trace illustrating constitutive activity of rASIC1a-MDS. Amiloride (1 mm) and substitution of Na+ by the large cation NMDG+ reduced the background current, revealing some constitutive activity of rASIC1a-MDS at pH 7.4.
We then substituted in sASIC1b the three amino acids just upstream of the PFM triplet, 109MDS, by those of rASIC1a, 80VAA. sASIC1b-VAA desensitized rapidly with a time constant τ < 100 ms (n = 16), but strikingly it no longer had a sustained current at pH 6.4 nor a slow current at pH 5 (Fig. 4A), clearly showing that the MDS triplet in sASIC1b is necessary for sustained sASIC1b opening. Combined substitution of both triplets, MDS and PFM, by VAASQL yielded a channel that had no sustained openings and desensitized with a time constant τ = 160 ± 11 ms (n = 23), similar to chimera srs2 (p = 0.13; Fig. 4A). These results demonstrate that both triplets, MDS and PFM, contributed to the kinetics of desensitization of the sASIC1b current but that only the MDS triplet was necessary for sustained opening of sASIC1b.
To further define the role of individual amino acids within the MDS triplet for sASIC1b desensitization, we substituted the three amino acids individually with those found in rASIC1a (V, A, A, respectively). sASIC1b with any of the three individual substitutions, M109V, D110A, and S111A, desensitized very rapidly (τ < 50 ms, n = 8; Fig. 4A). sASIC1b-M109V had no sustained currents at pH 6.4 but did at pH 5 (7 ± 4% of the peak current amplitude, n = 6). In contrast, sASIC1b-D110A had no sustained currents at pH 6.4 or 5 (n = 6; Fig. 4A), and not even at pH 4 (not shown). sASIC1b-S111A still had sustained openings at pH 6.4 and slow openings at pH 5 (2 ± 1% of the peak current amplitude at pH 6.4 and 8 ± 3% at pH 5, n = 11; Fig. 4A). Collectively, these results show that the MDS triplet in the proximal ECD is necessary for sustained sASIC1b opening and that Asp110 has an especially crucial role within the triplet.
To further corroborate these findings, we did inverse substitutions in rASIC1a (Fig. 4B). rASIC1a-PFM desensitized with a time course similar to rASIC1a wild type (τ = 1.2 ± 0.14 s, n = 10, compared with τ = 1.8 ± 0.16 s, n = 13; p = 0.12) and had no sustained or slow current (Fig. 4B), showing that this triplet is not sufficient for sustained opening of rASIC1a. In sharp contrast, rASIC1a-MDS desensitized much faster than rASIC1a wild type (τ < 50 ms, n = 10; p < 0.001) and displayed robust sustained currents at pH 5 (13 ± 2% of the peak current amplitude, n = 8; Fig. 4B). Combined substitution of the two triplets, VAA and SQL, by MDSPFM had similar effects as substitution of VAA by MDS alone: fast desensitization of the transient current (τ < 50 ms, n = 7) and sustained openings at pH 6.4 and 5 (8 ± 2% of the peak current amplitude at pH 6.4 and 12 ± 2% at pH 5, n = 8; Fig. 4B). These findings clearly show that the MDS triplet is not only necessary for sustained opening of sASIC1b but also sufficient to introduce sustained opening into rASIC1a. Because Asp110 had an especially prominent role for sustained opening of sASIC1b, we substituted the corresponding amino acid in rASIC1a, Ala81, with an Asp. rASIC1a-A81D desensitized significantly faster (τ = 340 ± 50 ms, n = 11; p < 0.001) than rASIC1a wild-type and showed sustained opening with an amplitude of 2% of the transient current at pH 5 (n = 4; Fig. 4B), showing that am Asp at this position is a determinant of fast desensitization and is sufficient to introduce sustained opening in rASIC1a.
Sustained sASIC1b currents are unselective, whereas transient currents are Na+-selective (18). Similarly, reversal potentials of leak-subtracted sustained rASIC1a-MDS currents were also significantly shifted by 20 mV to the left compared with transient currents (1 ± 2 mV compared with 21 ± 3 mV; n = 10; p < 0.001; Fig. 4C), showing that the MDS triplet introduces unselective sustained currents into rASIC1a. Some sustained activity is even induced at pH 7.4, as revealed by amiloride block and sensitivity of the background current to exchange by the large cation NMDG+ (Fig. 4C). Constitutive ASIC1a activity will load the oocytes with Na+ and shift the Na+ equilibrium potential to the left, explaining the reversal potential of the transient current ∼20 mV (instead of the usual 50–60 mV; Fig. 4C).
