

Abstract
UDP-glucose dehydrogenase (UGDH) catalyzes the formation of UDP-glucuronate. Glucuronate represents an integral component of the glycosaminoglycan, hyaluronan, which accumulates in orbital Graves disease. Here we report that orbital fibroblasts express higher levels of UGDH than do those from skin. This is a consequence of greater UGDH gene promoter activity and more abundant steady-state UGDH mRNA. Six Sp1 sites located in the proximal 550 bp of the UGDH gene promoter appear to determine basal promoter activity, as does a previously unrecognized 49-bp sequence spanning −1436 nucleotides (nt) and −1388 nt that negatively affects activity. Nuclear Sp1 protein is more abundant in orbital fibroblasts, and its binding to specific sites on DNA is greater than that in dermal fibroblasts. Mutating each of these Sp1 sites in a UGDH gene promoter fragment, extending from −1387 to +71 nt and fused to a luciferase reporter, results in divergent activities when transfected in orbital and dermal fibroblasts. Reducing Sp1 attenuated UGDH gene promoter activity, lowered steady-state UGDH mRNA levels, and reduced UGDH enzyme activity. Targeting Sp1 and UGDH with specific siRNAs also lowered hyaluronan synthase-1 (HAS-1) and HAS-2 levels and reduced hyaluronan accumulation in orbital fibroblasts. These findings suggest that orbital fibroblasts express high levels of UGDH in an anatomic-specific manner, apparently the result of greater constitutive Sp1. These high UGDH levels may underlie susceptibility of the orbit to localized overproduction of hyaluronan in Graves disease.
Keywords: Extracellular Matrix, Fibroblast, Glycosaminoglycan, Hyaluronate, Inflammation, Promoters, Sp1, Autoimmune
Introduction
UDP-glucose dehydrogenase (UGDH,3 EC1.1.1.22) catalyzes the conversion of UDP-d-glucose to UDP-d-glucuronate (UDP-GlcUA) (1) as defined by the chemical reaction
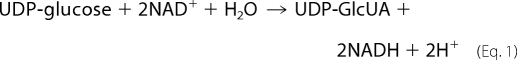 |
The Km values for human UGDH are 11.2 and 355 μm, whereas those for Vmax are 118 and 128 nmol/min/mg for UDP-glucose and NAD+, respectively (2), in agreement with those for the bovine enzyme (3, 4). Many of this enzyme's functional residues have been identified. Sommer et al. (2) reported that Cys-276 is a catalytic residue essential for the second run oxidation performed by UGDH, whereas Lys-279 acts as a positioning residue for the active site and participates in hexameric quaternary structural integrity. Efficient NAD+ binding requires Gly-13 (5). The gene structures for both mouse and human enzymes have been reported (6), and UGDH is encoded by single genes (6, 7), localizing to mouse chromosome 5 and human 4p15.1 (8). Mouse and bovine cDNAs exhibit greater than 97% sequence identity with their human counterpart (6, 9). Human UGDH comprises 12 exons and spans 26 kb. The full-length cDNA spans 2.35 kb and encodes 494 amino acids. UGDH functions as a homohexamer. The protein subunit migrates on SDS-PAGE at ∼55 kDa (10, 11), whereas the mRNA resolves on Northern blot analysis as a doublet of ∼2.4 and 2.7 kb (12, 13). The use of alternate transcription start sites results in the generation of these two transcripts (14). Vatsyayan et al. (14) and Bontemps et al. (15) reported cloning the UGDH gene promoter and began to characterize its regulation resulting from specific Sp1 binding. Basal UGDH promoter activity in HepG2 and HeLa cells requires Sp1 binding to a GC-rich region located at −548 nt of this promoter (14). Furthermore, Sp1 binding sites within the first 500 bp of the UGDH promoter are responsible for the up- and down-regulation of this gene caused by TGF-β and hypoxia, respectively (15). The absence of either TATA or CAAT sequences in this promoter suggests the potential for Sp1 serving as a particularly important determinant of promoter activity and in defining the transcriptional start site (16).
Little is currently known about the anatomic distribution of UGDH expression, yet the pattern of GlcUA utilization within human connective tissues could underlie important aspects of macromolecular biogenesis. UGDH activity produces UDP-GlcUA, which in turn serves as a substrate for multiple, abundant glucuronosyltransferases. These catalyze the glucuronidation of xeno- and endobiotics, opioids, steroid hormones, and heme proteins. GlcUA constitutes an integral component of several glycosaminoglycans, including hyaluronan, chondroitin, heparan, and heparin sulfates. These molecules play important roles in mammalian development (17–19). UGDH activity increases as cells rapidly proliferate in log phase and decreases as they become quiescent (20).
Graves disease (GD) represents an autoimmune disease where increased localized glycosaminoglycan content plays an important role in tissue dysfunction (21, 22). Thyroid-associated ophthalmopathy (TAO) is a manifestation of GD where orbital tissue accumulates hyaluronidase-sensitive glycosaminoglycan and becomes infiltrated by mast cells, and T and B lymphocytes (21–23). The cytokines they generate activate orbital fibroblasts to produce excessive hyaluronan, which leads to tissue expansion and proptosis (24). Orbital fibroblasts exhibit a peculiar phenotype, especially when treated with cytokines (25). When activated by IL-1β, they express all three hyaluronan synthase (HAS) isozymes (26), UGDH mRNA is up-regulated (6), and substantially more hyaluronan is produced than in extra-orbital fibroblasts (25–28). The expression of UGDH and the production of GlcUA in the orbit have yet to be explored, yet glycosaminoglycan accumulation within the orbit in TAO could reflect an increased availability of UDP-GlcUA.
In the present study we report that UGDH expression and activity in orbital fibroblasts are substantially greater than those found in dermal fibroblasts and appear related to higher levels of UGDH gene promoter activity. Furthermore, we find that levels of nuclear Sp1 protein and binding to 6 Sp1 recognition sites in the gene promoter are considerably greater in orbital fibroblasts. Knocking down Sp1 and UGDH expression diminishes levels of HAS-1 and HAS-2 and reduces cell layer-associated hyaluronan in orbital fibroblasts. Thus, this enzyme may underlie, at least in part, the peculiar phenotype of orbital fibroblasts that renders the orbit susceptible to TAO.
EXPERIMENTAL PROCEDURES
Materials
Dr. Andrew Spicer (Texas A&M University, Houston, TX) kindly provided a full-length human cDNA encoding UGDH. An affinity-purified polyclonal antibody against UGDH was custom-generated by Zymed Laboratories Inc. (South San Francisco, CA) using the peptide sequence SGEIPKFSLQDPPN. siRNA for UGDH was custom synthesized by Qiagen (Hilden, Germany) according to its cDNA sequence, whereas siRNA targeting Sp1 was from Applied Bioscience (Carlsbad, CA, catalog #AM16706). 5,6-Dichlorobenzimid-azole (catalog #D1916), dexamethasone (catalog #D4902), cycloheximide (catalog #01810), UDP-glucose (catalog #U4625), and NAD+ (catalog #N8285) were purchased from Sigma.
Cell Culture
Orbital fibroblast cultures were initiated from tissue explants obtained from the deep orbit as surgical waste during decompression surgery for severe TAO or from normal orbital tissues after obtaining informed consent. Dermal fibroblasts were from healthy volunteers or were purchased from American Type Culture Collection (Manassas, VA) as were HeLa cells. A total of nine orbital and four dermal strains were used in these studies, each from a different donor. These activities have been approved by the Institutional Review Boards of the Harbor-UCLA Medical Center, Center for Health Sciences at UCLA, and the University of Michigan Medical Center. Explants were disrupted and placed in a culture dish, and fibroblasts were allowed to outgrow. Dermal fibroblasts were initiated from skin biopsies. They were covered with Dulbecco's medium supplemented with 10% fetal bovine serum (FBS) as described previously (29). Monolayers were maintained in a humidified, 5% CO2 incubator at 37 °C. Culture strains were utilized between the 2nd and 12th passage. Fibroblast phenotype, including hyaluronan production, does not change during that interval. Cultures are essentially free of contaminating endothelial, epithelial, and smooth muscle cells (30). Medium was changed every 3–4 days. For most studies, cultures were allowed to proliferate to confluence. The exceptions were those involving immunostaining and cell transfections. These were conducted on 80% confluent monolayers. Where indicated, test compounds were added to medium containing 1% FBS and were incubated for 16 h. Otherwise, cultures remained in medium with 10% FBS throughout the study.
Western Blot Analysis
Cellular proteins were solubilized from fibroblasts in ice-cold lysis buffer containing 0.5% Nonidet P-40, 50 mm Tris-HCl (pH 8.0), and Halt protease inhibitor mixture (Pierce, catalog #87786) after various treatments as indicated. Nuclear proteins were prepared using the NE-PER extraction kit (Pierce, catalog #78833). Cell protein was quantified (Bio-Rad, catalog #500-0001), and samples were boiled in Laemmli SDS sample buffer, separated with SDS-PAGE, and transferred to PVDF membranes (Schleicher and Schuell). Primary antibodies against UGDH (Zymed or Santa Cruz Biotechnology, Santa Cruz, CA, catalog #sc-67137) and Sp1 (Abcam, Inc, MA, catalog #ab77441) were diluted 1:1000 and incubated with the membranes at room temperature for 2 h and at 4 °C overnight, respectively. Washed membranes were then incubated with a horseradish peroxidase-conjugated secondary Ab from DAKO (Carpinteria, CA, catalog #p0447) and Cell Signaling (Danvers, MA, catalog #7074). Control mouse IgG was from Santa Cruz (catalog #sc-2762). ECL reagent (Amersham Biosciences, catalog #RPN2109) was used for signal generation. Protein bands were analyzed with a densitometer and normalized against respective β-actin bands.
