

Abstract
Temporins are a group of closely related short antimicrobial peptides from frog skin. Lipopolysaccharide (LPS), the major constituent of the outer membrane of Gram-negative bacteria, plays important roles in the activity of temporins. Earlier studies have found that LPS induces oligomerization of temporin-1Tb (TB) thus preventing its translocation across the outer membrane and, as a result, reduces its activity on Gram-negative bacteria. On the other hand, temporin-1Tl (TL) exhibits higher activity, presumably because of lack of such oligomerization. A synergistic mechanism was proposed, involving TL and TB in overcoming the LPS-mediated barrier. Here, to gain insights into interactions of TL and TB within LPS, we investigated the structures and interactions of TL, TB, and TL+TB in LPS micelles, using NMR and fluorescence spectroscopy. In the context of LPS, TL assumes a novel antiparallel dimeric helical structure sustained by intimate packing between aromatic-aromatic and aromatic-aliphatic residues. By contrast, independent TB has populations of helical and aggregated conformations in LPS. The LPS-induced aggregated states of TB are largely destabilized in the presence of TL. Saturation transfer difference NMR studies have delineated residues of TL and TB in close contact with LPS and enhanced interactions of these two peptides with LPS, when combined together. Fluorescence resonance energy transfer and 31P NMR have pointed out the proximity of TL and TB in LPS and conformational changes of LPS, respectively. Importantly, these results provide the first structural insights into the mode of action and synergism of antimicrobial peptides at the level of the LPS-outer membrane.
Keywords: Antibiotics, Antimicrobial Peptides, Bacterial Toxins, Membrane, NMR
Introduction
The recent surge of antibiotic-resistant bacterial pathogens has generated an intense interest toward achieving host defense antimicrobial peptides (AMPs)4 (1–7). AMPs, being a fundamental component of the innate immunity, are bestowed with a broad-spectrum of antimicrobial activities, killing bacteria, both Gram-negative and Gram-positive, viruses, and fungi (2, 3, 8–10). Many AMPs are also active against multidrug-resistant pathogens (11–13). Thus, AMPs are thought to be an important source of new antibiotics (4, 14–16). The cell lytic activities of AMPs stem from their ability to permeabilize or disintegrate the microbial membrane (2, 3, 17–19). The amino acid sequences of AMPs are rich in hydrophobic and cationic residues, enabling the peptides to initially interact with the anionic membrane of pathogens and subsequently to insert into its hydrophobic core (17–19). In this regard, several AMPs are less active against Gram-negative bacteria as compared with Gram-positive bacteria (20–23). Gram-negative bacteria are protected from antimicrobial agents by the lipopolysaccharide (LPS)-outer membrane (24–26). In particular, the highly anionic LPS layer at the outer leaflet of the outer membrane acts as a permeability barrier against a number of molecules including cationic AMPs (25, 27–29). The LPS structure can be divided into three regions: (a) the conserved lipid A, consisting of fatty acid chains linked to two phosphorylated glucosamine residues; (b) the core oligosaccharide covalently linked to lipid A moiety; and (c) a highly variable hydrophilic polysaccharide named O-antigen (30, 31). The initial attachment of AMPs to LPS might occur through ionic interactions between the cationic peptides and the anionic LPS molecules, leading to “self-promoted” uptake via displacement of divalent cations stabilizing adjacent LPS molecules (14, 26). Most of the highly active AMPs contain a number of positively charged, on average, four to six, amino acids (32). The high cationicity and amphipathic structure formation of AMPs in membrane environments are crucial features for their cell lysis activity (2, 7, 8). To achieve outer membrane permeabilization, AMPs may acquire specific structures in LPS (14, 33–35).
LPS, also termed endotoxin, is well known for pathogenesis of septic shock syndromes in humans, killing 120,000 people annually in the United State alone (36, 37). LPS is intercepted by host immune cells, macrophages, and monocytes, through a specific Toll-like 4 pattern recognition receptor (38, 39). Activation of Toll-like 4 receptor triggers the release of cytokines, e.g. TNF-α, IL-6, IL-1, and others, which are required to clear infections. However, overstimulation of the immune system, induced by excess LPS, may produce an overwhelming amount of cytokines that damage host tissues and organs (40–42). Therefore, structures and interaction studies of AMPs with LPS are vital to better understand their mechanism of antibacterial activities and to facilitate the development of new antiendotoxic/antibacterial agents (43–45).
Temporins are among the smallest (8–14 amino acids) naturally occurring AMPs found in frogs (46–49). The amino acid sequences of temporins are notably characterized by the presence of a larger proportion of non-polar residues and few cationic residues, giving a net charge of 0 to +3 at neutral pH. Temporins are mainly active on Gram-positive bacteria (47). It was recently discovered that LPS is actively involved in regulating the translocation of temporin-1Ta (TA) and temporin-1Tb (TB) (47) through the outer membrane (48, 49). The high minimal inhibitory concentrations of TA and TB, against a number of Gram-negative bacterial strains, could be correlated to self-association of TA and TB in LPS (47–49). By contrast, temporin-1Tl (TL), rich in aromatic residues and bearing two positively charged amino acids, is also active on Gram-negative bacteria (47–49). TL has also been shown to exhibit antiendotoxic activity under both in vitro and in vivo conditions (50). Interestingly, synergistic effects in inhibiting the growth of Gram-negative bacteria and in neutralizing the toxic effect of LPS have been found when TL is combined either with TA or TB (48, 49). It has been demonstrated that TL can inhibit self-association of TA or TB in LPS, presumably rendering facile translocation into the cytoplasmic membrane (48, 49).
In this work, by use of NMR spectroscopy and fluorescence, new insights of temporin-LPS interactions and the mechanisms underlying the synergistic effect of TL+TB were elucidated. In particular, we have utilized LPS either from Escherichia coli 0111:B4 or E. coli 055:B5 containing long polysaccharide chains at the O-antigen domain (Fig. 1A). Our results have delineated a dimeric helical structure of TL in complex with LPS micelles. On the other hand, we have found that LPS induces oligomerization of the TB peptide, involving residues from the N and C termini. The dimeric structure of TL may interact with the lipid A domain of LPS through charge-charge complementarity and hydrophobic packing interactions. This would explain the outer membrane permeabilization and anti-endotoxic activities of TL. In addition, our data revealed that the synergistic activity between TL and TB could be manifested by a complex interplay involving structural changes of LPS, selective stabilization of helical conformation (or destabilization of oligomeric species) of TB, and enhanced proximities of both peptides to LPS micelles.
FIGURE 1.

Interactions and binding affinity of TL and TB with LPS. Panel A, schematic structure of the smooth type LPS, containing a long polysaccharide chain, of E. coli 0111:B4 and E. coli 055:B5 strains, used in this study. Glc, glucose; Hep, heptose; Gal, galactose; Kdo, ketodeoxyoctonic acid; GlcN, N-acetylglucosamine; P, phosphate. Panel B, intrinsic Trp fluorescence emission spectra of TL; panel C, dansyl fluorescence emission spectra of TB in the presence of different concentrations of LPS. Panels D and E, determination of LPS binding affinity of TL (panel D) and TB (panel E) peptides following changes in emission maxima (λmax) with LPS concentrations.
EXPERIMENTAL PROCEDURES
Reagents
LPS from E. coli 0111:B4, fluorescein isothiocyanate (FITC)-conjugated LPS from E. coli 055:B5 and 1,6-diphenyl-1,3,5-hexatriene (DPH) and Triton X-100 were purchased from Sigma. TL (FVQWFSKFLGRIL-amide) and TB (LLPIVGNLLKSLL-amide) were synthesized as described previously (48, 49). Fluorescently labeled, dansylated at the N termini, TL and TB peptides were synthesized commercially by GL Biochem (Shanghai, China) and were further purified by a Waters reverse-phase HPLC using a C18 column (300 Å pore size, 5-μm particle size) by a linear gradient of an acetonitrile/water mixture. The molecular weight of the peptides was confirmed by mass spectrometry.