Accessibility of Residue 110 Is State-dependent
By which mechanism does an aspartate at position 110 (in sASIC1b) keep the ASIC1 pore constitutively open? One possibility is that this residue moves during the gating transitions of ASIC1 and that it sterically destabilizes the desensitized conformation. To get evidence for movement of this residue during gating of ASIC1, we assessed the accessibility of residue 110 of sASIC1b to modification by cysteine-reactive MTSEA, a positively charged MTS reagent. For wild-type sASIC1b, MTSEA had no effect on the peak amplitude or on the appearance of the sustained current, when applied at pH 7.4 and 5 (Fig. 5A). Introduction of a cysteine at position 110 resulted in channels that desensitized rapidly and did not have a sustained current (Fig. 5B), similar to the introduction of an alanine at the same position (Fig. 4A). Different reactivity of Cys110 with MTSEA in different states of the channel (closed versus desensitized) would detect a movement of this residue relative to surrounding residues. We first investigated MTS modification in the closed state (Fig. 5B). Application of a low concentration (0.5 μm) of MTSEA at pH 7.4 in the interval between applications of pH 6 dramatically altered the currents of sASIC1b-D110C: peak amplitude gradually increased 2.3-fold, and a robust sustained current appeared that had an amplitude of 28 ± 4% of the peak current (n = 6). After five applications of 10-s duration, the modification of the current was complete (Fig. 5B).
FIGURE 5.

MTS modification of sASIC1b-D110C leads to sustained opening. A, 1 mm MTSEA, when applied at pH 7.4 and 5.0, did not change the current of wt-sASIC1b. B, 500 nm MTSEA, applied for 5 s in the interval between activation (at pH 7.4), induced robust sustained currents for sASIC1b-D110C. Activation was with pH 6 for 5 s. C, a higher concentration of MTSEA (1 mm) than in B, applied at pH 6 when channels were desensitized, also induced robust sustained currents. Note also the different time scales in B and C. The sustained currents could be reversed by application of 10 mm DTT. D, increase in sustained currents of sASIC1b-D110C as a function of exposure time (time exposed × concentration MTSEA) when MTSEA was applied at pH 7.4 (open squares) or at pH 6 (open circles), in experiments like those shown in B and C. The symbols represent the mean amplitudes of the sustained currents, normalized to the maximal sustained current, and quantified every 5 s (n = 8); the lines represent exponential fits. E, activation curves (circles) and SSD curves (squares) for peak currents of sASIC1b-D110C before (open symbols) and after MTSEA modification (100 μm for 60 s at pH 7.4; filled symbols). For activation curves, the channels were activated with different acidic solutions, as indicated, from a conditioning pH 7.4 (n = 8, without MTSEA; n = 9, with MTSEA). For SSD curves, channels were activated with pH 5 with different conditioning pH, as indicated (n = 8, without MTSEA; n = 6, with MTSEA). Peak current amplitudes were normalized to the peak current amplitude at pH 5.0 (activation curves) or with conditioning pH 7.4 (SSD curves), respectively. The dotted lines illustrate the expected increase in peak current at pH 6 by MTS modification, which is too small to explain the increase in peak current amplitude observed in experiments like in B and C.
Application of a 2,000-fold higher MTSEA concentration (1 mm) at pH 6.0 (instead of pH 7.4) similarly led to the slow appearance of a sustained current (18 ± 6% of the initial peak current amplitude, n = 11; Fig. 5C), suggesting that MTSEA also modified the channel when it was in the desensitized conformation, albeit with a dramatically smaller reaction rate (see below). After recovery at pH 7.4, the amplitude of the transient current also was increased, further suggesting MTSEA modification of sASIC1b-D110C at pH 6.0. Application of the reducing agent DTT (10 mm) reversed the increase in sustained current (Fig. 5C). Together these results suggest that MTSEA covalently modified Cys110 and that this modification strongly increased peak amplitude and enables sustained opening of sASIC1b. MTSEA modification was faster at pH 7.4 than at pH 6, although a 2,000-fold smaller concentration was used to modify the channel. Consequently, exponential fits of the fractional increase of the sustained current versus exposure time (in mm × seconds; Fig. 5D) yielded a reaction rate of 100,000 m−1 s−1 at pH 7.4 that was 22,000-fold higher than at pH 6 (4.6 m−1 s−1; p < 0.005). Thus, at pH 7.4, Cys110 was modified by MTSEA with a rate comparable with mercaptoethanol in solution (∼37,000 m−1 s−1) (24), suggesting unhindered access to this residue in the closed conformation.
The increase of the peak current amplitude after MTSEA modification suggested an increased apparent H+ affinity. However, MTSEA modification (100 μm for 60 s) shifted the apparent H+ affinity of activation only slightly, showing that an increased apparent H+ affinity cannot explain the large increase in peak current amplitude. We speculate that MTSEA modification increased the open probability at any given pH. In contrast, steady state desensitization (SSD) curves were significantly shifted to the right by ∼0.3 pH units (p < 0.001; Fig. 5E), so that larger concentrations of H+ were needed to induce SSD. Similar to the appearance of a sustained current, this requirement for larger concentrations of H+ to induce SSD after MTSEA modification further suggests that MTSEA destabilized the desensitized state of sASIC1b.