Immunostaining
Fibroblasts were allowed to proliferate to subconfluence on coverslips, washed with phosphate-buffered saline (PBS), and fixed with 4% paraformaldehyde in PBS. They were then permeated with 0.2% Triton X-100 in PBS at room temperature for 10 min and blocked with 10% normal goat serum. Cells were stained with anti-UGDH Ab (1:100) in blocking solution at 4 °C overnight. After extensive rinsing, they were incubated with Oregon Green 488-conjugated goat anti-rabbit antibody (1:500, Molecular Probes, Eugene, OR, catalog #O6381) for 1 h at room temperature, washed, air-dried, and mounted with Vectashield containing DAPI (Vector, Burlingame, CA, catalog #H-1200). They were visualized under an Axioskop40 microscope (Zeiss, New York, NY).
RNA Isolation and Real-time PCR
Total cellular RNA was extracted by the method of Chomczynski and Sacchi (31) using ULTRASPEC RNA isolating reagent (Biotecx, Houston, TX, catalog #BL-10050) or an RNeasy mini-kit (Qiagen, catalog #74106), and samples were treated with RNase-free DNase I in a spin column (Qiagen, catalog #79254). 2 μg RNA was reverse-transcribed in a reaction with oligo(dT) primer (Invitrogen, catalog #AM5730G) for 1 h at 37 °C using the Omniscript RT kit (Qiagen, catalog #205111). Real-time PCR was performed in an Applied Biosystems instrument using a QuantiTect SYBR Green PCR kit (Qiagen, catalog #204143). Primer sequences for amplifying UGDH cDNA were 5′-CATCCAGGTGTTTCAGAGGATGAC-3′ (forward) and 5′-GAATGCGTTCATAATCCAATTCC-3′ (reverse). Sample values were generated against a standard curve created with the same primer pair and normalized to the values of GAPDH generated from the same cDNA samples. For quantifying HAS transcripts, the following primer sets were used: HAS1 forward (5′-TGTGTATCCTGCATCAGCGGT-3′) and reverse (5′-CTGGAGGTGTACTTGGTAGCATAACC-3′); HAS2 forward (5′-GTGTTATACATGTCGAGTTTACTTCC-3′) and reverse (5′-GTCATATTGTTGTCCCTTCTTCCGC-3′); HAS3 forward (5′-GGTACCATCAAGTTCCTAGGCAGC-3′) and reverse (5′-GAGGAGAATGTTCCAGATGC-G'3′); hAPRT forward (5′-GCTGCGTGCTCATCCGAAAG-3′) and reverse (5′-CCTTAAGCGAGGTCAGCTCC-3′).
RNA Stability Assay
Cells were shifted to serum-free medium overnight before the addition of 5,6-dichlorobenzimidazole (20 μg/ml), an inhibitor of gene transcription. After treatment, RNA was harvested at the graded intervals described. UGDH mRNA levels were quantified by real-time RT-PCR.
Promoter Construct, Deletion, and Site-directed Mutation Analysis
A 1.7-kb fragment spanning −1583 to +71 nt upstream from the initiation codon was cloned using the Human Genome Walker kit (Clontech, catalog #638901) according to the manufacturer's instructions. This was denoted as Fragment 7 and was generated in nested PCR reactions using adaptor primer 1 and nested primer 2 provided in the kit and reverse primers located in the cDNA as follows: 5′-GATCGTTCGGACAGCACCTTCCTACGG-3′ and 5′-GCTCGATCTGAGCTCCCTCTCGGC-3′. The amplified fragment was sequenced and subcloned from pCR2.1-TOPO vector (Invitrogen, catalog #K455001) into a promoter-less pGL2 luciferase reporter vector (Promega, Madison, WI, catalog #E1641). A series of deletion mutations was constructed using a similar strategy; each was generated using various PCR primers from both directions according to the promoter sequence. The reverse primer common to fragments 1, 2, 3, 8, 11, and 12 was 5′-GCTCGATCTGAGCTCCCTCTC-3′. Forward primers for these deletion mutations were: Fragment 1, 5′-CAGTGTCCGCGCAGCCCTAAAG-3′; Fragment 2, 5′-CACGCACGGCACTTACATGTTG-3; Fragment 3, 5′-GTTGTGGGCGCCTGTAATCCC-3′; Fragment 8, 5′-CATAGCAAAACCCGGTCTC-3′; Fragment 11, 5′-GATCACCTGAGGTCAGGAG-3′; Fragment 12, 5′-CTGCATAGAATGCAGAAAGCC-3′. For Fragments 4, 5, and 6, reverse primer was replaced with 5′-CTTTAGGGCTGCGCGGACACTG-3′, and forward primers were those used to generate Fragments 2, 3, and 7, respectively. Fragment 9 was generated using the same reverse primer as for fragments 1, 2, 3, 8, 11, and 12 and forward primer 5′-CACTGCAACAGGGAGCCTCCTG-3′. Deletions of Fragment 3 resulted in the generation of Fragments 14 and 15 using the respective forward primers: 5′-GGGAGGCTGAGGCAGGCAAACTGTTTCATCC-3′ and 5′-GGAGGCTGAGGCAGGCACCTGATGTAATCC-3′. The corresponding reverse primers were 5′-GGATGAAACAGTTTGCCTGCCTCAGCCTCCC-3′ and 5′-GAATTACATCAGGTGCCTGCCTCAGCCTCC-3′. Fragment 16 was generated from Fragment 3 using forward 5′-GGGGGAGCTAGGAAGCGTGCGCCGAGAGGGAGC-3′ and reverse 5′-GCTCCCTCTCGGCGCACGCTTCCTAGCTCCCCC-3′.Fragment 13 was generated by using forward primer 5′-GGTTGTGGGCGGGAGCGTGAAGGAAATAGGG-3′ and reverse primer 5′-CCCTATTTCCTTCACGCTCCTCGCCCACAACC-3′. Site-directed mutagenesis of the six putative Sp1 binding sites found in Fragment 3 was conducted using the QuikChange site-directed mutagenesis kit (Stratagene, Santa Clara, CA, catalog #200518) following the manufacturer's instructions and confirmed by sequencing. These mutations are identified in Table 2. Promoter constructs were subcloned into pGL2 and transiently transfected into cells using Lipofectamine PLUS system (Invitrogen, catalog #15338100). 0.75 μg of pGL2 promoter DNA with 0.05 μg of pRL-TK vector DNA (Promega, catalog #E2241), serving as a transfection efficiency control, was mixed with PLUS reagent for 15 min at room temperature before being combined with Lipofectamine for another 15 min. The DNA-lipid mixture was added to culture medium of 80% confluent cells for 3 h at 37 °C. Medium containing 10% FBS replaced the transfection mixture overnight. Cellular material was harvested in lysis buffer provided (Promega) and stored at −80 °C. Luciferase activity was monitored with a Dual-Luciferase Reporter Assay System (Promega, catalog #E1980) and an FB12 tube luminometer (Zylux, Huntsville, AL). Values were normalized to internal controls, and each experiment was performed at least three times.
TABLE 2.
Oligonucleotide sequences containing six Sp1 sites harbored in the human UGDH gene promoter
Mutations were introduced into Fragment 3 using QuikChange site-directed mutagenesis kit (Stratagene) and fused to a reporter construct. The resulting constructs underwent confirmatory sequencing. Mutated nucleotides are italicized. Wild type sequences appear above mutated bases. Putative recognition sequences are underlined.

Electrophoresis Mobility Shift Assay (EMSA) and NoShift Assays
Nuclear protein was extracted with NE-PER nuclear and cytoplasmic extraction reagents (Pierce) using the supplier's protocol. Protein concentrations were determined with the Bradford Bio-Rad assay. Sp1 binding consensus oligonucleotide (5′-CCCTTGGTGGGGGCGGGGCCTAAGCTGCG-3′) was used in all EMSA reactions designed to characterize the protein-DNA complex. Two complementary strands of the oligonucleotide were annealed and end-labeled with [γ-32P]ATP (Amersham Biosciences, catalog #25001748) using T4 kinase (Promega, catalog # M4101). Nuclear protein (5 μg/reaction) and labeled DNA probes were incubated in gel-shift reaction buffer containing poly(dI-dC) (Promega, catalog #E3050) in a total volume of 10 μl for 30 min at room temperature. For competition experiments, a 100-fold excess of unlabeled oligonucleotides was incubated with nuclear proteins before the addition of labeled probes. Reaction complexes were loaded on 5% non-denaturing acrylamide gels and electrophoresed at 4 °C. Gels were dried and exposed to an x-ray film with double intensifier screens at −80 °C. For Sp1 supershift assays, the anti-Sp1 mAb (1 μg/10 μl reaction) was added to the DNA-protein complexes and incubated for another 30 min at room temperature before loading samples on the gels. NoShift assays (EMD Biosciences, Gibbstown, NJ) were performed to detect Sp1 binding activity according to the supplier's instructions using a biotinylated Sp1-specific probe and a competing non-biotinylated Sp1-specific probe and the Sp1 recognition sequences identified in Table 1. The blank contained all assay components except nuclear protein extracts.
TABLE 1.