Fluorescence Studies
Fluorescence experiments were carried out using a Cary Eclipse fluorescence spectrophotometer (Varian, Inc.) equipped with dual monochromators. All measurements were performed using a 0.1-cm path length cuvette and a slit width of 5 nm. TL-LPS binding interactions were recorded by titrating 5 μm peptide with increasing concentrations of LPS. The intrinsic tryptophan of TL was excited at 280 nm and emission was monitored from 300 to 400 nm. TB-LPS binding interactions were monitored by titrating 5 μm dansylated TB with increasing concentrations of LPS. The dansyl group was excited at 335 nm and emission was monitored from 400 to 560 nm. The changes in emission maxima were further used to determine the binding affinity of peptides with LPS using a standard single-site binding equation. Fluorescence resonance energy transfer (FRET) experiments, between Trp of TL, either with the dansylated TL or dansylated TB, were carried out by monitoring the quenching of Trp fluorescence emission intensity upon addition of dansylated peptides. Typically, unlabeled or native TL (5 μm) bound to LPS (5 μm) was titrated with specific concentrations of dansylated TB or TL peptides in 10 mm sodium phosphate buffer, pH 6.8. Samples were excited at 280 nm wavelength. The normalized quenching values (F/F0), F and F0, representing the intensity of fluorescence emission of Trp in the presence and absence of dansylated peptides, respectively, were plotted as a function of mole fraction (Pa) (where Pa = molar concentration of dansylated peptides/molar concentrations of total peptides) of dansylated peptides. An inter-chromophore distance, between Trp and the dansyl group, was determined using the equation r = R0 (E−1 − 1)1/6, where R is the distance between the chromophores, R0 represents the Föster distance, R0 is 23 Å for the Trp-dansyl pair, and E is the efficiency of FRET. E(1−Q) was determined from the linear extrapolation of the quenching plot (F/F0 versus Pa) of Trp fluorescence emission, where Q is the maximum quenching value (51, 52).
Disaggregation studies of FITC-LPS were performed with 0.5 μm FITC-LPS titrated with increasing concentrations of either TB or TL or the TL+TB mixture. All experiments were carried out in 10 mm sodium phosphate buffer, pH 6.8. For DPH binding to LPS and LPS-peptide complexes, fluorescence emission spectra of DPH were obtained either for free DPH (1 μm) or in the presence of LPS alone (5 μm) and in complexes of LPS and peptides (TL and TB) at 1:1 and 1:4, LPS:peptide ratios, in 10 mm sodium phosphate buffer, pH 6.8. The excitation wavelength of DPH was fixed to 358 nm and the emission spectra were collected from 400 to 600 nm (53). The molar concentration of LPS was estimated using a molecular mass of 10 kDa, disregarding the state of aggregations (54).
Dynamic Light Scattering
Dynamic light scattering experiments were carried out in BI-9000AT with a digital autocorrelator (Brookhaven Instruments Corp., Holtsville, NY). Solutions containing either LPS or LPS/peptide mixtures were filtered through 0.45-μm filters (Whatman Inc.) prior to measurements. 1 μm LPS and the samples containing 1:1 and 1:20 ratios of LPS:peptides were scattered at 90° and particle sizes were analyzed through the standard CONTIN method supplied with the instrument. All experiments were done in 10 mm sodium phosphate buffer, pH 6.0.
NMR Spectroscopy
All NMR spectra were recorded either on BRUKER DRX 600 or 800 MHz spectrometers equipped with a cryo-probe and pulse field gradients. Data acquisition and processing were performed with Topspin software (BRUKER) running on a Linux work station. Two-dimensional TOCSY and NOESY spectra of free TL and TB peptides were acquired in aqueous solutions containing 10% D2O at pH 4.5 with a peptide concentration of 0.5 mm and mixing times of 80 and 150 ms, respectively. The measurements were performed at 298 K using 2,2-dimethyl-2-silapentane-5-sulfonate sodium salt as an internal standard (0.0 ppm). Two-dimensional TOCSY and two-dimensional NOESY spectra for free TL were also obtained at 290 K to avoid resonance overlap between residues Val2 and Leu13. A series of one-dimensional 1H NMR spectra were recorded for TL, TB, and a mixture of TL+TB with various concentrations, ranging from 9 to 18 μm, of LPS. Two-dimensional tr-NOESY experiments were performed for TL and TB peptides, at a 0.5 mm peptide concentration, in the presence of 14.5 and 5 μm LPS, respectively, with four different mixing time: 50, 100, 150, and 200 ms. Two-dimensional tr-NOESY spectra were acquired for an equimolar mixture of TL and TB in the presence of 7.5 μm LPS in aqueous solutions with mixing time of 50, 100, and 150 ms. tr-NOESY spectra were also obtained in D2O solutions for TL and TL+TB at a 100-ms mixing time. The natural abundance 13C-1H heteronuclear single quantum coherence spectra for TL and TB, at 1 mm concentration, were recorded with a spectral width of 12 (t1) × 80 (t2) ppm with 400 scans. The 13C chemical shifts were indirectly referenced to 2,2-dimethyl-2-silapentane-5-sulfonate sodium salt. NMR data processing and analysis were carried out using the programs Topspin (BRUKER) and SPARKY (Goddard, T. D., and Kneller, D. G., University of California, San Francisco), respectively.
For saturation transfer difference (STD) experiments, peptide samples, TL (0.5 mm), TB (0.5 mm), and TL+TB together (0.5 mm each), were dissolved in 550 μl of D2O and pH was adjusted to 4.5. A stock solution (1 mm) of LPS from E. coli 0111:B4 was prepared in D2O solution, pH 4.5. STD experiments were performed at 298 K in the presence either of 5.4 μm LPS (for TL and TB) or 15 μm LPS (for TL + TB) using standard STD pulse sequences (55) and the WATERGATE 3-9-19 sequence for water suppression. One-dimensional STD experiments were carried out as described previously (56, 57). Initially, to identify a safe on-resonance frequency, a series of one-dimensional STD NMR experiments were performed at different on-resonance frequencies in the range of 0 to −10 ppm, with an interval of 0.5 ppm for TL and TB in free solution. Finally, on-resonance frequencies were fixed to either at −2.5 or −3.5 ppm, for TL and TB, respectively. An on-resonance frequency of −3.5 ppm was further used for TL+TB samples. At these on-resonance frequencies, −2.5 or −3.5 ppm, peptides were not saturated in the absence of LPS micelles; however, STD signals were detected in the presence of LPS micelles. It is likely that broad NMR signals, due to the high molecular weight of LPS micelles, were responsible for selective saturation of LPS at these on-resonance frequencies as well as the observed STD effects (58–60). The off-resonance frequency for STD experiments was set at 40 ppm (56, 57). Two-dimensional STD-TOCSY spectra were recorded with 350 increments in t1 and 80 transients using a MLEV-17 spin lock field of 80 ms (34). The relaxation delay was set to 2.1 s. Saturation transfer was achieved by using 40 selective Gaussian 270 pulses with a duration of 50 ms.
A series of proton-decoupled 31P NMR spectra were recorded on a Bruker DRX machine operating at a proton frequency of 400 MHz to monitor LPS-peptide interactions. LPS samples were prepared by dissolving 5 mg or 1 mm LPS from E. coli 0111:B4 into 550 μl of water, 50 μl of D2O containing 100 mm Triton X-100 at pH 4.5. LPS samples were titrated with 1 mm TB, TL individually, and with an equimolar mixture, 1 mm each of TL and TB. 31P NMR spectra were acquired with 3072 scans, a spectral width of 9746 Hz, and 16,384 data points at 298 K.
Calculation of NMR-derived Structures
Two-dimensional tr-NOESY spectra either of TL or TL+TB obtained at 150 ms mixing time were utilized to generate the upper bound distance constraints. The volume of NOE cross-peaks was estimated from SPARKY. The upper bound NOE-driven distance constraints were obtained with reference to NOE intensity between the well separated geminal methylene protons of Ser6 and CβH2 of Pro3 for TL and TB, respectively. The lower distance was fixed to 2.0 Å. NOESY spectra of peptides in free solution showed only a few sequential and intra-residue NOEs. These NOEs were also present in the tr-NOESY spectra in the presence of LPS micelles. We have estimated an average value for the upper limit of the distance constrains, ranging from 3.5 to 3.0 Å, for these NOEs based on cross-peak intensities in NOESY and tr-NOESY spectra. It may be noted that determination of three-dimensional structures would be largely driven by the medium and long-range NOEs, exclusively present in the LPS-bound states of the peptides.
The backbone dihedral angle φ was varied from −30 to −120 for non-glycine residues to minimize conformational searching. NMR structures of TL were iteratively refined from an extended polypeptide chain, using a protocol supplied with the CNS 1.1 program (61). The homodimeric structure of TL was generated by making two non-covalent identical copies of TL. To maintain the axial symmetry, a “non-crystallographic symmetry” restraint was applied with a weight of 10 along with the intermolecular NOEs between the two monomers of TL. An ensemble of monomeric helical structures of TB was determined using the DYANA 1.5 program (62). Of 100 structures, 20 structures with the lowest target function were selected to represent the ensemble. Structures were visualized using MOLMOL, PyMOL, and Insight II software. The quality of structures was determined using Procheck (63) and the Protein Structure Validation Suite (64). The completeness among NOE distance restraints and NMR-derived structures were assessed by residue contact plots using the Protein Structure Validation Suite (64).