The profound difference in reaction rate at pH 7.4 versus 6.0 could be due to a dependence of the modification on the state of the channel (closed versus desensitized) or on the pH (pH 7.4 versus 6). To differentiate between these possibilities, we used the fact that increased extracellular Ca2+ concentrations shift SSD to lower pH (25) to find a single pH at which channels were predominantly in the desensitized or in the closed state, respectively, depending solely on the extracellular Ca2+ concentration (but not on pH). We determined SSD curves in the presence of 1 and 20 mm Ca2+ to strongly shift the SSD curve; for both Ca2+ concentrations, SSD curves were determined with and without MTS modification. SSD curves were shifted by ∼0.4 pH units by the high Ca2+ concentration and by approximately the same amount by MTS modification (Fig. 6A). We chose pH 7.0 to determine the reaction rate of MTS modification because at this pH and 1 mm Ca2+, all unmodified channels were in the desensitized conformation, whereas at 20 mm Ca2+, ∼50% of the unmodified channels were desensitized, and the other 50% were closed (Fig. 6A). We argued that the partial desensitization with 20 mm Ca2+ might not strongly affect the reaction rate, because MTS modification happened so quickly at pH 7.4 that it should quickly absorb modified channels into the closed state (because of the shift of the SSD curve; Figs. 5E and 6A). In fact, the reaction rate at pH 7, 20 mm Ca2+, was not significantly different (35,000 m−1 s−1, n = 4; p = 0.2) than at pH 7.4 and 1.8 mm Ca2+ but ∼100-fold higher than with 1 mm Ca2+ (320 m−1 s−1, n = 4; p < 0.05; Fig. 6, B and C), clearly demonstrating state dependence of the MTS modification. The reaction rate at pH 7, 1 mm Ca2+, was ∼70-fold higher than at pH 6, suggesting some pH dependence of the modification (26). In any case, however, the significantly faster reaction rate at pH 7 with 20 mm Ca2+ than with 1 mm Ca2+ clearly shows that the modification of Cys110 by MTSEA was state-dependent and suggests that residue 110 is easily accessible for MTSEA in the open conformation and moves upon desensitization to become less accessible.
FIGURE 6.

MTS modification of sASIC1b-D110C is state-dependent. A, SSD curves for peak currents of sASIC1b-D110C in 1 mm Ca2+ (circles) and 20 mm Ca2+ (squares) before (open symbols) and after MTSEA modification (100 μm for 60 s at pH 7.4; filled symbols). The channels were activated with pH 5 with different conditioning pH levels, as indicated (n = 8). Peak current amplitudes were normalized to the peak current amplitude with conditioning pH 7.8 (1 mm Ca2+) or pH 7.4 (20 mm Ca2+), respectively. The dotted line illustrates the partial desensitization at pH 7 and in 20 mm Ca2+ (before MTS modification). B, 600 nm MTSEA was applied in 20 mm Ca2+ for 10 s in the interval between activation (pH 6, 5 s) of the channel. C, a higher concentration of MTSEA (20 μm) than in B was applied in 1 mm Ca2+ for 30 s in the interval between activation (pH 6, 5 s) of the channel. Note also the different time scales in B and C. D, increase in sustained currents as a function of exposure time (time exposed × concentration MTSEA) when MTSEA was applied in 20 mm Ca2+ (closed squares) or in 1 mm Ca2+ (closed circles) in experiments like those shown in B and C. For both Ca2+ concentrations, conditioning pH was 7.0. The symbols represent the mean amplitudes of the sustained currents (sust), normalized to the maximal sustained (sust,max) current and quantified every 30 s with 1 mm Ca2+ and every 10 s with 20 mm Ca2+ (n = 4); the lines represent exponential fits.
The introduction at residue 110 of a long side chain with a positive charge had dramatic effects on sASICb currents. To confirm this result, we introduced a permanent positive charge at this position. The side chain of a lysine closely resembles the side chain of a cysteine modified by MTSEA (Fig. 7). As expected from this close resemblance, the kinetics of currents carried by sASIC1b-D110K was virtually identical to those carried by sASIC1b-D110C after MTSEA modification: the transient current was followed by a large sustained current that was much more pronounced than for wild-type sASIC1b (50 ± 11% of the peak current amplitude at pH 6.4, n = 3, 56 ± 11% at pH 6.0, n = 2, and 400 ± 240% at pH 5, n = 4; Fig. 7). This result confirms that, at position 110, a long side chain with a positive charge strongly favors sustained opening of sASIC1b.
FIGURE 7.