The biotinylated and unlabeled capture probes used to detect Sp1 binding activity in the NoShift Assay
Putative recognition sequences are underlined.
| Oligoucleotides | Sequence (5′ to 3′) |
|---|---|
| −253 | GCCGCTCGCGGTAGGGGCGGAGCGCTGAGCTGG |
| −276 | CGCCACGCACGCAGCCCCACCCAGCGCCGCTCGC |
| −312 | CTCCTACACCTGCGGCGCGCCCCCGTCCCCTGGTCCCAG |
| −490 | TCCGGTCGCCTCGGCCCACCACCC |
| −515 | GACCGGAAAGGCGGCCAGGACCTT |
| −548 | GAAACCCGAGGGCGGCGCTCCAGG |
| Sp1-consensus | CCCTTGGTGGGGGCGGGGCCTAAGCTGCG |
siRNA Transfection
UGDH siRNA was synthesized by Qiagen using the cDNA target sequence 5′-CCGGCTCGTGACCATTTCCAA-3′. This siRNA (300 ng) was transiently transfected into orbital fibroblasts using Oligofect or HiPerFect reagents (Qiagen, catalog #301705) following the manufacturer's instructions. Another siRNA was used to target Sp1 (Applied Bioscience). After transfections, medium supplemented with 10% FBS was replaced, and cell layers were harvested as indicated. Cell material was taken up either in lysis buffer for Western blot analysis or using the RNeasy mini-kit for real-time RT-PCR analysis. In other experiments, transfected cells were assayed for UGDH enzymatic activity (described below), or extracted RNA was subjected to PCR analysis of HAS1, HAS2, and HAS3 mRNA levels. In others, cell layers were solubilized in 0.1 n NaOH and analyzed for hyaluronan content with a specific assay (Echelon, Salt Lake City, UT, catalog #K-1200). In some studies, medium was also analyzed for glycosaminoglycan content.
UGDH Enzyme Activity Assay
Enzyme activity in cell lysates was measured by quantifying the reduction of NAD+ in the absence or presence of UDP-glucose. Confluent monolayers were washed in cold PBS, harvested, and stored at −20 °C. Thawed cells were homogenized in 0.15 m NaCl and centrifuged at 12,000 × g for 15 min. Supernatants were collected, and enzyme activity was assayed according to the method of Axelrod et al. (32) and Tomida et al. (33). Briefly, reactions were performed in a total volume of 0.5 ml with 50 μg of cell lysate protein, 0.2 μmol of UDP glucose, and 0.4 μmol of NAD+ in 0.1 m glycine buffer (pH 8.7). Reactions were conducted at room temperature for 1 h, and the conversion to NADH from NAD+ was detected as absorbance at 340 nm. Data are presented as the mean ± S.D. of triplicate determinations from one experiment, representative of five performed.
Statistics
Results were reported as the mean ± S.D. of replicates described in the text and figure legends. Statistical significance was determined using a two-tailed Student's t test.
RESULTS
Divergent Expression of UGDH Protein and mRNA in Orbital and Dermal Fibroblasts
Little information currently exists concerning the relative levels of UGDH protein expressed in human cells. Using a polyclonal anti-human UGDH Ab directed at the carboxyl terminus, Western blot analysis revealed a single 55-kDa band expressed under basal culture conditions in several orbital fibroblast strains, each derived from a different donor (Fig. 1A). This protein band could be completely attenuated by preincubating with the target peptide used for Ab generation. In contrast, the protein band was virtually absent in dermal fibroblasts. Images from immunofluorescent microscopic examination demonstrate specific staining of the enzyme in orbital fibroblasts (Fig. 1B). UGDH protein localized to the cell membrane and was also detected diffusely in the cytoplasm. In some fibroblasts, perinuclear staining was observed. Staining in dermal fibroblasts was minimal and resembled that found in the orbital fibroblasts where anti-UGDH Ab had been pre-adsorbed. Abundant constitutive UGDH mRNA was detected by real-time PCR in orbital fibroblasts. These levels were higher than those in dermal fibroblasts (Fig. 1C). Transcript abundance was examined in a total of 6 orbital strains where levels were 0.73 ± 0.13-fold (mean ± S.D.) compared with 0.385 ± 0.12-fold in 4 dermal strains (p < 0.01).
FIGURE 1.

UGDH protein and mRNA expression in orbital and dermal fibroblasts. A, immunoblot analysis of UGDH protein using an anti-UGDH polyclonal Ab. Cellular proteins from confluent orbital and dermal fibroblasts were solubilized, and equivalent amounts were loaded onto SDS-PAGE, transferred to a PVDF membrane, blocked, and probed with anti-UGDH Ab in a 1:1000 dilution. Pretreatment with UGDH peptide confirmed the Ab specificity by adsorbing any detectable immunoreactivity. A negative isotype control is shown on the far right. Membranes were then probed with β-actin as a loading control. B, UGDH was localized in both orbital and dermal fibroblasts by immunostaining with anti-UGDH Ab. Sub-confluent fibroblasts were grown on coverslips, fixed, blocked, and stained with anti-UGDH Ab without or with the UGDH peptide. Cells were then stained with an Oregon Green-conjugated goat anti-rabbit Ab, counter-stained, and mounted with Vectashield containing DAPI. Monolayers were visualized with an Axioskop40 microscope (Carl Zeiss, ×200). C, steady-state UGDH mRNA in orbital and dermal fibroblasts was quantified by real-time PCR. Confluent fibroblasts were shifted overnight to medium containing 1% FBS. RNA was extracted and reverse-transcribed. cDNA was subjected to quantitative real time-PCR. Each reaction was performed three times, and the values were normalized to their respective GAPDH signals and are expressed as the mean ± S.D. (**, p < 0.01).
De Luca et al. (34, 35) have suggested that fibroblast density influences levels of UGDH. A recent report by Ren et al. (36) suggested that nutrients such as serum and glucose might also influence hyaluronan production in rat mesangial cells through a cyclin D3-related stress mechanism. Varying fibroblast density from 80 to 100% confluent failed to alter UGDH mRNA levels in fibroblasts (1.25- versus 1.23-fold, respectively, not significant). Reducing serum concentration in medium from 10 to 1% overnight increased these levels by less than 5% in either orbital or dermal fibroblasts (not significant).
High Level UGDH Expression in Orbital Fibroblasts Derives from UGDH Gene Promoter Activity
Analysis of the UGDH gene promoter was next undertaken to gain insight into the pattern of expression found in fibroblasts. Initially, 1.7-kb Fragment 7, spanning −1583 to +71 nt, was cloned, fused to a luciferase reporter pGL2 basic and transfected into orbital fibroblasts (Fig. 2A). In addition, a series of deletion mutants was also generated from the parent fragment, and the relative activities were determined. The highest luciferase activity in orbital fibroblasts of all promoter fragments tested was that associated with Fragment 3, comprising 1458 bp and spanning from −1387 to +71 nt (Fig. 2A). The relative activities of the various fragments suggest that an important positive regulatory element(s) might be found in the sequence spanning −900 to −1387 nt of the gene promoter. Fragments 4, 5, and 6 uniformly lack the first 547 bp surrounding and immediately upstream from the transcriptional start site and exhibit substantially lower luciferase activity than does Fragment 3. That finding suggests this span of sequence contains crucial basal promoter elements, consistent with the report of Bontemps et al. (15) who found that the first 644 bp represented the “core promoter.” The relatively low activities of Fragment 7 (−1583 to +71 nt) and Fragment 8 (−1436 to +71 nt) suggest that a repressor element(s) localizes between −1436 and −1388 nt. The addition of this 49-bp sequence to Fragment 3 results in Fragment 8 with its ∼70% reduction in promoter activity. To further characterize this repressor, a chimeric gene was constructed by fusing the sequence to pGL2, the expression of which is driven by the SV40 promoter. The additional 49-bp sequence reduced activity by 70% when transfected into orbital fibroblasts (Fig. 2B). This is consistent with −1436 to −1387 nt repressing UGDH gene promoter activity in orbital fibroblasts.
FIGURE 2.

Analysis of human UGDH gene promoter activity in orbital fibroblasts. A, sequences of the promoter spanning −1858 to +71 nt (Fragment 9) and the deletion mutants indicated were generated as described under “Experimental Procedures.” They were fused to the fruit fly luciferase gene in pGL2 basic vector and transfected into orbital fibroblasts concurrently with an internal control pRLTK vector. Deletion constructs are diagramed schematically. Rectangular boxes represent the regions upstream from the UGDH coding sequence. Luciferase activity was normalized to that of pRLTK used as a transfection efficiency control and are shown as bar graphs on the right. B, the sequence spanning −1436 to −1388 bp of the UGDH gene promoter exhibits repressor activity. The schematic diagram of the chimeric construct including the putative repressor fused to an SV40 promoter driven vector is shown. This was transfected into orbital fibroblasts. C, the sequence extending from −598 to −148 nt of Fragment 3 was deleted, resulting in Fragment 13, removing several Sp1 binding sites. D, Fragment 3 was subjected to the deletions indicated. Data represent the mean ± S.D. of two experiments, each performed in triplicate.
We performed additional deletions to further locate the sequence(s) conveying high activity of Fragment 3 in orbital fibroblasts (Fig. 2, C and D). Removing the sequence spanning −598 to −148 nt from Fragment 3 and thus deleting all 6 recognizable Sp1 binding sites resulted in Fragment 13 exhibiting activity that was reduced by 86%. This substantial reduction suggests that the Sp1 sites are particularly important to the high promoter activity observed in orbital fibroblasts. When Fragment 3 was truncated by removing the 141-bp sequence extending from −1387 to −1202 nt (Fragment 14), activity was reduced by 65% when transfected into orbital fibroblasts. A further deletion from −1202 to −906 nt (Fragment 15) reduced activity by 47% compared with intact Fragment 3. In contrast, these same deletions increased activities of 44% and 36%, respectively, when transfected into dermal fibroblasts. Deleting −87 to +71 nt (Fragment 16) reduced reporter activity by 74% in orbital fibroblasts but only a 16% decrease in dermal cultures. Taken in aggregate, these findings suggest that basal UGDH expression in orbital fibroblasts derives from multiple sequences along the putative gene promoter. Moreover, the influence of these sequences appears to be fibroblast-type specific.