RESULTS
Interactions of TL and TB with 0111:B4 LPS by Fluorescence
Intrinsic Trp and dansyl fluorescence were monitored to determine the binding affinities of TL and TB with LPS, respectively. The amino acid sequence of TL contains a Trp at position 4; however, because of the absence of intrinsic fluorophore, the N terminus of TB was labeled with a dansyl group. Upon addition of LPS, the emission maxima of Trp and dansyl experienced a blue shift or shifted toward the shorter wavelength, indicating their incorporation into the nonpolar milieu of LPS micelles (Fig. 1, panels B and C). The intensity of Trp emission spectra of TL showed a concomitant increase with increased LPS concentrations (Fig. 1B), whereas dansyl fluorescence of TB became quenched with additional LPS (Fig. 1C). An enhanced intensity of emission of a fluorophore in a non-polar environment is expected to occur as a result of higher quantum yield. However, a diminished intensity of fluorescence emission, observed for dansyl, may result from self-association or aggregation of TB in LPS micelles. Previous studies demonstrated aggregation of TB in LPS lipids (48, 49). The changes in fluorescence emission maxima of TL and TB, as a function of LPS concentrations, have yielded an equilibrium dissociation constant (Kd) of 4.4 ± 0.4 and 3.0 ± 0.5 μm (Fig. 1, panels D and E), respectively. These data indicate that despite the differences in amino acid sequences, both peptides interacted with LPS with comparable affinity. However, TL has been shown to have much higher antiendotoxic and antibacterial activity against Gram-negative bacteria when compared with TB (48, 49). Therefore, the mere LPS binding ability cannot be correlated with the antiendotoxic and antimicrobial activities of the temporins.
Furthermore, we investigated whether interactions of TL and TB caused changes or dissociation of LPS micelles, using DPH fluorescence (53) and DLS studies (supplemental Fig. S1). DPH is characterized by a weak fluorescence emission in aqueous solution. However, an enhanced emission intensity of DPH can be observed in binding to lipid micelles (53). A high-intensity fluorescence emission of DPH was noted for both LPS micelles and LPS-peptide complexes at two different LPS:peptide ratios (1:1 and 1:4, supplemental Fig. S1), indicating micelle states of LPS. DLS experiments also showed that LPS micelles and LPS-peptide complexes formed aggregates of high molecular weight with average diameters ranging from 1400 nm (for LPS alone) to 500 nm (for LPS-peptide complexes) (supplemental Fig. 1). Therefore, these results demonstrate that LPS retains aggregated or micelle states in complex with TL or TB peptides. Hence, according to previous work (49) in the presence of temporins LPS retains micelle organization, although with a reduced size.
NMR Studies of TL, TB, and TL+TB in 0111:B4 LPS Micelles
Sequence-specific resonance assignments of TL and TB were achieved by combined analyses of two-dimensional TOCSY and two-dimensional NOESY spectra (65). NOESY spectra of free peptides were predominantly characterized by intra and sequential NOE connectivities, indicating random conformations in free solutions (supplemental Fig. S2). A limited deviation of 13Cα chemical shifts of amino acids from the random coil values of TL and TB also indicated largely random conformations in free solutions (supplemental Fig. S3).
Three-dimensional structures of AMPs and LPS-interacting peptides could be obtained in complex with LPS micelles by use of transferred nuclear Overhauser effect spectroscopy (tr-NOESY) (14, 41). LPS readily forms micelles of high molecular weight at significantly low concentrations (54). The critical micelle concentration of E. coli 0111:B4 LPS, used in this work, has been estimated to be ∼1.3 to 1.6 μm (54). Peptides undergoing a fast chemical exchange between free and LPS-bound states may generate tr-NOEs enabling determination of bound conformations (33–35, 66, 67). Typically, at the fast chemical exchange regime, NMR spectra of peptide ligands would exhibit binding-induced resonance broadening (68–70). Thus, one-dimensional proton NMR spectra of TL, TB, and a 1:1 mixture of TL+TB were obtained in the presence of increasing concentrations of LPS. Proton NMR spectra of TL, TB, and TL+TB exhibited broadening of almost all the resonances upon addition of LPS micelles (supplemental Fig. S4). The chemical shifts of TL and TB did not show any significant changes in the presence of LPS micelles (supplemental Fig. S4). These observations indicated a fast exchange of peptide ligands between their free and LPS-bound states. At a saturating concentration of LPS, NMR resonances of peptides were highly broadened as a result of high molecular weight or slow tumbling of LPS micelle-peptide complexes, precluding structural studies under such conditions (supplemental Fig. S4).
Interestingly, we found clear differences regarding the extent of LPS-induced resonance broadening for TB alone and for TB in the TL+TB mixture (supplemental Fig. S4). Resonances of TB displayed markedly enhanced broadening in response to LPS binding (supplemental Fig. S4). On the other hand, restricted LPS-induced broadening of resonances of TB was detected in the presence of TL (supplemental Fig. S4). These data may indicate that interactions of TB with LPS micelles have been significantly influenced by the presence of the TL peptide (see below).
The two-dimensional tr-NOESY spectra of TL and TB obtained in the context of LPS micelles have indicated a large number of NOE connectivities (supplemental Fig. S2). Fig. 2 (panels A and B) illustrates diagnostic backbone-mediated NOEs of TL peptide in LPS micelles. Sequential (i to i+1) and medium-range (i to i+2) NH/NH NOEs (supplemental Fig. S5) and medium-range CαH/NH (i to i+2, i+3, and i+4) NOEs can be identified for almost all the residues of TL in complex with LPS (Fig. 2A). All of the medium-range and sequential NOEs of TL identified while bound to LPS micelles are summarized in supplemental Fig. S5. In the presence of LPS micelles, side chain-side chain interactions of TL were also well defined (Fig. 2B). TL contained a number of aromatic residues, inter-side chain NOEs involving aromatic ring proton resonances with aliphatic side chain proton resonances can be seen for TL (Fig. 2B). Strikingly, analyses of tr-NOESY spectra of TL revealed a number of long-range NOE interactions among the aromatic ring proton resonances of Trp4 with aliphatic side chain resonances of Leu9 and Ile12 (Fig. 2B). In addition, NOEs could be identified from Leu13CαH/Val2NH and Leu9CαH/Phe5NH (Fig. 2A). These types of NOE contacts are qualitatively indicative of an anti-parallel dimerization of the helical structure of TL in LPS micelles. Taken together, the LPS-bound structure of TL was well defined, as manifested by the large number of backbone/backbone, backbone/side chain, and side chain-side chain NOEs (supplemental Fig. S5). Trp4 of TL delineated as many as ∼34 NOEs, including long-range NOEs, in LPS (supplemental Fig. S5).
FIGURE 2.

Analyses of tr-NOESY spectra of TL, TB, and TL+TB in LPS micelles. Selected sections of two-dimensional 1H-1H tr-NOESY spectra of TL (panels A and B), TB (panels C and D), and TL+TB (panel E). Panels A and B, NOE connectivities between α proton (CαH) and amide proton (NH) resonances (panel A) and the aromatic ring proton resonances with the up-field shifted aliphatic side chain resonances (panel B) for TL peptide. Panels C and D, NOE connectivities between α proton (CαH) and amide proton (NH) resonances; panel C, down-field shifted side chain resonances of residues Asn7 and Lys10 with the up-field shifted aliphatic side chain resonances for TB peptide. All the long-range NOE contacts are boldface and italicized. Panel E, superposition of a section of tr-NOESY spectrum of TB (in gray contour) and TL + TB (in black contour) correlating NOE connectivities from amide proton resonances with aliphatic proton resonances highlighting the absence of long-range NOE contacts for TB in TL+TB tr-NOESY spectra.
tr-NOESY spectra of TB and TL+TB, obtained in LPS micelles, displayed remarkable differences in NOE connectivities for TB peptide (Fig. 2, panels C–E). In the absence of TL, tr-NOESY spectra of TB displayed medium-range CαH/NH (i to i+2, i+3, and i+4) (Fig. 2C) and sequential NH/NH NOEs (supplemental Fig. S6). The side chain-side chain NOEs (i to i+3/i+4) were detected from the NγH2 of residue Asn7 with residues Pro3, Ile4, and Lys10 (Fig. 2D). These types of NOE patterns are largely compatible with the helical conformations (65). In addition, a number of long-range NOE contacts were observed among residues located at the N and C termini of the TB peptide. In particular, there were backbone-mediated NOEs, of medium to weak intensity, between NH/NH (supplemental Fig. S6 and supplemental Table S1) and CαH/HN resonances of residues Leu2/Leu12, Leu2/Leu13, and Ile4/Leu13 (Fig. 2C and supplemental Table S1). The side chain resonances of Ile4 and Pro3 delineated several NOEs with NHs of Leu12 and Leu13 (Fig. 2C and supplemental Table S1). Taken together, these long-range NOEs indicated that oligomerization of TB occurred in LPS micelles involving residues at the N and C termini of the molecule.