A Lys at position 110 mimics MTSEA modification. Left panels, chemical structures of an MTSEA-modified cysteine and a lysine. Right panels, representative current traces for MTSEA modification of sASIC1b-D110C (top panel) and for sASIC1b-D110K (bottom panel). The currents for sASIC1b-D110K are shown at three different pH values. The current rise time is also shown on an expanded time scale. The current trace at the bottom illustrates amiloride sensitivity (1 mm) of the sustained sASIC1b-D110K current.
Residue 110 Is in Close Contact with Residue 428 in the β11-β12 Linker
The results so far suggest that certain residues at position 110 destabilize the desensitized conformation of sASIC1b, leading to constitutive opening of the channel. Amino acids with short, neutral side chains (Ala and Cys) do not destabilize the desensitized conformation, whereas the desensitized conformation becomes slightly destabilized by a negatively charged side chain (Asp) and strongly destabilized by a large, positively charged side chain (MTSEA-modified Cys or Lys). In addition, state dependence of MTS modification shows that residue 110 becomes less accessible in the desensitized conformation, suggesting that in this conformation its side chain gets buried in a pocket of the protein.
What is the structural basis for destabilization of the desensitized conformation and sustained opening of sASIC1b by large, charged residues? Given the high homology of sASIC1b with cASIC1 (70% amino acid identity) and the fact that cASIC1 was probably crystallized in the desensitized state, we considered the crystal structure of cASIC1 as a good model for the desensitized state of sASIC1b and modeled an aspartate at the position corresponding to Asp110 of sASIC1b (Ala82). Structural analysis showed that an Asp residue at position 82 of cASIC1 is not easily accommodated because this residue is indeed buried. An Asp could form an H-bond with Asn415 but at the same time there would be unfavorable interactions with the main chain oxygens of Ala413, which would probably trigger some local structural changes.
The unfavorable interaction of Asp82 with the main chain oxygen atoms of Ala413 in the β11-β12 linker (Fig. 3) suggested that such an interaction might destabilize the desensitized conformation and lead to sustained opening of sASIC1b. To test this prediction, we substituted the residue corresponding to chicken Ala413 in sASIC1b (A428) by a negatively charged amino acid. The resulting channels (sASIC1b-A428D) had a normal transient current, followed by a sustained current that was much larger than the sustained current of wild-type sASIC1b (at pH 6.4, 42 ± 14% of the peak current amplitude, n = 5, and at pH 5.0, 111 ± 49%, n = 8; Fig. 8), confirming that this exchange further destabilized the desensitized state of sASIC1b. Because sASIC1b-D110A had no sustained current, we made the double substitution D110A/A428D. The resulting channels had a phenotype that resembled that of wild-type sASIC1b except that the slow current desensitized faster (amplitude of the sustained current was 7 ± 1% of the peak current at pH 6.4, n = 4, and 12 ± 2% at pH 5, n = 5; Fig. 8), providing further support for an interaction of amino acid 110 and 428. If both amino acids have short side chains, no sustained currents develop; if one has a negatively charged side chain, robust sustained currents develop; and if both have a negatively charged side chain, the resulting channels have a strong sustained current with an amplitude similar to the transient current.
FIGURE 8.

Representative current traces at pH 6.4 and 5.0 for sASIC1b-wt, -D110A, -A428D, and -D110A/A428D, respectively. The D110A substitution abolished sustained currents, whereas the A428D substitution strongly increased sustained currents. Combined substitutions (D110A/A428D) conferred sustained currents that resembled those of wild type.
Cross-linking of Residues 110 and 428 Traps sASIC1b in the Desensitized State
We searched for further evidence for a tight interaction of residues 110 and 428 in the desensitized conformation of the protein. When we modeled two cysteines at the corresponding residues in the cASIC structure and selected the best side chain rotamer conformations, the two S-atoms were at a distance of 2.9 Å (Fig. 9A), which would be compatible with the formation of an intramolecular disulfide bond. The discrepancy of ∼1 Å between the observed and the ideal distance (2.05 Å) together with an imperfect dihedral angle of ∼120° (ideal is 90°) predicted a strained disulfide bond between Cys82 and Cys413.
FIGURE 9.

Cross-linking of residues 110 and 428 traps sASIC1b in the desensitized state. A, left panel, detail from the cASIC1 crystal structure, in which two Cys residues had been modeled: one at position 82 and one at position 413 (corresponding to positions 110 and 428 of sASIC1b). Right panel, schematic representation with the two cysteines as blue bars. B, reducing and oxidizing conditions had no effect on sASIC1b-wt currents. C, left panel, sASIC1b-D110C/A428C current amplitude gradually decreased with repeated stimulation by ligand (pH 5.0). Switching the pH to 7.8 or reducing conditions strongly increased current amplitude. Right panel, oxidizing conditions strongly reduced current amplitude of sASIC1b-D110C/A428C.