From the results of studies depicted in Fig. 1, it became clear that basal UGDH expression might prove greater in orbital fibroblasts than in those derived from skin. To determine whether activity of the UGDH gene promoter differed in multiple strains of the two types of fibroblasts, Fragment 3 was transfected into five orbital and two dermal fibroblast strains. The fragment/reporter construct exhibited substantially higher activity in orbital fibroblast strains, each from a different donor, compared with the dermal fibroblasts (Fig. 3A). Moreover, no clear-cut differences in activity could be discerned in those strains from diseased and control orbits. The stability of UGDH mRNA was then determined by incubating cultures of orbital and dermal fibroblasts in the presence of 5,6-dichlorobenzimidazole (20 μg/ml), an inhibitor of gene transcription (37), and assessing steady-state UGDH mRNA levels at graded intervals. As Fig. 3B demonstrates, initial transcript levels in the dermal fibroblasts are considerably lower than those from the orbit. The apparent half-life of the transcript is 5–9 h in orbital fibroblasts and somewhat longer in dermal cultures, suggesting that greater mRNA stability in orbital fibroblasts does not account for differences in transcript abundance observed in the two cell types. Thus, differential gene transcription is a likely determinant of the divergent levels of UGDH expression found in orbital and dermal fibroblasts.
FIGURE 3.

UGDH promoter activity and mRNA decay in orbital and dermal fibroblasts. A, orbital and dermal fibroblasts were transfected with a reporter containing Fragment 3 (−1387 to +71 nt). Transfections were conducted as described under “Experimental Procedures.” Data represent the mean of six to nine independent replicates. B, differences in UGDH mRNA decay do not explain the differential expression of UGDH in orbital and dermal fibroblasts. Confluent cultures were incubated in medium supplemented with 5,6-dichlorobenzimidazole (20 μg/ml) and harvested at the graded intervals indicated along the abscissa. Cellular RNA was extracted and subjected to reverse transcription and real-time quantitative PCR using the UGDH primer set described under “Experimental Procedures.” These values were normalized to their respective GAPDH levels and are expressed as the mean ± S.D. Each reaction was performed in triplicate. UGDH mRNA was reduced by 61.5 ± 16.9% in orbital fibroblasts and 18.4 ± 31.7% in dermal cultures after 7 h incubations in the 4 experiments performed (p < 0.01 versus their respective controls; orbital versus dermal p < 0.05).
Involvement of Sp1 in UGDH Gene Promoter Activity
Sp1 plays a critical role in UGDH gene expression in HepG2, MRC5, and HeLa cells (14, 15, 38). The UGDH gene promoter region spanning −899 to +71 nt (Fragments 2, 3, 7, and 8) contains 6 recognizable Sp1 sites. Their potential involvement in UGDH gene promoter activity was assessed by treating promoter/reporter transfected orbital and dermal fibroblasts with bisanthracyclin (WP-631, 1 μm), a selective Sp1 inhibitor (39), for 16 h. Luciferase activity from Fragment 3 was attenuated by WP-631 (Fig. 4A). The effects on promoter activity were considerably greater in orbital fibroblasts (79%) compared with those from the skin (39%). To further establish the importance of Sp1, knocking it down with a specific siRNA also substantially reduced UGDH protein levels in orbital fibroblasts (Fig. 4B). The effects of Sp1 siRNA were mediated at the level of UGDH mRNA (Fig. 4C). Levels were reduced by 57% in orbital fibroblasts treated with Sp1 siRNA (p < 0.001). In a total of 3 different experiments, Sp1 siRNA reduced UGDH mRNA by 55.5 ± 2.1% (p < 0.01) in orbital fibroblasts compared with 19.5 ± 9.9% (p < 0.05) in dermal cultures.
FIGURE 4.

Impact of interrupting Sp1 on UGDH gene promoter activity, UGDH protein, and UGDH mRNA levels. A, WP-631 (1 μm), a selective inhibitor of Sp1, reduces the activity of UGDH promoter Fragment 3 in orbital and dermal fibroblasts. The compound or diluent (control) was added for 16 h to the medium of transfected monolayers. Cells were disrupted, and luciferase activity was detected as described under “Experimental Procedures.” Data are expressed as the mean ± S.D. of three independent reactions. B, orbital fibroblasts were transfected with either scrambled (control) siRNA or one directed against Sp1for 5 days as described under “Experimental Procedures.” Cell layers were solubilized and subjected to immunoblot analysis with an Ab directed against UGDH. The membrane was then stripped and re-probed with anti-β-actin (5 independent replicates, p < 0.01). C, orbital fibroblasts were transfected with either control or Sp1-specific siRNA as in panel B. At the end of the incubation, RNA was extracted, and UGDH mRNA levels were quantified by real-time PCR. Data are expressed as the mean ± S.D. of triplicate determinations (***, p < 0.001). Results from a total of three different experiments reveal 55.5 ± 2.1% reduction in orbit (p < 0.01 versus controls).
Given the importance of Sp1 in supporting constitutive UGDH expression, we next determined whether the transcription factor was differentially expressed in orbital and dermal fibroblasts. Sp1 was abundant in the nuclear extract from four different orbital fibroblast strains but was undetectable by Western blot in two dermal strains (Fig. 5A). An EMSA detected Sp1 binding to its target DNA using nuclear protein extracts from orbital fibroblasts but was absent in extracts from two dermal strains tested (Fig. 5B). Using the NoShift assay, Sp1 binding to nuclear protein from three additional orbital and three dermal fibroblast strains was assessed. Orbital binding was 12.1 ± 3.4 compared with 3.5 ± 1.6 arbitrary units in dermal fibroblasts (p < 0.05). The obvious shift in reactions containing orbital nuclear extract and 32P-labeled consensus Sp1 fragment was completely quenched with a 100-fold molar excess unlabeled Sp1 consensus oligonucleotide (Fig. 5C). On the other hand, an NF-κB probe, used here as a control, failed to influence binding, attesting to the specificity of protein/DNA interactions. The addition of an anti-Sp1 Ab to the reaction formed a super-shifted complex (Fig. 5D), again supporting the authenticity of the Sp1 interactions. HeLa cell extracts, utilized as a positive control, displayed higher levels of binding activity than did reactions with the fibroblast proteins. The substantially higher levels of Sp1 could explain, at least in part, the differences in UGDH expression found in orbital compared with dermal fibroblasts.
FIGURE 5.

Sp1 levels and specific DNA binding are substantially greater in orbital compared with dermal fibroblasts. A, nuclear protein from four orbital and two dermal fibroblast strains were subjected to Western immunoblot for Sp1 protein using a specific Ab against Sp1, as described under “Experimental Procedures.” Filters were re-probed with an Ab directed against β-actin. B, nuclear proteins were extracted from isolated nuclei of three orbital and two dermal fibroblast strains and subjected to EMSA using a 32P-labeled oligonucleotide conforming to the consensus sequence of human Sp1 as a probe. Reactions were loaded onto native polyacrylamide gels, electrophoresed, and exposed to x-ray film. C, orbital fibroblast nuclear extract was subjected to EMSA using the 32P-labeled consensus Sp1 binding sequence as a probe and competing the reaction with excess unlabeled Sp1 or a control (NF-κB) (100× molar excess each). D, nuclear protein extracts from orbital fibroblasts and HeLa cells were subjected to EMSA and “supershift” analysis using an anti-Sp1 monoclonal Ab. Studies were repeated at least three times.
In another set of studies, the NoShift assay was used to determine whether potential differences in binding of nuclear proteins from orbital and dermal fibroblasts to the six individual Sp1 sites identified in the UGDH gene promoter could contribute to the divergent UGDH levels. As the results in Fig. 6 demonstrate, in every case binding to oligonucleotides containing a single Sp1 site (Table 1) was substantially greater with nuclear proteins from orbital fibroblasts than with their dermal counterparts (p < 0.01). This was also true for binding to the Sp1 consensus sequence. In general, good agreement with regard to rank order was found between orbital and dermal nuclear protein binding to a particular site in that the highest binding occurred at the −276-nt site, whereas the lowest was found at site −490 nt.
FIGURE 6.

Six recognizable Sp1 binding sites in the human UGDH gene promoter exhibit higher binding activity to orbital compared with dermal nuclear proteins. NoShift assays were performed on nuclear protein extracts from orbital and dermal fibroblasts according to the manufacturer's instructions (EMD Biosciences). Equivalent amounts of nuclear protein were subjected to each reaction with an oligonucleotide containing one of the six binding sites or the consensus sequence described in Table 1. The data are presented as the mean ± S.D. of three independent replicates. Binding to the orbital nuclear protein exceeded that found with the dermal extract for all sites (p < 0.01).
To determine whether binding to each Sp1 site might play a functional role in regulating UGDH expression, a series of mutations (described in Table 2) was generated in Fragment 3, the fragment exhibiting the highest activity level in orbital fibroblasts. As Fig. 7, A and B, demonstrate, mutating the site at −515 nt resulted in a substantial reduction in both orbital and dermal fibroblasts. In contrast, mutations at −276, −312, and −490 nt also reduced activity in orbital fibroblasts but increased reporter activity in dermal cultures. Mutating the −253-nt site increased promoter activity in orbital while attenuating activity in dermal fibroblasts. These findings suggest that specific Sp1 sites in the UGDH gene promoter exert divergent influence on activity in orbital and dermal fibroblasts, perhaps accounting for the cell type-specific pattern of expression exhibited by the enzyme. Moreover, it would appear that although binding to some Sp1 sites enhances UGDH promoter activity, Sp1 protein might be exerting a negative influence on gene activity at other sites. We next determined what impact mutating multiple sites exerted on promoter activity. In that study, introducing single mutations at −253 and −515 nt again resulted in enhanced and reduced levels of reporter activity, respectively, compared with the wild type (Fig. 7C). When Fragment 3 was mutated at both −253 and −515 nt, the positive influence of the single −276-nt mutation was completely abrogated, and the activity of the double-mutated fragment was intermediate to that provoked by the single mutations. These results suggest that each Sp1 site impacts UGDH gene promoter activity in a distinct manner. The net influence of Sp1 on UGDH gene promoter activity derives from the aggregate of effects mediated at multiple sites.