We were able to analyze NOEs in the tr-NOESY spectra of TL+TB in LPS micelles. The chemical shifts of amino acids of TL and TB were found to be largely non-degenerate in the mixture (supplemental Fig. S7). The NOE pattern assigned for TL alone was preserved in the tr-NOESY spectra obtained for the TL+TB mixture (supplemental Fig. S8), indicating that conformation of TL was unaffected by the presence of TB peptide. Strikingly, the long-range tr-NOE contacts of TB were found to be either absent or having significantly reduced intensity upon inclusion of TL peptide (Fig. 2E). As presented in Fig, 2E, NOEs from Pro3 to Lys10, Leu12, Leu13, and Lys10 were not detectable. NOEs among residues Ile4CγHs/Leu12NH, Ile4CγHs/Leu13NH, Leu2NH/Lys10CβHs, and Leu2NH/Leu13CαH of TB were absent when TL and TB were combined together in LPS micelles (Fig. 2E and supplemental Fig. S9). The long-range NH/NH NOEs, involving residues Leu2/Leu12, Leu13, and Ile4/Leu13 of TB were not observed in the tr-NOESY spectra of TL+TB, in the context of LPS micelles (supplemental Fig. S9). However, there were sequential NH/NH NOEs (supplemental Fig. S9) and medium-range CαH/HN NOEs and medium-range side chain-side chain NOEs (supplemental Fig. S9) for TB residues, in the TL+TB mixture. Collectively, analyses of tr-NOESY spectra of TL+TB strongly suggested that in the presence of TL, the TB peptide predominantly assumed monomeric helical conformations in LPS micelles. The disappearance of long-range NOE contacts among residues of TB in the TL+TB samples demarcated that oligomeric states detected in LPS micelles were mostly destabilized in the presence of TL peptide.
Three-dimensional Structure of TL in 0111:B4 LPS Micelles
An ensemble of dimeric structures of TL in complex with LPS micelles was determined based on 156 medium-range and 22 long-range tr-NOE-driven distance constraints (Tables 1 and 2). The correspondence between NOEs utilized for structure determination and that expected from the three-dimensional structure of TL was validated using contact plots (supplemental Fig. S10). Fig. 3A shows a superposition of all backbone atoms (Cα, N, and C′) of the 20 lowest energy structures of TL in LPS micelles. As can be seen, TL assumed an anti-parallel helical structure in LPS micelles (Fig. 3B). The individual helical subunit, spanning Gln3 to Ile12, is amphipathic, whereby the basic and polar residues, e.g. Gln3, Ser6, Lys7, and Arg11, occupy one face of the helix (Fig. 3B). The non-polar and aromatic residues are positioned at the other face of the helical structure (Fig. 3B). The dimeric structure of TL could be sustained by packing interactions among residues at the non-polar faces of two helical subunits (Fig. 3, panels B and C). Intimate packing interactions were found between the aromatic side chain of Trp4 and the aliphatic side chain of Ile12 (Fig. 3B). In addition, at the center of the dimer, residues Phe5, Leu9, and Phe8 from two helical subunits had mutual contacts (Fig. 3C). At the outer surface of the dimeric structure of TL, residues Lys7/Ser6 and Gln3 may be engaged in potential hydrogen bond interactions (Fig. 3B). The electrostatic surface of the dimeric structure of TL was characterized by a predominantly positively charged region at one face of the molecule, whereas the opposite face was rich in non-polar residues (Fig. 3D). The dimerization of TL in LPS micelles delineated a unique amphipathic topology. All of the hydrophobic and aromatic side chains were positioned toward one surface, whereas four basic residues of the dimer were aligned to the opposite face, resembling a “basket” shape (Fig. 3E). The dimeric structure of TL may lie parallel to the micelle surface of LPS, whereby the two positively charged side chains, Lys7 and Arg11, of each monomer could simultaneously interact with the negatively charged phosphate groups of the lipid A moiety of LPS (Fig. 3E). The hydrophobic and aromatic side chains of the dimer could have favorable packing interactions with the acyl chains of LPS (Fig. 3E).
TABLE 1.
Inter-helical NOEs used to determine dimeric structure of TL in LPS micelles
| Inter-helical NOEs |
|---|
| Leu13CαH—Val2HN |
| Leu9CαH—Phe5HN |
| Leu9CβHs—Phe5HN |
| Leu9CδHs—Phe5HN |
| Leu9CδHs—Trp4H4 |
| Leu9CδHs—Trp4H7 |
| Ile12CγHs—Trp4H5 |
| Ile12CδHs—Trp4H5 |
| Ile12CβHs—Trp4H5 |
| Val2CαH—Leu13CαH |
| Phe5CαH—Leu9CαH |
TABLE 2.
Summary of structural statistics for the 20 lowest energy structures of dimeric TL and monomeric TB (in presence of TL) in LPS micelles
| TL | TB | |
|---|---|---|
| Distance restraints | ||
| Intraresidue (i − j = 0) | 56 | 38 |
| Sequential (|i − j| = 1) | 110 | 53 |
| Medium-range (2 ≤ |i − j| ≤ 4) | 156 | 50 |
| Inter-helical NOE | 22 | 0 |
| Total NOE constraints | 344 | 141 |
| Dihedral angle constraints | ||
| φ | 22 | 10 |
| Constraint violations | ||
| Minimum NOE violation (Å) | 0.12 | 0.17 |
| Maximum NOE violation (Å) | 0.34 | 0.34 |
| Minimum dihedral angle violation (°) | 7 | 2 |
| Maximum dihedral angle violation (°) | 14 | 3 |
| Deviation from mean structure | ||
| Backbone atoms (N, Cα, C′) (Å) | 0.46 ± 0.12 | 0.79 ± 0.5 |
| Heavy atoms (Å) | 1.27 ± 0.23 | 1.37 ± 0.6 |
| Ramachandran plot Analysisa | ||
| % Residues in the most favorable region | 80 | 96.7 |
| % Residues additionally allowed region | 20 | 3.3 |
| % Residues in the generously allowed region | 0 | 0 |
| % Residues in the disallowed region | 0 | 0 |
a based on Procheck.
FIGURE 3.

Structure of TL in LPS micelles. Panel A, superposition of backbone atoms (N, Cα, C′) of the 20 lowest energy conformers of dimeric TL in the context of LPS micelles. Panel B, a representative structure of the anti-parallel dimer of TL showing side chain orientation and backbone topology. Panel C, a space-filling representation of the side chain-side chain contacts between two helical monomers of TL involving aromatic residues Trp4, Phe5, Phe8, and aliphatic residue Leu9. Panel D, electrostatic potential surface of the dimeric structure of TL in LPS micelles showing the distribution of charge and non-polar residues. Panel E, disposition of side chains of aromatic and aliphatic amino acid residues and cationic and polar amino acid residues of the dimeric structure of TL. Backbone and side chain atoms of the dimer are represented as a stick.
Self-association of TL in 0111:B4 LPS Micelles by Fluorescence Resonance Energy Transfer
The specificity of self-association into dimerization of TL peptide in LPS micelles was further probed using FRET experiments. In these assays, an N-terminal dansylated TL was used as an acceptor fluorophore. Otherwise, residue Trp4 of native or unlabeled TL acted as a donor moiety. An energy transfer from Trp of unlabeled TL to the dansyl group of the labeled TL may occur, whereas peptides are located within 30 Å. This would result in quenching of the fluorescence intensity of the donor fluorophore (52). Fig. 4 shows Trp emission spectra of the unlabeled TL in the presence of various concentrations of dansylated TL. Note that fluorescence emission of Trp from the dansylated TL was highly quenched because of the proximity of Trp4 to the dansyl group. Therefore, the dansylated peptide did not affect the emission intensity of Trp fluorescence of the unlabeled temporin (Fig. 4). The Trp fluorescence of TL underwent a marked quenching with the addition of increasing concentrations of dansylated TL, indicating FRET between the unlabeled TL and the dansylated TL, in the context of LPS micelles (Fig. 4). A linear relationship was found between the quenching of Trp fluorescence and the acceptor peptide concentrations (Fig. 4, inset). Such a linear decrease in intensity of donor fluorescence upon titrations with acceptor peptides has been considered a signature of dimerization of peptides in lipid micelles (51, 71). The maximum efficiency of FRET can be estimated by extrapolating the straight line, enabling determination of an approximate inter-chromophore distance (51). Here, an extrapolation of the line yielded a value of 0.78 ± 0.02 for maximal quenching (Fig. 4, inset). The efficiency of FRET thus translated into a distance of 28.5 Å between Trp and the danysl groups of unlabeled and labeled TL peptides, respectively (see “Experimental Procedures”). The estimated distance from FRET analysis was in line with an anti-parallel arrangement of the dimeric structure of TL. Note that the NMR-derived structure indicated a distance of 22.5 Å between the nitrogen atom of the NϵH group Trp of one helix and the nitrogen atom of the NH2 of the N terminus of the other helix of the dimer. However, the slightly higher distance obtained from FRET experiments may be attributed to motional freedom of the N terminus region of the dimeric structure of TL. Such deviations of distances estimated from FRET and atomic resolution structures had been noted before and were thought to occur because of the random orientation of the chromophores (51). Nevertheless, FRET studies have supported the notion that in LPS micelles TL peptide self-associates into a dimeric state, orientated in an anti-parallel manner.