First, we confirmed that neither the oxidizing reagent H2O2 nor the reducing reagent DTT had an effect on sASIC1b wt (Fig. 9B) nor on sASIC1b with individual cysteine substitutions at positions 110 or 428 (D110C and A428C, respectively; results not shown). Then to test the prediction of disulfide bond formation between residues 110 and 428 of sASIC1b, we engineered two cysteines at these positions. After stimulation with pH 5, the resulting channels (sASIC1b-D110C/A428C) showed transient and sustained currents, both of comparatively small amplitude (Fig. 9C). Repeated activation with pH 5 further reduced current amplitudes (Fig. 9C), which is expected if a disulfide bond formed rapidly when the channels are desensitized in the presence of H+ and if this disulfide bond trapped channels in the desensitized conformation. Because the pH in the secretory pathway gradually decreases from ER to Golgi to secretory vesicles (from 7.4 to 5.5) (27), we reasoned that the disulfide bond also might have formed spontaneously during trafficking of sASIC1b and that spontaneous trapping explains the small current amplitudes when channels were activated the first time. In fact, switching the conditioning pH from 7.4 to 7.8 strongly increased the amplitude of the currents elicited by pH 5 (5.4 ± 0.7-fold increase, p < 0.01, n = 11; Fig. 9C), which is expected if the putative disulfide bond were under considerable strain and spontaneously hydrolyzes when the pH is slightly raised and the desensitized state is further destabilized. Alternatively, the two Cys substitutions might have simply shifted the SSD curve. Application of the oxidizing reagent H2O2 (0.15%) at pH 7.8 strongly reduced current amplitudes (4-fold decrease, p = 0.05, n = 5; Fig. 9C), which is not expected if Cys substitutions had shifted the SSD curve and thus clearly argues for formation of a disulfide bond. Moreover, application of the reducing reagent DTT (10 mm) at pH 7.4 strongly increased current amplitudes (5.6 ± 1-fold increase, p < 0.01, n = 8; Fig. 9C), which is in agreement with hydrolysis of a disulfide bond and ensuing recovery from desensitization. Together, these results strongly suggest the spontaneous formation of a strained disulfide bond between Cys110 and Cys428, which trapped sASIC1b in the desensitized conformation. It follows that (i) in the desensitized conformation both residues are in tight contact with each other and (ii) both residues have to move a considerable distance away from each other for recovery from desensitization and the transition into the closed state.
We wanted to confirm the relevance of these findings for rASIC1a and introduced Cys residues at the corresponding positions of rASIC1a (residues 81 and 412; Fig. 3). For rASIC1a-A81C/V421C, at a conditioning pH 7.4, application of pH 6.4 induced currents of comparatively small amplitude. Repetitive activation further reduced current amplitudes (Fig. 10B). Such a sequential reduction is also known for ASIC1a-wt (28) but was significantly more pronounced for the double-cysteine mutant (Fig. 10C), suggesting that a disulfide bond formed in the desensitized state at pH 6.4 and cumulatively trapped channels in this state. At conditioning pH 7.8 amplitudes were only slightly increased (n = 6; Fig. 10B), suggesting that if a disulfide bond had formed it was under less strain than for sASIC1b. Application of 10 mm DTT, however, strongly increased the current amplitude (2.6 ± 0.4-fold increase, p < 0.01, n = 5; Fig. 10B), an effect that was not seen with rASICa wt (Fig. 10A) and that therefore strongly suggests formation of a disulfide bond that trapped channels in the desensitized state. In agreement with this conclusion, application of 0.15% H2O2 strongly decreased current amplitudes (4-fold decrease, p = 0.04, n = 6; Fig. 10B). Thus, rASIC1a-A81C/V421C reproduced the basic findings from sASIC1b-D110C/A428C, suggesting that the two critical residues also are in close apposition in rASIC1a.
FIGURE 10.

Cross-linking of residues 81 and 412 traps rASIC1a in the desensitized state. A, reducing and oxidizing conditions had no effect on rASIC1a-wt currents. B, left panel, rASIC1a-A81C/V412C current amplitude gradually decreases with repeated stimulation by ligand (pH 6.4). Switching the pH to 7.8 has no effect on current amplitude, whereas reducing conditions dramatically increased current amplitude. Right panel, oxidizing conditions reduced current amplitude of rASIC1a-A81C/V412C. C, quantification of tachyphylaxis for rASIC1a-wt and -A81C/V412C (2C). The channels were repeatedly activated, and current amplitudes were normalized to the first amplitude. Repeated activation reduced rASIC1a-A81C/V412C currents significantly more strongly than wt currents. Absolute values of the initial amplitudes were 14.3 ± 2.5 μA (wt; n = 6) and 1.0 ± 0.2 μA (A81C/V412C; n = 6), respectively. The lines represent fits to a mono-exponential function. ***, p < 0.001.