FIGURE 7.
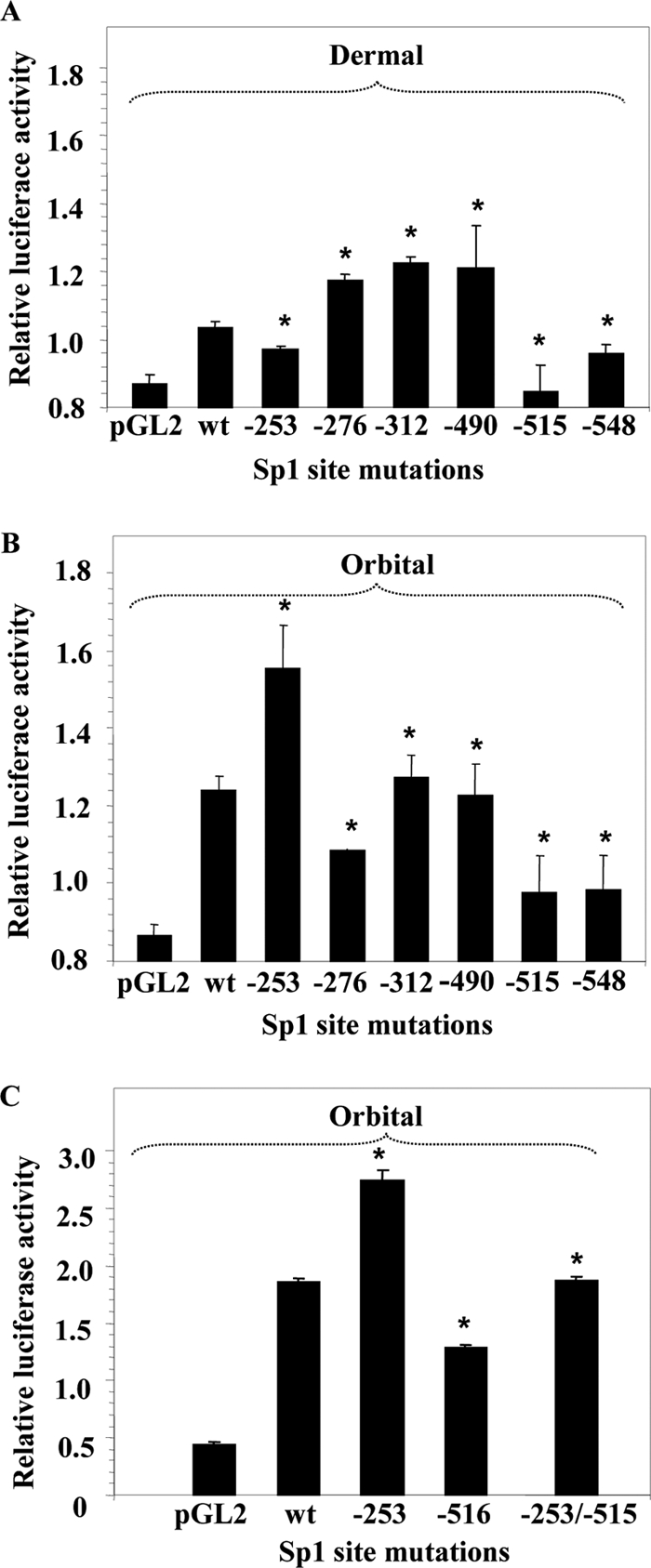
Site-directed mutations in six Sp1 binding sites alters Fragment 3 activity. Dermal (A) and orbital fibroblasts (B) were transfected with a luciferase reporter containing either wild type (wt) Fragment 3 of the UGDH gene promoter or that fragment harboring a single mutation at the Sp1 binding site indicated along the abscissa. Mutations are described in Table 2. C, orbital fibroblasts were transfected with a reporter containing wild type Fragment 3, that fragment mutated at site −253, −515, or a double mutation at both sites. After 24 h, the cell monolayers were harvested, and luciferase activities were determined as described under “Experimental Procedures.” Data are expressed as the mean ± S.D. of three independent determinations. * denotes a significant difference in activity between wt Fragment 3 and that fragment harboring the mutation(s) indicated, p < 0.05.
Orbital Fibroblasts Exhibit High Levels of UGDH Activity
We next determined whether the UGDH expressed by human orbital fibroblasts is catalytically active. An assay was performed on extracts from eight orbital (three Graves and five healthy controls) and three normal dermal fibroblast strains, each from a different donor (Table 3). After a 60-min reaction, the reduction of NAD attributable to UDP-Glc-dependent UGDH activity was 31.26 ± 0.28 nmol/min/μg of protein in orbital fibroblasts compared with 8.66 ± 0.12 nmol/min/μg of protein in dermal fibroblasts (p < 0.01). Thus, the higher level UGDH found in orbital fibroblasts appears to represent active enzyme and therefore is of potential biological importance.
TABLE 3.
Higher levels of UGDH activity are exhibited by orbital compared to dermal fibroblasts
Fibroblast lysates were homogenized and taken up in glycine buffer (pH 8.7) NAD+ (0.4 μmole) without or with UDP-glucose (0.2 μmole). After a 60-min incubation at room temperature, levels of reduced NAD (NADH) were quantified in a spectrophotometer at A340 nm. Data are expressed as the mean ± S.D. of triplicate independent determinations.
| Strain | UGDH activity |
|
|---|---|---|
| Without UDP-Glc | With UDP-Glc | |
| nmol/min/μg | ||
| Orbital 1 | 29.2 ± 1.5 | 56.4 ± 1.5 |
| Orbital 2 | 31.3 ± 1.0 | 60.4 ± 0.6 |
| Orbital 3 | 64.3 ± 1.2 | 70.8 ± 1.1 |
| Orbital 4 | 32.2 ± 1.4 | 74.0 ± 1.3 |
| Orbital 5 | 28.3 ± 0.8 | 78.4 ± 1.5 |
| Orbital 6 | 34.7 ± 0.6 | 72.1 ± 0.1 |
| Orbital 7 | 24.0 ± 0.3 | 58.4 ± 0.4 |
| Orbital 8 | 17.4 ± 1.2 | 41.2 ± 0.1 |
| Dermal 1 | 8.7 ± 1.4 | 17.3 ± 0.9 |
| Dermal 2 | 10.5 ± 1.5 | 16.9 ± 0.7 |
| Dermal 3 | 16.2 ± 0.9 | 27.2 ± 1.4 |
UGDH siRNA Interrupts Not Only UGDH Expression but Also Abolishes UGDH Enzymatic Activity, Reduces Basal Levels of HAS-1 and HAS-2 Expression, and Diminishes Cell-associated Hyaluronan Levels
Knocking down Sp1 could attenuate UGDH expression in orbital fibroblasts (Fig. 4). We next assessed the consequences of knocking down enzyme expression in orbital fibroblasts with siRNA targeting UGDH. Control (scrambled) and UGDH-specific siRNAs were transfected into orbital fibroblasts, and cellular RNA and protein were harvested. From the real time RT-PCR and Western blot analyses shown in Fig. 8, A and B, respectively, UGDH siRNA effectively reduced UGDH mRNA and protein by greater than 80% when assessed 5 days after cell transfection. This interruption of UGDH expression was reflected in decreased enzyme activity compared with cells undergoing transfection with control siRNA (Fig. 8C). To determine whether interfering with Sp1 could reduce UGDH activity, orbital and dermal fibroblast cultures were transfected with Sp1 siRNA. This reduced enzyme activity dramatically in orbital cultures (p < 0.01), whereas activity in the dermal fibroblasts was unaffected (Fig. 8D).
FIGURE 8.