FIGURE 4.

Dimerization of TL in LPS micelles by FRET. Changes in the intensity of the emission spectra of Trp fluorescence of unlabeled or native TL upon the addition of dansylated TL as a result of fluorescence resonance energy transfer from the donor Trp of unlabeled TL to the acceptor dansyl group of TL. The intrinsic Trp fluorescence of the dansylated TL was highly quenched because of the proximity between the Trp and dansyl group. Inset, the quenching of Trp fluorescence of TL was plotted as a function of the mole fraction of dansylated TL peptide, showing a linear relationship. All of the FRET studies were carried out in 10 mm sodium phosphate buffer, pH 6.8.
Structure of TB in 0111:B4 LPS Micelles
Conformational heterogeneity of independent TB peptide, as shown in tr-NOESY spectra of LPS micelles, precluded determination of three-dimensional structures. Structure determination, using the long-range NOEs, into a plausible dimer or higher-order oligomers generated a number of discrete conformations. Therefore, at present, the structural nature of the oligomeric states of TB in LPS micelles remains elusive.
Nevertheless, we have determined the structure of TB using tr-NOE-driven distance constraints obtained from tr-NOESY spectra of TL+TB (Table 2 and supplemental Fig. S10). Fig. 5A shows the superposition of all backbone atoms (Cα, N, and C′) of the 20 lowest energy structures of TB in LPS micelles. In the presence of TL, TB adopted a monomeric helical conformation in LPS micelles (Fig. 5B). The first two residues, Leu1 and Leu2, at the N terminus and the last residue, Leu13, at the C terminus, were in extended conformations (Fig. 5B). One face of the helical structure of TB was characterized by the presence of non-polar residues including Leu1, Ile4, Val5, Leu8, and Leu12 (Fig. 5B). The single cationic residue, Lys10, and the two polar residues, Asn7 and Ser11 of TB, were situated along the opposite face of the helical structure (Fig. 5B). The proximity of the side chains of Asn7 and Ser11 may indicate the possibility of hydrogen bonds at the polar surface (Fig. 5C). Several NOE contacts were indeed observed between Asn7NγH2 and Ser11CβHs, suggesting probable side chain-side chain interactions (Fig. 2D). The electrostatic surface potential of the helical structure of TB delineated a large non-polar face and a limited polar or basic region (Fig. 5D).
FIGURE 5.

LPS-bound helical structure of TB in the presence of TL. Panel A, superposition of backbone atoms (N, Cα, C′) of the 20 lowest energy structures of TB derived from an equimolar mixture of TL and TB in the context of LPS micelles. Panel B, ribbon representation of a selected structure of TB depicting the orientation of all the side chains. Panel C, plausible side chain-side chain interactions, mediated by hydrogen bond formation, between residue Asn7 and Ser11 in the helical structure of TB. Panel D, electrostatic potential surface of the helical structure of TB in LPS micelles showing a large hydrophobic face and a limited polar and positively charged region.
Interactions of TL, TB, and TL+TB with 0111:B4 LPS Micelles by STD NMR
Interactions of TL and TB with LPS were investigated by performing one-dimensional STD and two-dimensional STD-TOCSY experiments (Fig. 6 and supplemental Fig. S11). Due to better resonance separation, two-dimensional STD-TOCSY spectra were used to map LPS interacting residues of TL and TB. Fig. 6A shows STD-TOCSY spectrum of TL in LPS micelles, correlating backbone CαH resonances and up-field shifted aliphatic side chain proton resonances. As demonstrated in previous works, resonances displaying cross-peaks in the STD-TOCSY spectra were believed to achieve a high degree of saturation from the binding macromolecules because of their close contacts (34, 58, 72). The STD-TOCSY spectra of TL delineated a number of backbone-side chain and side chain-side chain cross-peaks (Fig. 6A) in parallel with the off-resonance (or reference) spectra (supplemental Fig. S11). More precisely, the polar and basic residues, e.g. Gln3, Ser6, Gly10, Lys7, and Arg11 displayed strong STD-TOCSY cross-peaks, indicating their proximity with LPS micelles (Fig. 6A). In addition, the STD effect could be seen for several aromatic and non-polar residues, namely, Phe5, Phe8, Val2, Ile12, and Leu13 (Fig. 6A). One-dimensional STD spectra displayed the ring protons of aromatic residues, namely, Phe1, Phe5, Phe8, and Trp4 of TL, which were found to be experiencing strong STD effects (supplemental Fig. S11). However, as a result of spectral overlaps among the ring proton resonances of aromatic residues, unambiguous estimation of STD effects was difficult except for ring proton resonances of C4H, C7H, and C5H of Trp4 typified by distinctly different chemical shifts (supplemental Fig. S11).
FIGURE 6.

Mapping contact residues of TL, TB, and TL+TB with LPS micelles by STD-NMR. Panel A, STD-TOCSY spectrum of TL, and panel B, STD-TOCSY spectrum of TB in the presence of LPS in D2O at pH 4.5, 298 K, showing correlations among aliphatic proton resonances. Cross-peak connectivities of residues exhibiting a strong STD effect are indicated by broken lines. Panel C, comparison among one-dimensional STD spectra of TL, TB, and TL+TB together for the down-field shifted aromatic resonances of TL. A higher STD effect from LPS micelles can be seen for the aromatic ring proton resonances of TL in the presence of TB. Panel D, an overlay of STD-TOCSY spectra, of a selected section, of TB (solid contour) and TL+TB (dotted contour) indicating a higher STD effect for the side chain resonances of the Pro3 residue of TB in the presence of TL. The STD-TOCSY spectra were acquired using a spin-lock MLEV17 sequence with a mixing time of 80 ms. Saturation of LPS was achieved by applying a cascade of 40 Gaussian pulses (50 ms each), resulting in a total saturation of ∼2 s.
By contrast to STD-TOCSY spectra of TL, the STD-TOCSY spectra of TB was characterized by a limited number of correlations among resonances, indicating that not all of the residues of TB were in close contact with LPS micelles (Fig. 6B). Notably, among the aliphatic amino acids, STD-TOCSY spectra exhibited correlations from CαH resonances to other side chain proton resonances, only for residues Leu1, Leu2, and Leu9. Much weaker cross-peaks, CαH/CβH, were visible for residues Ile4 and Val5 of TB (Fig. 6B). There were also fewer STD-TOCSY correlations at the up-field-shifted region, 0.8–1.6 ppm, among aliphatic side chain resonances (Fig. 6B). However, strong cross-peaks, from backbone CαH to the side chain proton resonances, in STD-TOCSY spectra were observed for residues Pro3, Asn7, Lys10, and Ser11 (Fig. 6B). Markedly, the single basic residue Lys10 of TB displayed intense cross-peaks correlating side chain resonances, CβHs/CγHs/CδHs/CϵHs, including a long-range correlation from the CαH resonance to CϵHs resonances (Fig. 6B). These data clearly suggested that polar residues Asn7, Ser11, and the positively charged Lys10 of TB were in proximity to LPS micelles. Among the non-polar amino acids of TB, only the terminal residues, e.g. Leu1, Leu2, and Pro3, provided significant STD effects with LPS. It is therefore highly likely that self-associations of independent TB might be responsible for the restricted interactions between the numerous non-polar residues and LPS micelles.
STD spectra were also acquired for TL+TB in the presence of LPS micelles. Remarkably, enhanced STD effects from LPS micelles could be seen for both TL and TB peptides (Fig. 6, C and D). Clearly, aromatic ring proton resonances of TL, in the presence of TB (TL+TB), reflected an overall increase in STD effects in comparison with that of TL alone (Fig. 6C). The CδHs of residue Pro3 of TB, exhibiting non-overlapping resonances, displayed additional STD-TOCSY cross-peaks with its CβHs (Fig. 6D). Enhanced STD effects were also detected for the up-field shifted aliphatic resonances belonging to TB and TL in the TL+TB mixture (supplemental Fig. S11). Taken together, STD analyses revealed that several residues of independent TL, including polar/basic, non-polar, and aromatic, were in intimate contact with LPS micelles. By contrast, a limited surface, predominantly formed by the polar and the single basic residue of TB, was involved in interactions with LPS micelles, whereas TL and TB together appeared to possess an increased proximity with LPS micelles.