DISCUSSION
In this paper, we present severalfold evidence that the interaction of the β1-β2 and β11-β12 linkers modulates desensitization gating of ASICs and that a pair of amino acids, each in one of the linkers, is especially crucial for this modulation. First, we identify a triplet of amino acids (109MDS) in the β1-β2 linker that determines sustained opening of sASIC1b (Figs. 2 and 4). Within this triplet, aspartate 110 has a predominant effect (Fig. 4). Second, we show that modification by MTS reagents of residue 110, when mutated to cysteine, strongly depends on the state of the channel: modification in the closed state happens at least 100-fold faster than in the desensitized state (Figs. 5 and 6), showing that residue 110 becomes partially buried in the desensitized state. Third, substitutions of residue 428 in the β11-β12 linker that tightly apposes with residue 110 in the desensitized conformation of cASIC also affect reopening of sASIC1b (Fig. 8). Fourth, cysteines engineered at positions 110 and 428 lead to spontaneous formation of a disulfide bond that traps the channel in the desensitized state (Fig. 9). All of these observations suggest the following model: β1-β2 and β11-β12 linkers are dynamic during ASIC gating and tightly appose in the desensitized conformation, occluding residue 110. Hindrance of this apposition destabilizes the desensitized state and leads to sustained reopening. During recovery from desensitization, β1-β2 and β11-β12 linkers “open up” again, exposing residue 110 to solvent. Occlusion in the desensitized state and exposition in the closed state readily explains the state dependence of MTS modification of residue 110.
We found that introduction of the MDS triplet into rASIC1a is sufficient for robust sustained opening of the rASIC1a pore (Fig. 4). Moreover, a disulfide bond also formed between two cysteines in rASIC1, engineered at positions corresponding to 110 and 428, trapping rASIC1a in the desensitized state (Fig. 10). Thus, there is evidence that the interaction of this pair of amino acids has a general role for desensitization gating of ASIC1.
What Is the Basis for Sustained Openings?
We propose that in sASIC1b the interaction of Asp110 and Αla428 destabilizes the desensitized state, leading to reopening of the ion pore. In this model, sustained currents would not reflect incomplete desensitization from the transient open state O1 but rather reopening from the desensitized state to a new open state O2. Evidence for this model is, first, the rebound of current that follows the transient current at low pH (for example pH 5.0, Fig. 1) (18), and, second, the different selectivity of the ion pore during transient and sustained openings (18). In the simplest case, it follows that a sustained current develops whenever channels enter the desensitized state. In fact, transient sASIC1b currents are always followed by sustained currents (18). This situation is different for rASIC3, an ASIC that shows transient currents at pH < 6.9 but sustained currents only at pH ≤ 5.0 (16, 29). For ASIC3, the development of sustained currents would thus require at least one additional H+ binding step. Such additional protonation steps could also explain the slow sASIC1b currents, which develop at low pH and also desensitize (Fig. 1) (18). We speculate that slow structural relaxation accommodates β1-β2 and β11-β12 linkers at low pH (e.g. pH 5) and leads to slow desensitization of sustained currents.
A completely different way of generating sustained currents is by window currents, which are generated by the overlap of the activation and desensitization curves (30). In the narrow window of overlap, sustained currents are generated that can be calculated by the multiplication of the fractional activation and desensitization at a given pH. Thus, sustained window currents are mediated by the transient open state O1 and do not require a separate open state O2. Window currents have first been described for rASIC3 (30, 31) and are also generated by sASIC1b but only in the small pH range from 7.4 to 6.6 (18). Below pH 6.6, sustained sASIC1b currents are entirely unselective sustained currents (18), which we propose are mediated by the second open state O2.
In contrast to homomeric ASIC3, heteromeric channels in which ASIC3 combines with ASIC2b have unselective sustained currents similar to sASIC1b (15), suggesting that ASIC2b introduces structural elements that induce sustained reopening of ASIC3/2b channels. Future experiments will show whether amino acids in the β1-β2 and β11-β12 linkers of ASIC2b also determine unselective sustained currents of heteromeric ASIC3/2b.
The Role of the β1-β2 and β11-β12 Linkers in Desensitization Gating
Results from previous studies that addressed desensitization gating of ASICs (reviewed in Ref. 21) largely agree with our own findings. In ASIC3, residue 79 at the end of β-strand 1 (corresponding to Glu108 in sASIC1b; Fig. 3), if replaced by a cysteine, cannot react with MTS reagents when channels are desensitized (32), suggesting that it is deeply buried. In fact, in the crystal structure of desensitized cASIC1, β-strands 1 and 12 tightly appose and Glu79 (Glu80 in cASIC1) forms a carboxyl-carboxylate pair with Glu417 (cASIC1) at the beginning of β-strand 12 (Fig. 3) (19). In contrast, in the closed state residue 79 can react with MTS reagents with a rate of ∼200 m−1 s−1, suggesting that the carboxyl-carboxylate pair forms only during desensitization and that the tight apposition between β-strands 1 and 12 is not maintained in the closed state. The comparatively low reaction rate (reaction rate was 100,000 m−1 s−1 for modification of Asp110 in the closed state; Figs. 5 and 6) indicates that residue 79 is partially buried also in the closed state of ASIC3. Moreover, MTS modification of ASIC3-E79C dramatically slowed desensitization (32), suggesting that volume on residue 79 destabilizes the desensitized state by hindrance of the tight apposition of β-strands 1 and 12. This is in agreement with our own results.