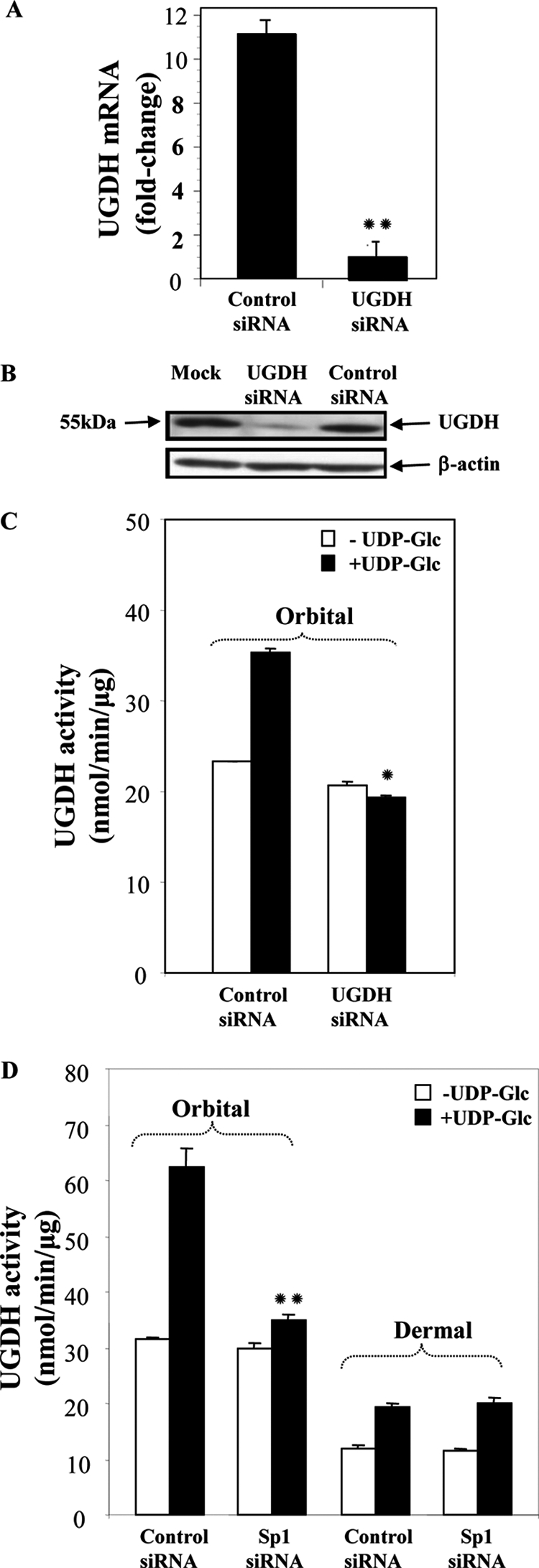
UGDH-specific siRNA down-regulates UGDH mRNA, protein, and enzyme activity levels in orbital fibroblasts. A, cultures reaching 80% confluence were transfected with scrambled (Control) or UGDH-specific siRNA using Oligofect reagent. Cells were disrupted, and cellular RNA was analyzed by real-time PCR. Data are expressed as the mean ± S.D., n = 3 independent determinations; **, p < 0.01 versus control siRNA. In all experiments, UGDH siRNA reduced UGDH levels by 86% (p < 0.01 versus control siRNA). B, UGDH protein levels were substantially reduced in orbital fibroblasts after transfection with UGDH-specific siRNA. Cells were transfected as described under “Experimental Procedures” and lysed, and cellular proteins were subjected to Western blot analysis with an anti-UGDH Ab. C, cultures were transfected with scrambled (Control) siRNA or UGDH-specific siRNA and lysed, and UGDH enzyme activity was assayed in the absence or presence of UDP-Glc as described under “Experimental Procedures.” *, p < 0.05. In three separate experiments, UGDH siRNA reduced UGDH activity by 41 ± 4% (p < 0.001 versus control). D, orbital and dermal fibroblasts were transfected with scrambled (Control) or Sp1-specific siRNA, cell layers were assayed for UGDH activity in the absence or presence of UDP-Glc. Data are expressed as the mean ± S.D. of triplicate determinants from one experiment. **, p < 0.01 versus control siRNA. When results of separate experiments were combined, Sp1 siRNA reduced activity in orbital fibroblasts by 40 ± 4% versus controls.
The impact of knocking-down UGDH on the expression of the terminal HAS enzymes was determined by assessing the levels of each isozyme transcript. As the results in Fig. 9A suggest, interrupting UGDH expression reduces HAS1 and HAS2 levels by 58 and 39%, respectively (p < 0.001 and p < 0.01 versus controls). In contrast, levels of HAS-3 transcript were unaffected by knocking-down UGDH expression. These findings suggest that HAS-1 and HAS-2 are functionally coupled to UGDH and that the basal expression of the two isozymes is dependent, at least in part, on that enzyme. Sp1 siRNA also reduced in orbital fibroblasts HAS1 mRNA (control siRNA, 1.91 ± 0.01-fold versus Sp1 siRNA, 1.53 ± 0.05-fold, p < 0.01) and HAS2 mRNA (control siRNA, 1.40 ± 0.01-fold versus Sp1 siRNA, 1.13 ± 0.05-fold, p < 0.01). In contrast, expression of none of the three isozymes was affected by Sp1 siRNA in dermal fibroblasts (<2% change, not significant).
FIGURE 9.

Impact of interrupting UGDH or Sp1 on the expression of HAS isozymes and hyaluronan production. A, orbital fibroblast cultures reaching 80% confluence were transfected with an siRNA targeting UGDH (30 μg) or scrambled (Control) siRNA using RNAiFect reagent, as described under “Experimental Procedures.” Cells were disrupted, and cellular RNA was subjected to real-time RT-PCR using HPRT as the reference gene. Data are expressed as the mean ± S.D. of triplicate, independent replicates. **, p < 0.01; ***, p < 0.001. B, orbital fibroblasts were transfected with control or UGDH-specific siRNA, solubilized, and subjected to an ELISA specific for hyaluronan. *, p < 0.05. In three separate experiments, UGDH siRNA reduced hyaluronan content by 54 ± 1.9% (p < 0.01). C, orbital and dermal fibroblasts were transfected with control or Sp1-specific siRNA. They were assayed for hyaluronan content. Data are expressed as the mean ± S.D. of triplicate determinants in one orbital and one dermal fibroblast strain. *, p < 0.05. In a total of three different strains of each, Sp1 siRNA reduced hyaluronan content by 71 ± 4.6% (p < 0.01) in orbital and 4.9 ± 2.1% (p < 0.001) in dermal fibroblasts.
The impact of Sp1 and UGDH interruption on hyaluronan production was next assessed. Orbital fibroblast cell layers transfected with control, UGDH-specific, or Sp1-specific siRNA were analyzed for hyaluronan content using a specific ELISA. As Fig. 9B demonstrates, reducing UGDH levels substantially lowers the cell layer content of hyaluronan. Three days after transfection, hyaluronan levels were reduced by 59% compared with controls (p < 0.05). Medium-associated levels were also reduced by 56%, from 271 ± 5.9 ng/10,000 cells in controls to 119.7 ± 12.2 ng/10,000 cells (p < 0.05). Hyaluronan production was dramatically higher in orbital compared with dermal fibroblasts (Fig. 9C). Sp1 siRNA reduced cell layer-associated levels in orbital fibroblasts from 115.4 ± 10.4 ng/10,000 cells to 28.3 ± 5.3 ng/10,000 cells (76% reduction, p < 0.05). In contrast, hyaluronan production was uninfluenced in dermal fibroblasts (12.6 ± 2.4 ng/10,000 cells versus 12.2 ± 1.4 ng/10,000 cells, not significant). Thus, interrupting Sp1 and UGDH expression resulted in attenuation of hyaluronan production in orbital fibroblasts. The relationship between Sp1 and hyaluronan production in dermal fibroblasts differs from that in orbital fibroblasts.
DISCUSSION
UGDH is widely expressed in nature, and its function and regulation have been characterized in bacteria (40, 41). Disruption of UGDH expression in mutant mice results in developmental arrest from altered migration of mesoderm and endoderm during gastrulation (17). In zebrafish, a jekyll mutation disrupts UGDH and dampens the differentiation of atrioventricular borders (18), suggesting that the enzyme may play a critical role in establishing the anatomic boundaries between the atrium and ventricle. A knock-out dfna5 mutation in zebrafish results in loss of UGDH expression and reduced hyaluronan production associated with impaired craniofacial development (19). Consistent with the findings of Busch-Nentwich et al. (19) were those reported by Vigetti et al. (42), who found coupling between UGDH and hyaluronan production in Xenopus laevis. The latter studies, conducted in human aortic smooth muscle cells, revealed that inhibiting UGDH expression with an siRNA failed to alter levels of any of the HAS isozyme mRNAs. In this regard, that earlier study is at variance with our findings in orbital fibroblasts (Fig. 9A) but is congruent with those involving dermal fibroblasts. The study of Vigetti et al. (42) also demonstrated that transfecting aortic smooth muscle cells with Xenopus UGDH resulted in increased UGDH activity, enhanced cellular UDP-GlcUA levels, selectively increased hyaluronan production and led to increased steady-state levels of HAS2 and HAS3 mRNAs. Thus, it would appear that the relationship between UGDH, HAS isozyme expression, and hyaluronan production is cell type-specific.
Our findings suggest that the regulation of the UGDH gene in connective tissue is complex and varies with anatomic region. They indicate that orbital fibroblasts exhibit relatively high basal levels of the enzyme. These observations are consistent with the pathogenesis of TAO, where excessive accumulation of hyaluronan underlies the clinical phenotype associated with Graves disease (21, 22). This divergence can be found in orbital fibroblasts from both control (healthy) tissues and those from patients with TAO. Thus, relatively high UGDH levels represent an inherent property of orbital fibroblasts and can thus be linked to disease susceptibility rather than to its pathogenesis per se.
Basal UGDH gene promoter activity is considerably higher in orbital fibroblasts than that found in dermal fibroblasts (Fig. 3A). The current studies link this activity with the relatively high level of hyaluronan production in orbital fibroblasts and with the more abundant Sp1 protein expressed by these cells. Previous studies by Monslow et al. (43) demonstrated that Sp1 and Sp3 support the constitutive expression of HAS2 in a human renal epithelial cell line. Interrupting Sp1 in orbital fibroblasts reduced not only UGDH expression (Fig. 4) but lowered its activity (Fig. 8D) and attenuated HAS1 and HAS2 expression. Reduced hyaluronan after this disruption (Fig. 9C) could thus be attributed to altered levels of several enzymes. In addition, the relatively high Sp1 levels in orbital fibroblasts could account, at least in part, for other divergent phenotypic attributes displayed by these fibroblasts.