Proximity of TL and TB in 0111:B4 LPS Micelles by FRET
We were unable to detect any NOE interactions between TL and TB in the tr-NOESY spectra obtained for TL+TB samples in LPS micelles. This may suggest that TL and TB do not achieve proximity close to 5 or 6 Å for NOE to occur. To gain insights into the proximal localization of TL and TB in LPS micelles and understand the underlying synergistic mechanisms, we utilized FRET experiments between Trp (as a donor) of TL and the N-terminal dansyl group (as an acceptor) of TB (Fig. 8). Additions of dansylated TB into solutions containing TL and LPS micelles gave rise to a decrease in the fluorescence emission Trp intensity of TL peptide (Fig. 8). This quenching of fluorescence intensity could be interpreted as energy transfer between TL and TB peptides, indicating their proximity in LPS micelles (Fig. 8). Note that the quenching plot (supplemental Fig. S12) showed deviations from linearity, in contrast to the results of FRET studies with TL (Fig. 4, inset), perhaps due to the presence of the dimeric TL and the monomeric TB in LPS micelles. Nevertheless, extrapolation of the quenching plot to the ordinate yielded a maximal quenching value of 0.48 ± 0.05 (supplemental Fig. S12). Therefore, a distance of 24 Å was estimated between the dansyl group of TB and the Trp of TL (see “Experimental Procedures”). Control experiments were carried out further, including unlabeled TB peptide into solutions containing TL and LPS micelles. Notably, neither the Trp emission maxima (λmax) nor its emission intensity showed any changes upon addition of native TB in the presence of LPS micelles (supplemental Fig. S12). These fluorescence results lend credence to the fact that there are presumably no non-covalently packed heterocomplex formations between TL and TB in LPS micelles, as judged by the lack of changes in intrinsic Trp fluorescence of TL (supplemental Fig. S12). The observed FRET data between TL and TB indicate that TL and TB can simultaneously bind to LPS micelles, presumably at different but closely located regions whereby two peptides are separated by ∼24 Å distance.
FIGURE 8.

Perturbation of LPS by TL, TB, and TL+TB. Panel A, enhancement of FITC fluorescence of FITC-LPS micelles upon the additions of TL, TB, and equimolar mixtures of TL and TB peptides. Fluorescence experiments were carried out in 10 mm sodium phosphate buffer, pH 6.8. Panel B, 31P NMR spectra of Triton X-100 dissolved LPS in the absence (black) and presence of TL (green), TB (red), and an equimolar mixture of TL and TB peptides (blue). (Inset), an expansion of the 31P NMR spectra of LPS highlighting changes in the up-field resonances belonging to the diphosphate groups of LPS in the presence of TL, TB, and TL and TB together.
Effect of TL, TB, and TL+TB on 0111:B4 and 055:B5 LPS Structure
We examined plausible structural changes of E. coli 055:B5 LPS micelles upon binding to TL, TB, and the TL+TB combination. FITC is a sensitive probe of the aggregation states of FITC-conjugated molecules (73). FITC fluorescence is quenched in FITC-conjugated LPS or FITC-LPS as a result of self-association of LPS molecules. Structural perturbation of LPS micelles may alleviate quenching of FITC fluorescence (34, 67, 73). As can be seen, TL and TB, independently, induced enhancement of FITC fluorescence of FITC-LPS in a concentration-dependent fashion, indicating perturbation of LPS micelles (Fig. 8A). TL peptide caused a slightly higher increase in the fluorescence intensity of FITC with respect to TB (Fig. 8A). In contrast, a significantly higher enhancement of FITC fluorescence was noted upon addition of TL and TB together (Fig. 8A). Thus, LPS micelles experienced greater structural disorganization in the presence of both TL and TB. Similarly, disaggregation of LPS micelles from E. coli 0111:B4 and E. coli 026:B6 by temporins TL, TB, and TA were investigated from FITC fluorescence experiments (47). Akin to the current observation, combining temporin peptides (TL + TB and TL + TA) caused greater perturbation of these LPS micelles; however, the TL peptide induced fewer effects in comparison with TB and TA (47). In our study, independent TL was found to dissociate LPS micelles with a slightly higher efficacy as compared with TB (Fig. 8A). This disparity may arise from the differences in chemical structures, in particular, LPS from E. coli 055:B5 has a longer polysaccharide chain, among these LPS systems.
Furthermore, we used 31P NMR to detect peptide-induced structural changes in the head group region of LPS. Fig. 8B shows an overlay of 31P NMR spectra of LPS in the presence of TL, TB, and TL+TB combined. In these experiments, LPS was dissolved in 100 mm Triton X-100, a non-ionic detergent solution to resolve 31P resonances belonging to monophosphate and diphosphate groups (74, 75). The down-field shifted 31P resonances observed at around −1.00 to −2.00 ppm correspond to the monophosphate groups of LPS (Fig. 8B) (74, 75). The up-field shifted 31P resonances were attributed to diphosphate or pyrophosphate groups of LPS (Fig. 8B) (74, 75). In the presence of TL peptide there were appreciable chemical shift changes of 31P resonances, particularly for the diphosphate groups of LPS (Fig. 8B, inset). The 31P resonances of diphosphate groups of LPS, at −9 to −10 ppm, showed an up-field shift in the presence of TL peptide (Fig. 8B, inset). The monophosphate resonances of LPS, at −1 to −2 ppm, appeared to be experiencing a lower degree of chemical shift changes (Fig. 8B). By contrast, inclusion of TB peptide did not cause any significant chemical shift changes in 31P resonances of LPS (Fig. 8B). These observations indicated that TL and TB peptides may be interacting with different regions of LPS. The lipid A and the inner sugar core of LPS contain many of the diphosphate groups; whereas the outer core of LPS is chiefly made of sugar residues (see Fig. 1A). Thus, TL might bind to the phosphate head groups of either lipid A and/or inner core of LPS. TB may preferentially interact with the outer core of LPS.
The 31P NMR spectra of LPS obtained in the presence of TL and TB revealed differences in comparison to those of the individual peptides (Fig. 8B and inset). Notably, the 31P resonances of the diphosphate groups of LPS, at −9 to −10 ppm, also experienced an up-field shift in the presence of TL and TB combined (Fig. 8B, inset). However, the extent of up-field shift was not the same as that observed with TL peptide alone (Fig. 8B, inset). The distinctly different chemical shifts for the diphosphate groups of LPS in the context TL and TB may be interpreted as disparate conformational states of LPS. In addition, the combined peptides TL and TB, gave a reduction in intensity of 31P signals due to a significant broadening of resonances (Fig. 8B and inset). Such broadening of resonances has usually been attributed to the chemical or conformational exchange processes occurring at the millisecond to microsecond time scale. DLS and FITC-LPS fluorescence experiments have indicated that temporin peptides TL, TB, and TL+TB would disaggregate LPS micelles, precluding any peptide-induced aggregation of LPS, thus leading to broadening of 31P NMR spectra. Taken together, the FITC and 31P NMR studies have suggested conformational changes of LPS upon binding to temporin peptides. Furthermore, the combination of TL and TB appeared to determine a different perturbation of LPS structure in comparison with that caused by each single temporin.
DISCUSSION
The LPS outer membrane of Gram-negative bacteria has been found to play important roles in the antibacterial activities of temporins (47). Several temporins including TA and TB (46, 47) exhibit very low activities against Gram-negative bacteria. Recent works of Mangoni and co-workers (48, 49) described the molecular basis for the limited or high activity of TA/TB or TL, respectively. In the presence of LPS micelles, TA and TB undergo self-association; by contrast, a limited self-association was detected for TL peptide (48, 49). Furthermore, these studies found a synergistic mechanism between TL and TA or TB in overcoming the LPS-outer membrane barrier (47–49). Specifically, in the presence of TL, the LPS-mediated self-association of TB was significantly abolished. Here, we have determined atomic resolution structures, interactions, and localization of TL, TB, and TL and TB in the presence of E. coli 0111:B4 LPS micelles. The LPS-bound structure of TL was defined by two antiparallel helices formed by close packing, as a central core, among aromatic residues and non-polar aliphatic residues (Fig. 3). The dimeric structure is amphipathic in nature, displaying a non-polar surface and a polar/positively charged region (Fig. 3D). The LPS-bound structure of TL provides molecular insights into the outer membrane permeabilization and endotoxin neutralization of this peptide. In the basket-shaped amphipathic dimeric structure of TL, the side chain of Arg11 from each helical subunit may form ionic and/or hydrogen bonding interactions with the bisphosphate groups of lipid A (Fig. 3E). The charged guanidinium groups of the two Arg residues (Arg11 from each monomer) of the dimeric structure of TL are separated by ∼12 to 13 Å, which is geometrically compatible with the inter-phosphate distance of the lipid A moiety (Fig. 3E). Two Lys residues from the helices of TL may further augment the LPS-peptide ionic interactions (Fig. 3E). STD studies had established intimate contacts between Lys7 and Arg11 with LPS micelles (Fig. 6A). Furthermore, 31P NMR experiments suggested plausible binding of TL with the diphosphate groups of lipid A or inner sugar core of LPS (Fig. 8B). From the non-polar surface of TL, aromatic side chains of Phe(s) and Trp4 could be embedded in the LPS micelles, along with the alkyl side chains of the terminus amino acids, e.g. Val2, Ile12, and Leu13 (Figs. 3E and 6A). The plausible disaggregation of LPS micelles, as suggested from FITC-LPS (Fig. 8A) and DLS experiments (supplemental Fig. S1) here and in previous DLS studies (50) could be correlated to the antiendotoxic activities of TL peptide. Noteworthy, the dimeric structure of TL, deduced here, in complex with LPS micelles, is a novel finding for an active AMP. Indeed, LPS-bound structures of AMPs determined so far have either revealed monomeric helical conformations (57) or helix-loop-helix structures (33, 34). It is also noteworthy that helical structures of TL have been determined in SDS and DPC micelles (76). However, fluorescence and CD studies in zwitterionic lipid vesicles have proposed plausible oligomerization of TL, which might be responsible for its toxic activity in eukaryotic cells (77, 78).