In rASIC1, the 83SQL triplet in the β1-β2 linker (the triplet just following the MDS triplet; Fig. 3) is one determinant of desensitization kinetics (13), which suggests that it determines the energy barrier between open and desensitized states. This conclusion perfectly agrees with the notion that movement of the β1-β2 linker accompanies desensitization gating. In Xenopus ASIC1, the last residue of this triplet (Met114 in sASIC1b), if replaced by a cysteine, is modified by MTS reagents with an equal rate (∼100 m− s−1) in the closed and the desensitized states, suggesting that this residue is partially buried in both conformations of the protein (33). Equal reaction rates do not provide evidence for rearrangement of this residue during desensitization. It was proposed that this residue interacts with a residue (corresponding to Asn430 in sASIC1b; Fig. 3) in the β11-β12 linker (33). No direct interaction was found, however, and both residues contribute independently to the stability of the desensitized state (33). Thus, although those previous results confirm a general role of the β1-β2 and β11-β12 linkers for desensitization gating, our results highlight a tight interaction between Asp110 and Ala428 in those linkers of sASIC1b and show that this interaction controls sustained reopening.
Very recently, it was shown that small molecules containing a guanidium group and a heterocyclic ring, as exemplified by 2-guanidine-4-methylquinazoline (GMQ), activate ASIC3 at neutral pH (34). Of note, GMQ-induced currents did not desensitize and were less Na+-selective than H+-induced currents. Because it was shown that GMQ binds to a cavity around Glu79 and Glu423 (34) at the edges of β-strands 1 and 12, respectively, our results suggest a molecular explanation for GMQ activation of ASIC3: GMQ slips between β-strands 1 and 12 and prevents apposition of β1-β2 and β11-β12 linkers, thereby preventing desensitization gating of ASIC3 and inducing sustained opening to an unselective open state O2. Opening by GMQ at neutral pH is reminiscent to the small sustained opening of rASIC1a-MDS at pH 7.4 (Fig. 4C).
In summary, our results add to a better understanding of desensitization gating of ASICs and to the molecular basis of sustained ASIC currents. They identify the β1-β2 and β11-β12 linkers as a possible target for pharmacological modulation of ASIC activity, as recently exemplified by the discovery of nonproton ligands for ASIC3 that bind to this region (34).
Acknowledgment
We thank H. Schindelin for helpful discussions.
This work was supported by Deutsche Forschungsgemeinschaft Grant GR1771/3-5 (to S. G.).
- ASIC
- acid-sensing ion channel
- ECD
- extracellular domain
- MTSEA
- 2-aminoethyl methanethiosulfonate
- TMD
- transmembrane domain
- r
- rat
- s
- shark
- c
- chicken
- MES
- 2-(N-morpholino)ethanesulfonic acid
- SSD
- steady state desensitization
- GMQ
- 2-guanidine-4-methylquinazoline.