The structure of the UGDH gene promoter has been examined previously (14, 15). Two transcriptional start sites were mapped to 165 and 325 bp upstream from the AUG initiation codon, in good agreement with the lengths of the two UGDH mRNAs (14, 44). It includes an inverted TATA box but no consensus CAAT sequence (15). Multiple GC-rich regions appear crucial for the regulation of UGDH transcription through Sp1 (14, 15). Sp1 binding at −548 bp plays an essential role as an enhancer in HepG2 and HeLa cells (14). Moreover, its binding to two GC-rich sequences located in the proximal region of the promoter is critical to the up-regulation of UGDH promoter activity by TGF-β. This region exerts a reciprocal influence in states of hypoxia that diminish Sp1 binding (15). When compared in two different fibroblast types, the promoter exhibits a cell-specific pattern of activity. Six potential GC-rich elements for Sp1 binding reside in Fragment 3. We have demonstrated the essential contribution of Sp1 in supporting UGDH promoter activity with a specific inhibitor of this transcription factor (Fig. 4A). In addition, knocking down Sp1 expression with a specific siRNA attenuates UGDH expression (Fig. 4, B and C). Different levels of nuclear Sp1 protein, its DNA binding activity, and impact on UGDH promoter activity were observed in orbital and dermal fibroblasts. To determine which GC-rich sites might be crucial to orbital fibroblast-specific basal promoter function, we employed site-directed mutagenesis (Fig. 7). Those studies revealed that each of the six Sp1 binding sites might either enhance or reduce promoter activity. Moreover, mutating particular Sp1 sites affected promoter reporter activity divergently in orbital and dermal fibroblasts. Loss of the three Sp1 sites closest to the start site (−253, −276, and −312 nt), which occurs when Fragment 3 is truncated into Fragment 5, results in a substantial drop-off in activity. Because the site at −253 nt appears to reduce promoter activity (Fig. 7), our findings suggest that −276 and −312 play important roles in supporting promoter activity in the orbital fibroblasts. Furthermore, removing the sequence containing all six sites (Fragment 13) dramatically reduces promoter activity (Fig. 2C).
Sp1 is known to form complexes with other transcription factors. Interplay between these factors and Sp1 determines whether they co-activate or repress Sp1 function. An example is the estrogen receptor (45). From this interaction Sp1 sites can mediate estrogen responses (46). Thus, the basis for the orbital fibroblast-specific pattern of promoter activity may result from other transcription factors acting at the GC-rich sites to which Sp1 binds. In addition, flanking sequences influence Sp1 activity and the elements contained within these might determine whether a particular Sp1 site acts as a repressor or an activator (47). This might provide an explanation for our findings that mutating each of the Sp1 sites can yield divergent levels of UGDH promoter activity (Fig. 7). Moreover, it might explain the loss of activity found when Fragment 3 is truncated to Fragment 14, the latter of which has lost the sequence extending from −1387 to −1202 nt.
In addition to regulation through Sp1 sites, a potential repressor element(s) localizes to the promoter sequence between nt −1436 and −1388 bp. The chimeric construct containing this 49-bp sequence fused to the SV40 promoter/luciferase reporter exhibited substantially reduced activity when transfected into orbital fibroblasts (Fig. 2B). An NF-E2 consensus site could be identified within this sequence. Heterodimers of NF-E2 and small Maf proto-oncoprotein exert positive regulation, whereas homodimers of Maf proteins act to negatively regulate transcription (48). Both proteins act at NF-E2 sites, regulating gene expression in erythroid cells. Future studies will focus on further characterizing the activity of this sequence and its potential regulatory role in orbital fibroblasts and determining whether this repressor activity accounts for any aspect of cell-specific UGDH expression.
GlcUA represents a sugar residue found in several abundant glycosaminoglycans (49). Terminal transferases catalyze the rate-limiting steps in the synthesis of these macromolecules (34, 35, 49). In the case of hyaluronan, three mammalian HAS isoforms have been identified and cloned (50–54). Each has a characteristic pattern of tissue expression and regulation (55, 56). A number of diseases are associated with hyaluronan overproduction, including neoplasms, where this macromolecule influences growth and metastatic potential (57–61). Moreover, attenuation of hyaluronan synthesis in some tumors by HAS specific inhibition can diminish angiogenesis (62).
In TAO, the orbit becomes infiltrated with hyaluronan, associated with characteristic tissue remodeling and expansion, which results in substantial morbidity (24). Activated orbital fibroblasts appear to be responsible for abnormal production of hyaluronan in TAO (23, 27, 28). Once generated, hyaluronan influences orbital tissue function as a consequence of its rheological properties, including viscosity and extraordinary hydrophilic nature (22). Besides these traditional attributes, short chain hyaluronan can promote inflammation through a variety of actions (63–65) and thus could influence intra-orbital immunity.
The appreciable differences in UGDH expression found in orbital and dermal fibroblasts could reflect the specialized roles of each cell type in generating hyaluronan and other glycoconjugates. These are entirely consistent with their putative roles in health and disease (29, 30). IL-1β, leukoregulin, and CD154 (CD40 ligand) up-regulate hyaluronan synthesis in orbital fibroblasts, effects that are substantially greater than those found in other fibroblast types (25–28). Insight into the mechanisms involved in the regulation by cytokines of hyaluronan synthesis has been provided very recently by Vigetti et al. (66), who implicated the NF-κB pathway in the effects of tumor necrosis factor-α and IL-1β in endothelial cells. A recent report suggests that the thyrotropin receptor, the central self-antigen in Graves disease, might drive hyaluronan synthesis in orbital fibroblasts (67). Another report suggests that prostaglandin D2 can up-regulate hyaluronan synthesis in orbital fibroblasts through the D prostanoid receptor, an induction that is attributable to HAS2 activity (68). IL-1β induces HAS 1, 2, and 3 mRNA levels (26) as well as UGDH expression (6) in orbital fibroblasts. From our current observations, it is apparent that differential expression of multiple enzymes in the hyaluronan biosynthetic pathway may underlie the particularly high levels of production in TAO. This may represent a consequence of increased availability of UDP-GlcUA in the orbit. In keeping with this concept, Jokela et al. (69) have demonstrated previously that reducing the availability of UDP sugars can attenuate hyaluronan synthesis. Thus, the current findings have substantial clinical implications in that they identify a potential therapeutic target.
This work was supported, in whole or in part, by National Institutes of Health Grants EY008976, EY011708, and DK063121. This work was also supported by a Research to Prevent Blindness Award and by the Bell Charitable Trust.
- UGDH
- UDP-glucose dehydrogenase
- UDP-GlcUA
- uridine diphosphate-glucuronate
- TAO
- thyroid-associated ophthalmopathy
- HAS
- hyaluronan synthase
- GD
- Graves disease
- Sp1
- specific protein 1
- Ab
- antibody
- nt
- nucleotide(s).
REFERENCES
- 1. Zalitis J., Feingold D. S. (1968) Biochem. Biophys. Res. Commun. 31, 693–698 [DOI] [PubMed] [Google Scholar]
- 2. Sommer B. J., Barycki J. J., Simpson M. A. (2004) J. Biol. Chem. 279, 23590–23596 [DOI] [PubMed] [Google Scholar]
- 3. Hempel J., Perozich J., Romovacek H., Hinich A., Kuo I., Feingold D. S. (1994) Protein Sci. 3, 1074–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Strominger J. L., Kalckar H. M., Axelrod J., Maxwell E. S. (1954) J. Am. Chem. Soc. 76, 6411–6412 [Google Scholar]
- 5. Huh J. W., Yoon H. Y., Lee H. J., Choi W. B., Yang S. J., Cho S. W. (2004) J. Biol. Chem. 279, 37491–37498 [DOI] [PubMed] [Google Scholar]
- 6. Spicer A. P., Kaback L. A., Smith T. J., Seldin M. F. (1998) J. Biol. Chem. 273, 25117–25124 [DOI] [PubMed] [Google Scholar]