Our results indicated that TB undergoes oligomerization in complex with LPS micelles. The oligomerization is predominantly mediated by contacts among hydrophobic residues located at the N and C termini of the molecule (Fig. 2C). However, the precise structural characteristics of these oligomeric states of TB in LPS could not be ascertained because of heterogeneity. Regardless, independent TB was able to impart disaggregation of LPS micelles (Fig. 8A and supplemental Fig. S1). Furthermore, STD experiments delineated that the polar residues of TB are primarily involved in binding with LPS micelles (Fig. 6B). FRET (Fig. 7) and 31P NMR (Fig. 8B) data had suggested that TL and TB interacted with different regions of LPS.
FIGURE 7.
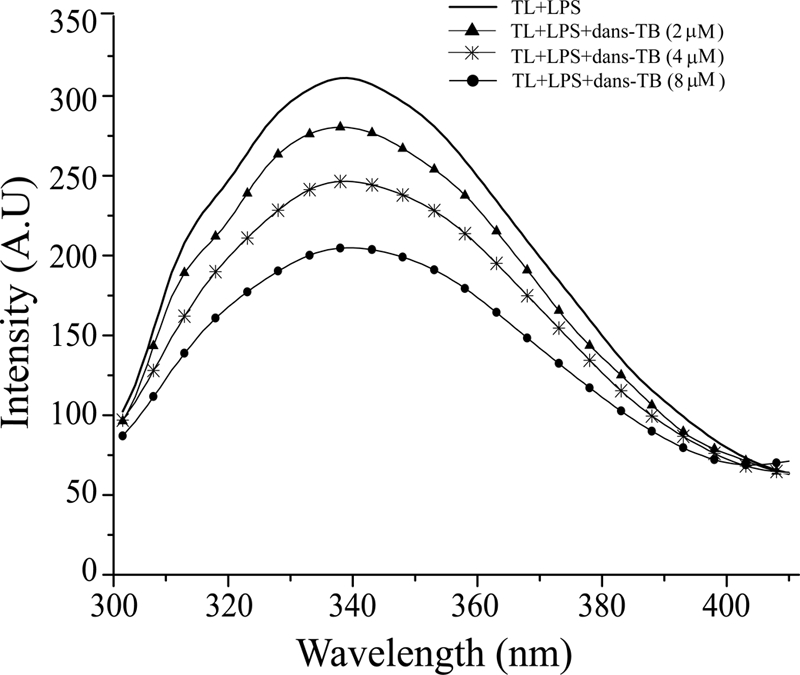
Proximity of TL and TB in LPS micelles from FRET. Changes in the intensity of the emission spectra of Trp fluorescence of TL upon the addition of dansylated TB as a result of fluorescence resonance energy transfer from the donor Trp of TL to the acceptor dansyl group of TB.
The present study elucidates important molecular insights into the mechanisms underlying the synergistic activity of temporins. In the presence of TL, oligomerization of TB in LPS is largely destabilized. However, the monomeric helical conformation of TB in LPS micelles is sustained by TL (Fig. 5). The dimeric helical structure of TL in LPS micelles is preserved in the presence of TB peptide. STD studies have established that when present together, TL and TB make enhanced contacts with LPS micelles (Fig. 6, C and D) and are able to generate larger perturbation to LPS micelles (Fig. 8A). The synergistic interplay of TL and TB may result from the structures and proximity of the peptides in the environment of LPS micelles and from changes in the structural states of LPS. As stated above, the key event in the antimicrobial synergism of TL+TB is the inhibited aggregation of TB in LPS in the presence of TL. This phenomenon may be mediated by structural changes of LPS, upon binding of the dimeric TL, thus preventing oligomerization of TB. The higher degree of structural changes and larger contacts of the combination TL and TB with LPS micelles would efficiently disrupt the LPS outer membrane, followed by concomitant translocation of both peptides en route to the inner membrane.
Supplementary Material
This work was supported by Grants 06/1/22/19/446 and 08/1/22/19/556 from A*Star Biomedical Research Council, Singapore.
The atomic coordinates and NMR constraints of TL and TB have been deposited to Biological Magnetic Resonance Bank data bank under the accession codes 21008 and 21005, respectively.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Figs. S1–S12.
- AMPs
- antimicrobial peptides
- tr-NOE
- transferred nuclear Overhauser effect
- NOESY
- nuclear Overhauser effect spectroscopy
- TOCSY
- total correlation spectroscopy
- NOE
- nuclear Overhauser enhancement
- STD
- saturation transfer difference
- TA
- temporin-1Ta
- TB
- temporin-1Tb
- TL
- temporin-1Tl
- DPH
- 1,6-diphenyl-1,3,5-hexatriene
- dansyl
- 5-dimethylaminonaphthalene-1-sulfonyl.
REFERENCES
- 1. Ganz T. (2003) Nat. Rev. Immunol. 3, 710–720 [DOI] [PubMed] [Google Scholar]
- 2. Zasloff M. (2002) Nature 415, 389–395 [DOI] [PubMed] [Google Scholar]
- 3. Brogden K. A. (2005) Nat. Rev. Microbiol. 3, 238–250 [DOI] [PubMed] [Google Scholar]
- 4. Hancock R. E., Scott M. G. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 8856–8861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boman H. G. (1995) Annu. Rev. Immunol. 13, 61–92 [DOI] [PubMed] [Google Scholar]
- 6. Brown K. L., Hancock R. E. (2006) Curr. Opin. Immunol. 18, 24–30 [DOI] [PubMed] [Google Scholar]
- 7. Epand R. M., Vogel H. J. (1999) Biochim. Biophys. Acta 1462, 11–28 [DOI] [PubMed] [Google Scholar]
- 8. Shai Y. (2002) Biopolymers 66, 236–248 [DOI] [PubMed] [Google Scholar]
- 9. Yeaman M. R., Yount N. Y. (2003) Pharmacol. Rev. 55, 27–55 [DOI] [PubMed] [Google Scholar]
- 10. Bulet P., Stöcklin R., Menin L. (2004) Immunol. Rev. 198, 169–184 [DOI] [PubMed] [Google Scholar]
- 11. Weinstein R. A. (2001) Emerg. Infect. Dis. 7, 188–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gottler L. M., Ramamoorthy A. (2009) Biochim. Biophys. Acta 1788, 1680–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Egal M., Conrad M., MacDonald D. L., Maloy W. L., Motley M., Genco C. A. (1999) Int. J. Antimicrob. Agents 13, 57–60 [DOI] [PubMed] [Google Scholar]
- 14. Bhattacharjya S., Ramamoorthy A. (2009) FEBS J. 276, 6465–6473 [DOI] [PubMed] [Google Scholar]
- 15. Mor A. (2009) FEBS J. 276, 6474–6482 [DOI] [PubMed] [Google Scholar]
- 16. Hancock R. E., Brown K. L., Mookherjee N. (2006) Immunobiology 211, 315–322 [DOI] [PubMed] [Google Scholar]
- 17. Haney E. F., Hunter H. N., Matsuzaki K., Vogel H. J. (2009) Biochim. Biophys. Acta 1788, 1639–1655 [DOI] [PubMed] [Google Scholar]
- 18. Shai Y. (1999) Biochim. Biophys. Acta 1462, 55–70 [DOI] [PubMed] [Google Scholar]
- 19. Bechinger B. (1999) Biochim. Biophys. Acta 1462, 157–183 [DOI] [PubMed] [Google Scholar]
- 20. Taubes G. (2008) Science 321, 356–361 [DOI] [PubMed] [Google Scholar]
- 21. Steiner H., Hultmark D., Engström A., Bennich H., Boman H. G. (1981) Nature 292, 246–248 [PubMed] [Google Scholar]
- 22. Dathe M., Wieprecht T. (1999) Biochim. Biophys. Acta 1462, 71–87 [DOI] [PubMed] [Google Scholar]
- 23. Rosenfeld Y., Shai Y. (2006) Biochim. Biophys. Acta 1758, 1513–1522 [DOI] [PubMed] [Google Scholar]
- 24. Nikaido H. (1994) Science 264, 382–388 [DOI] [PubMed] [Google Scholar]
- 25. Nikaido H. (2003) Microbiol. Mol. Biol. Rev. 67, 593–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hancock R. E. (1984) Annu. Rev. Microbiol. 38, 237–264 [DOI] [PubMed] [Google Scholar]
- 27. Snyder D. S., McIntosh T. J. (2000) Biochemistry 39, 11777–11787 [DOI] [PubMed] [Google Scholar]
- 28. Allende D., McIntosh T. J. (2003) Biochemistry 42, 1101–1108 [DOI] [PubMed] [Google Scholar]