REFERENCES
- 1. Wemmie J. A., Price M. P., Welsh M. J. (2006) Trends Neurosci. 29, 578–586 [DOI] [PubMed] [Google Scholar]
- 2. Baron A., Waldmann R., Lazdunski M. (2002) J. Physiol. 539, 485–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Askwith C. C., Wemmie J. A., Price M. P., Rokhlina T., Welsh M. J. (2004) J. Biol. Chem. 279, 18296–18305 [DOI] [PubMed] [Google Scholar]
- 4. Xiong Z. G., Zhu X. M., Chu X. P., Minami M., Hey J., Wei W. L., MacDonald J. F., Wemmie J. A., Price M. P., Welsh M. J., Simon R. P. (2004) Cell 118, 687–698 [DOI] [PubMed] [Google Scholar]
- 5. Wemmie J. A., Chen J., Askwith C. C., Hruska-Hageman A. M., Price M. P., Nolan B. C., Yoder P. G., Lamani E., Hoshi T., Freeman J. H., Jr., Welsh M. J. (2002) Neuron 34, 463–477 [DOI] [PubMed] [Google Scholar]
- 6. Weng J. Y., Lin Y. C., Lien C. C. (2010) J. Neurosci. 30, 6548–6558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Deval E., Noël J., Lay N., Alloui A., Diochot S., Friend V., Jodar M., Lazdunski M., Lingueglia E. (2008) EMBO. J. 27, 3047–3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Deval E., Gasull X., Noël J., Salinas M., Baron A., Diochot S., Lingueglia E. (2010) Pharmacol. Ther. 128, 549–558 [DOI] [PubMed] [Google Scholar]
- 9. Vukicevic M., Kellenberger S. (2004) Am. J. Physiol. Cell Physiol. 287, C682–C690 [DOI] [PubMed] [Google Scholar]
- 10. Friese M. A., Craner M. J., Etzensperger R., Vergo S., Wemmie J. A., Welsh M. J., Vincent A., Fugger L. (2007) Nat. Med. 13, 1483–1489 [DOI] [PubMed] [Google Scholar]
- 11. Sluka K. A., Winter O. C., Wemmie J. A. (2009) Curr. Opin. Drug Discovery Dev. 12, 693–704 [PMC free article] [PubMed] [Google Scholar]
- 12. Bässler E. L., Ngo-Anh T. J., Geisler H. S., Ruppersberg J. P., Gründer S. (2001) J. Biol. Chem. 276, 33782–33787 [DOI] [PubMed] [Google Scholar]
- 13. Coric T., Zhang P., Todorovic N., Canessa C. M. (2003) J. Biol. Chem. 278, 45240–45247 [DOI] [PubMed] [Google Scholar]
- 14. Paukert M., Sidi S., Russell C., Siba M., Wilson S. W., Nicolson T., Gründer S. (2004) J. Biol. Chem. 279, 18783–18791 [DOI] [PubMed] [Google Scholar]
- 15. Lingueglia E., de Weille J. R., Bassilana F., Heurteaux C., Sakai H., Waldmann R., Lazdunski M. (1997) J. Biol. Chem. 272, 29778–29783 [DOI] [PubMed] [Google Scholar]
- 16. Waldmann R., Bassilana F., de Weille J., Champigny G., Heurteaux C., Lazdunski M. (1997) J. Biol. Chem. 272, 20975–20978 [DOI] [PubMed] [Google Scholar]
- 17. Hesselager M., Timmermann D. B., Ahring P. K. (2004) J. Biol. Chem. 279, 11006–11015 [DOI] [PubMed] [Google Scholar]
- 18. Springauf A., Gründer S. (2010) J. Physiol. 588, 809–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jasti J., Furukawa H., Gonzales E. B., Gouaux E. (2007) Nature 449, 316–323 [DOI] [PubMed] [Google Scholar]
- 20. Gonzales E. B., Kawate T., Gouaux E. (2009) Nature 460, 599–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gründer S., Chen X. (2010) Int. J. Physiol. Pathophysiol. Pharmacol. 2, 73–94 [PMC free article] [PubMed] [Google Scholar]
- 22. Madeja M., Musshoff U., Speckmann E. J. (1995) J. Neurosci. Methods 63, 211–213 [DOI] [PubMed] [Google Scholar]
- 23. Chen X., Paukert M., Kadurin I., Pusch M., Gründer S. (2006) Neuropharmacology 50, 964–974 [DOI] [PubMed] [Google Scholar]
- 24. Stauffer D. A., Karlin A. (1994) Biochemistry 33, 6840–6849 [DOI] [PubMed] [Google Scholar]
- 25. Babini E., Paukert M., Geisler H. S., Grunder S. (2002) J. Biol. Chem. 277, 41597–41603 [DOI] [PubMed] [Google Scholar]
- 26. Karlin A., Akabas M. H. (1998) Methods Enzymol. 293, 123–145 [DOI] [PubMed] [Google Scholar]
- 27. Wu M. M., Grabe M., Adams S., Tsien R. Y., Moore H. P., Machen T. E. (2001) J. Biol. Chem. 276, 33027–33035 [DOI] [PubMed] [Google Scholar]
- 28. Chen X., Gründer S. (2007) J. Physiol. 579, 657–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Salinas M., Lazdunski M., Lingueglia E. (2009) J. Biol. Chem. 284, 31851–31859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yagi J., Wenk H. N., Naves L. A., McCleskey E. W. (2006) Circ. Res. 99, 501–509 [DOI] [PubMed] [Google Scholar]
- 31. Benson C. J., Eckert S. P., McCleskey E. W. (1999) Circ. Res. 84, 921–928 [DOI] [PubMed] [Google Scholar]
- 32. Cushman K. A., Marsh-Haffner J., Adelman J. P., McCleskey E. W. (2007) J. Gen. Physiol. 129, 345–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li T., Yang Y., Canessa C. M. (2010) J. Biol. Chem. 285, 31285–31291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yu Y., Chen Z., Li W. G., Cao H., Feng E. G., Yu F., Liu H., Jiang H., Xu T. L. (2010) Neuron 68, 61–72 [DOI] [PubMed] [Google Scholar]