- 7. Wegrowski Y., Perreau C., Bontemps Y., Maquart F. X. (1998) Biochem. Biophys. Res. Commun. 250, 206–211 [DOI] [PubMed] [Google Scholar]
- 8. Marcu O., Stathakis D. G., Marsh J. L. (1999) Cytogenet. Cell Genet. 86, 244–245 [DOI] [PubMed] [Google Scholar]
- 9. Lind T., Falk E., Hjertson E., Kusche-Gullberg M., Lidholt K. (1999) Glycobiology 9, 595–600 [DOI] [PubMed] [Google Scholar]
- 10. Franzen J. S., Marchetti P. S., Feingold D. S. (1980) Biochemistry 19, 6080–6089 [DOI] [PubMed] [Google Scholar]
- 11. Jaenicke R., Rudolph R., Feingold D. S. (1986) Biochemistry 25, 7283–7287 [DOI] [PubMed] [Google Scholar]
- 12. Peng H. L., Lou M. D., Chang M. L., Chang H. Y. (1998) Proc. Natl. Sci. Counc. Repub. China B 22, 166–172 [PubMed] [Google Scholar]
- 13. Lapointe J., Labrie C. (1999) Endocrinology 140, 4486–4493 [DOI] [PubMed] [Google Scholar]
- 14. Vatsyayan J., Peng H. L., Chang H. Y. (2005) J. Biochem. 137, 703–709 [DOI] [PubMed] [Google Scholar]
- 15. Bontemps Y., Vuillermoz B., Antonicelli F., Perreau C., Danan J. L., Maquart F. X., Wegrowski Y. (2003) J. Biol. Chem. 278, 21566–21575 [DOI] [PubMed] [Google Scholar]
- 16. Pugh B. F., Tjian R. (1990) Cell 61, 1187–1197 [DOI] [PubMed] [Google Scholar]
- 17. García-García M. J., Anderson K. V. (2003) Cell 114, 727–737 [DOI] [PubMed] [Google Scholar]
- 18. Walsh E. C., Stainier D. Y. (2001) Science 293, 1670–1673 [DOI] [PubMed] [Google Scholar]
- 19. Busch-Nentwich E., Söllner C., Roehl H., Nicolson T. (2004) Development 131, 943–951 [DOI] [PubMed] [Google Scholar]
- 20. Grubb M. F., Kasofsky J., Strong J., Anderson L. W., Cysyk R. L. (1993) Biochem. Mol. Biol. Int. 30, 819–827 [PubMed] [Google Scholar]
- 21. Hufnagel T. J., Hickey W. F., Cobbs W. H., Jakobiec F. A., Iwamoto T., Eagle R. C. (1984) Ophthalmology 91, 1411–1419 [DOI] [PubMed] [Google Scholar]
- 22. Smith T. J., Bahn R. S., Gorman C. A. (1989) Endocr. Rev. 10, 366–391 [DOI] [PubMed] [Google Scholar]
- 23. Smith T. J. (2005) Clin. Exp. Immunol. 141, 388–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kazim M., Goldberg R. A., Smith T. J. (2002) Arch. Ophthalmol. 120, 380–386 [DOI] [PubMed] [Google Scholar]
- 25. Cao H. J., Wang H. S., Zhang Y., Lin H. Y., Phipps R. P., Smith T. J. (1998) J. Biol. Chem. 273, 29615–29625 [DOI] [PubMed] [Google Scholar]
- 26. Kaback L. A., Smith T. J. (1999) J. Clin. Endocrinol. Metab. 84, 4079–4084 [DOI] [PubMed] [Google Scholar]
- 27. Smith T. J., Wang H. S., Evans C. H. (1995) Am. J. Physiol. 268, C382–C388 [DOI] [PubMed] [Google Scholar]
- 28. Smith T. J., Hoa N. (2004) J. Clin. Endocrinol. Metab. 89, 5076–5080 [DOI] [PubMed] [Google Scholar]
- 29. Smith T. J. (1987) J. Clin. Invest. 79, 1493–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smith T. J., Sempowski G. D., Wang H. S., Del Vecchio P. J., Lippe S. D., Phipps R. P. (1995) J. Clin. Endocrinol. Metab. 80, 2620–2625 [DOI] [PubMed] [Google Scholar]
- 31. Chomczynski P., Sacchi N. (1987) Anal. Biochem. 162, 156–159 [DOI] [PubMed] [Google Scholar]
- 32. Strominger J. L., Maxwell E. S., Axelrod J., Kalckar H. (1957) J. Biol. Chem. 224, 79–90 [PubMed] [Google Scholar]
- 33. Tomida M., Koyama H., Ono T. (1974) Biochim. Biophys. Acta 338, 352–363 [Google Scholar]
- 34. De Luca G., Rindi S., Rizzotti M. (1981) Basic Appl. Histochem. 25, 67–69 [PubMed] [Google Scholar]
- 35. Rizzotti M., Cambiaghi D., Gandolfi F., Rindi S., Salvini R., De Luca G. (1986) Basic Appl. Histochem. 30, 85–92 [PubMed] [Google Scholar]
- 36. Ren J., Hascall V. C., Wang A. (2009) J. Biol. Chem. 284, 16621–16632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dreyer C., Hausen P. (1978) Nucleic Acids Res. 5, 3325–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vatsyayan J., Lee S. J., Chang H. Y. (2005) J. Biochem. Mol. Toxicol. 19, 279–288 [DOI] [PubMed] [Google Scholar]
- 39. Martín B., Vaquero A., Priebe W., Portugal J. (1999) Nucleic Acids Res. 27, 3402–3409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mijakovic I., Petranovic D., Deutscher J. (2004) J. Mol. Microbiol. Biotechnol. 8, 19–25 [DOI] [PubMed] [Google Scholar]
- 41. Grangeasse C., Obadia B., Mijakovic I., Deutscher J., Cozzone A. J., Doublet P. (2003) J. Biol. Chem. 278, 39323–39329 [DOI] [PubMed] [Google Scholar]
- 42. Vigetti D., Ori M., Viola M., Genasetti A., Karousou E., Rizzi M., Pallotti F., Nardi I., Hascall V. C., De Luca G., Passi A. (2006) J. Biol. Chem. 281, 8254–8263 [DOI] [PubMed] [Google Scholar]
- 43. Monslow J., Williams J. D., Fraser D. J., Michael D. R., Foka P., Kift-Morgan A. P., Luo D. D., Fielding C. A., Craig K. J., Topley N., Jones S. A., Ramji D. P., Bowen T. (2006) J. Biol. Chem. 281, 18043–18050 [DOI] [PubMed] [Google Scholar]
- 44. Bontemps Y., Maquart F. X., Wegrowski Y. (2000) Biochem. Biophys. Res. Commun. 275, 981–985 [DOI] [PubMed] [Google Scholar]
- 45. Krishnan V., Wang X., Safe S. (1994) J. Biol. Chem. 269, 15912–15917 [PubMed] [Google Scholar]
- 46. Schultz J. R., Petz L. N., Nardulli A. M. (2005) J. Biol. Chem. 280, 347–354 [DOI] [PubMed] [Google Scholar]
- 47. Zhang J., Wang S., Wesley R. A., Danner R. L. (2003) J. Biol. Chem. 278, 29192–29200 [DOI] [PubMed] [Google Scholar]
- 48. Igarashi K., Kataoka K., Itoh K., Hayashi N., Nishizawa M., Yamamoto M. (1994) Nature 367, 568–572 [DOI] [PubMed] [Google Scholar]
- 49. Kornfeld R., Kornfeld S. (1980) The Biochemistry of Glycoproteins and Proteoglycans, p. 38, Plenum Press, New York [Google Scholar]
- 50. Spicer A. P., Augustine M. L., McDonald J. A. (1996) J. Biol. Chem. 271, 23400–23406 [DOI] [PubMed] [Google Scholar]
- 51. Spicer A. P., Olson J. S., McDonald J. A. (1997) J. Biol. Chem. 272, 8957–8961 [DOI] [PubMed] [Google Scholar]
- 52. Spicer A. P., Seldin M. F., Olsen A. S., Brown N., Wells D. E., Doggett N. A., Itano N., Kimata K., Inazawa J., McDonald J. A. (1997) Genomics 41, 493–497 [DOI] [PubMed] [Google Scholar]
- 53. Spicer A. P., McDonald J. A. (1998) J. Biol. Chem. 273, 1923–1932 [DOI] [PubMed] [Google Scholar]
- 54. Itano N., Kimata K. (2002) IUBMB Life 54, 195–199 [DOI] [PubMed] [Google Scholar]
- 55. Itano N., Sawai T., Atsumi F., Miyaishi O., Taniguchi S., Kannagi R., Hamaguchi M., Kimata K. (2004) J. Biol. Chem. 279, 18679–18687 [DOI] [PubMed] [Google Scholar]
- 56. Yamada Y., Itano N., Hata K., Ueda M., Kimata K. (2004) J. Invest. Dermatol. 122, 631–639 [DOI] [PubMed] [Google Scholar]
- 57. Smith H. S., Stern R., Liu E., Benz C. (1991) Basic Life Sci. 57, 329–337 [DOI] [PubMed] [Google Scholar]
- 58. Yeo T. K., Nagy J. A., Yeo K. T., Dvorak H. F., Toole B. P. (1996) Am. J. Pathol. 148, 1733–1740 [PMC free article] [PubMed] [Google Scholar]
- 59. Bourguignon L. Y., Singleton P. A., Zhu H., Zhou B. (2002) J. Biol. Chem. 277, 39703–39712 [DOI] [PubMed] [Google Scholar]
- 60. Turley E. A. (1992) Cancer Metastasis Rev. 11, 21–30 [DOI] [PubMed] [Google Scholar]
- 61. Toole B. P. (2004) Nat. Rev. Cancer 4, 528–539 [DOI] [PubMed] [Google Scholar]
- 62. Kakizaki I., Kojima K., Takagaki K., Endo M., Kannagi R., Ito M., Maruo Y., Sato H., Yasuda T., Mita S., Kimata K., Itano N. (2004) J. Biol. Chem. 279, 33281–33289 [DOI] [PubMed] [Google Scholar]
- 63. Mahaffey C. L., Mummert M. E. (2007) J. Immunol. 179, 8191–8199 [DOI] [PubMed] [Google Scholar]
- 64. Hascall V. C., Majors A. K., De La Motte C. A., Evanko S. P., Wang A., Drazba J. A., Strong S. A., Wight T. N. (2004) Biochimica et Biophysica Acta 1673, 3–12 [DOI] [PubMed] [Google Scholar]
- 65. Noble P. W., McKee C. M., Cowman M., Shin H. S. (1996) J. Exp. Med. 183, 2373–2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Vigetti D., Genasetti A., Karousou E., Viola M., Moretto P., Clerici M., Deleonibus S., De Luca G., Hascall V. C., Passi A. (2010) J. Biol. Chem. 285, 24639–24645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang L., Bowen T., Grennan-Jones F., Paddon C., Giles P., Webber J., Steadman R., Ludgate M. (2009) J. Biol. Chem. 284, 26447–26455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Guo N., Baglole C. J., O'Loughlin C. W., Feldon S. E., Phipps R. P. (2010) J. Biol. Chem. 285, 15794–15804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jokela T. A., Jauhiainen M., Auriola S., Kauhanen M., Tiihonen R., Tammi M. I., Tammi R. H. (2008) J. Biol. Chem. 283, 7666–7673 [DOI] [PubMed] [Google Scholar]