- 29. Delcour A. H. (2009) Biochim. Biophys. Acta 1794, 808–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Raetz C. R. (1990) Annu. Rev. Biochem. 59, 129–170 [DOI] [PubMed] [Google Scholar]
- 31. Raetz C. R., Whitfield C. (2002) Annu. Rev. Biochem. 71, 635–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tossi A., Sandri L., Giangaspero A. (2000) Biopolymers 55, 4–30 [DOI] [PubMed] [Google Scholar]
- 33. Bhunia A., Ramamoorthy A., Bhattacharjya S. (2009) Chemistry 15, 2036–2040 [DOI] [PubMed] [Google Scholar]
- 34. Bhunia A., Domadia P. N., Torres J., Hallock K. J., Ramamoorthy A., Bhattacharjya S. (2010) J. Biol. Chem. 285, 3883–3895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Japelj B., Pristovsek P., Majerle A., Jerala R. (2005) J. Biol. Chem. 280, 16955–16961 [DOI] [PubMed] [Google Scholar]
- 36. Martin G. S., Mannino D. M., Eaton S., Moss M. (2003) N. Engl. J. Med. 348, 1546–1554 [DOI] [PubMed] [Google Scholar]
- 37. Angus D. C., Wax R. S. (2001) Crit. Care Med. 29, S109–S116 [DOI] [PubMed] [Google Scholar]
- 38. Cohen J. (2002) Nature 420, 885–891 [DOI] [PubMed] [Google Scholar]
- 39. Miller S. I., Ernst R. K., Bader M. W. (2005) Nat. Rev. Microbiol. 3, 36–46 [DOI] [PubMed] [Google Scholar]
- 40. Fink P. F. (1990) in Handbook of Critical Care (Berk J. L., Sampliner J. E. eds) pp. 619, Little, Brown and Co., Boston, MA [Google Scholar]
- 41. Bhattacharjya S. (2010) Curr. Med. Chem. 17, 3080–3093 [DOI] [PubMed] [Google Scholar]
- 42. Jerala R., Porro M. (2004) Curr. Top. Med. Chem. 4, 1173–1184 [DOI] [PubMed] [Google Scholar]
- 43. Pristovsek P., Kidric J. (2004) Curr. Top. Med. Chem. 4, 1185–1201 [DOI] [PubMed] [Google Scholar]
- 44. Simmaco M., Mignogna G., Canofeni S., Miele R., Mangoni M. L., Barra D. (1996) Eur. J. Biochem. 242, 788–792 [DOI] [PubMed] [Google Scholar]
- 45. Mangoni M. L., Marcellini H. G., Simmaco M. (2007) J. Pept. Sci. 13, 603–613 [DOI] [PubMed] [Google Scholar]
- 46. Mangoni M. L. (2006) Cell Mol. Life Sci. 63, 1060–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mangoni M. L., Shai Y. (2009) Biochim. Biophys. Acta 1788, 1610–1619 [DOI] [PubMed] [Google Scholar]
- 48. Rosenfeld Y., Barra D., Simmaco M., Shai Y., Mangoni M. L. (2006) J. Biol. Chem. 281, 28565–28574 [DOI] [PubMed] [Google Scholar]
- 49. Mangoni M. L., Epand R. F., Rosenfeld Y., Peleg A., Barra D., Epand R. M., Shai Y. (2008) J. Biol. Chem. 283, 22907–22917 [DOI] [PubMed] [Google Scholar]
- 50. Giacometti A., Cirioni O., Ghiselli R., Mocchegiani F., Orlando F., Silvestri C., Bozzi A., Di Giulio A., Luzi C., Mangoni M. L., Barra D., Saba V., Scalise G., Rinaldi A. C. (2006) Antimicrob. Agents Chemother. 50, 2478–2486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Adair B. D., Engelman D. M. (1994) Biochemistry 33, 5539–5544 [DOI] [PubMed] [Google Scholar]
- 52. Lakowicz J. R. (2006) Principles of Fluorescence Spectroscopy, 3rd Ed., Springer, New York [Google Scholar]
- 53. Chattopadhyay A., London E. (1984) Anal. Biochem. 139, 408–412 [DOI] [PubMed] [Google Scholar]
- 54. Yu L., Tan M., Ho B., Ding J. L., Wohland T. (2006) Anal. Chim. Acta 556, 216–225 [DOI] [PubMed] [Google Scholar]
- 55. Mayer M., Meyer B. (1999) Angew. Chem. Int. Ed. Engl. 38, 1784–1788 [DOI] [PubMed] [Google Scholar]
- 56. Bhunia A., Domadia P. N., Bhattacharjya S. (2007) Biochim. Biophys. Acta 1768, 3282–3291 [DOI] [PubMed] [Google Scholar]
- 57. Bhunia A., Mohanram H., Bhattacharjya S. (2009) Biopolymers 92, 9–22 [DOI] [PubMed] [Google Scholar]
- 58. Bhunia A., Bhattacharjya S. (2011) Biopolymers 96, 273–287 [DOI] [PubMed] [Google Scholar]
- 59. Rademacher C., Krishna N. R., Palcic M., Parra F., Peters T. (2008) J. Am. Chem. Soc. 130, 3669–3675 [DOI] [PubMed] [Google Scholar]
- 60. Mayer M., Meyer B. (2001) J. Am. Chem. Soc. 123, 6108–6117 [DOI] [PubMed] [Google Scholar]
- 61. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Acta Crystallogr. D 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 62. Güntert P., Mumenthaler C., Wüthrich K. (1997) J. Mol. Biol. 273, 283–298 [DOI] [PubMed] [Google Scholar]
- 63. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 64. Huang Y. J., Powers R., Montelione G. T. (2005) J. Am. Chem. Soc. 127, 1665–1674 [DOI] [PubMed] [Google Scholar]
- 65. Wüthrich K. (1986) NMR of Protein and Nucleic Acids, John Wiley & Sons, New York [Google Scholar]
- 66. Bhattacharjya S., Domadia P. N., Bhunia A., Malladi S., David S. A. (2007) Biochemistry 46, 5864–5874 [DOI] [PubMed] [Google Scholar]
- 67. Bhunia A., Mohanram H., Domadia P. N., Torres J., Bhattacharjya S. (2009) J. Biol. Chem. 284, 21991–22004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Clore G. M., Gronenborn A. M. (1982) J. Magn. Reson. 48, 402–417 [Google Scholar]
- 69. Post C. B. (2003) Curr. Opin. Struct. Biol. 13, 581–588 [DOI] [PubMed] [Google Scholar]
- 70. Meyer B., Peters T. (2003) Angew. Chem. Int. Ed. Engl. 42, 864–890 [DOI] [PubMed] [Google Scholar]
- 71. Rath A., Melnyk R. A., Deber C. M. (2006) J. Biol. Chem. 281, 15546–15553 [DOI] [PubMed] [Google Scholar]
- 72. Haselhorst T., Weimar T., Peters T. (2001) J. Am. Chem. Soc. 123, 10705–10714 [DOI] [PubMed] [Google Scholar]
- 73. Tobias P. S., Soldau K., Gegner J. A., Mintz D., Ulevitch R. J. (1995) J. Biol. Chem. 270, 10482–10488 [DOI] [PubMed] [Google Scholar]
- 74. Strain S. M., Fesik S. W., Armitage I. M. (1983) J. Biol. Chem. 258, 13466–13477 [PubMed] [Google Scholar]
- 75. Wilkinson S. G. (1981) Biochem. J. 199, 833–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Carotenuto A., Malfi S., Saviello M. R., Campiglia P., Gomez-Monterrey I., Mangoni M. L., Gaddi L. M., Novellino E., Grieco P. (2008) J. Med. Chem. 51, 2354–2362 [DOI] [PubMed] [Google Scholar]
- 77. Zhao H., Kinnunen P. K. (2002) J. Biol. Chem. 277, 25170–25177 [DOI] [PubMed] [Google Scholar]
- 78. Zhao H., Rinaldi A. C., Di Giulio A., Simmaco M., Kinnunen P. K. (2002) Biochemistry 41, 4425–4436 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.