

Abstract
Network “guilt by association” (GBA) is a proven approach for identifying novel disease genes based on the observation that similar mutational phenotypes arise from functionally related genes. In principle, this approach could account even for nonadditive genetic interactions, which underlie the synergistic combinations of mutations often linked to complex diseases. Here, we analyze a large-scale, human gene functional interaction network (dubbed HumanNet). We show that candidate disease genes can be effectively identified by GBA in cross-validated tests using label propagation algorithms related to Google's PageRank. However, GBA has been shown to work poorly in genome-wide association studies (GWAS), where many genes are somewhat implicated, but few are known with very high certainty. Here, we resolve this by explicitly modeling the uncertainty of the associations and incorporating the uncertainty for the seed set into the GBA framework. We observe a significant boost in the power to detect validated candidate genes for Crohn's disease and type 2 diabetes by comparing our predictions to results from follow-up meta-analyses, with incorporation of the network serving to highlight the JAK–STAT pathway and associated adaptors GRB2/SHC1 in Crohn's disease and BACH2 in type 2 diabetes. Consideration of the network during GWAS thus conveys some of the benefits of enrolling more participants in the GWAS study. More generally, we demonstrate that a functional network of human genes provides a valuable statistical framework for prioritizing candidate disease genes, both for candidate gene-based and GWAS-based studies.
Causal genes remain extraordinarily difficult to identify in most genetic diseases and, in particular, in highly polygenic disorders for which current approaches are most limited (Bonetta 2008), identifying causal genes is a major barrier to progress in understanding these diseases. More generally, traditional linkage analyses have mapped causal genes for many diseases, often using positional cloning, but these methods are difficult and time-consuming (Altshuler et al. 2008). However, genome-wide association studies (GWAS) have opened the way to unbiased discovery of large numbers of disease genes in a more efficient manner.
A typical GWAS analysis involves comparing case and control individuals at selected single nucleotide polymorphisms (SNPs) or, more recently, copy number variants (CNVs). SNPs representing common haplotype blocks are measured genome-wide (at approximately 500,000–1,000,000 locations), and the disease-associated genetic markers are identified (for review, see McCarthy et al. 2008). The SNPs that show association strong enough to surpass a genome-wide significance threshold are then analyzed for chromosomal proximity to genes that might cause the disease, or otherwise affect its etiology. However, even though the data from GWAS support a great number of loci involved in common diseases, it is hard to separate many of the causal genes from the background noise of the hundreds of thousands of SNPs in the assay. Consequently, GWAS suffer from a lack of statistical strength, requiring large test populations to overcome the large multiple hypothesis correction needed in evaluating hundreds of thousands of candidate loci.
The lack of sufficient statistical power forces GWAS studies to ignore weaker loci, focus primarily only on the strongest genetic effectors, and genotype thousands of individuals (e.g., see Wellcome Trust Case Control Consortium 2007). Moreover, the combinatorial effects of multiple disease genes are often not simply additive, but epistatic (Liew and Dzau 2004; Pomp et al. 2004; Hirschhorn and Daly 2005), further hampering their discovery. Simply considering pairs of interacting loci increases the strength of associations required by orders of magnitude so as to be able to overcome the multiple testing criteria, requiring tens of thousands of individuals (Visscher 2008). Rarely has genetic association to allele triplets (or higher) been examined by these or any other approaches. Linear additive models have been successfully built, most notably for 54 alleles useful for predicting human height (Gudbjartsson et al. 2008; Lettre et al. 2008; Weedon et al. 2008), one of the first quantitative human traits successfully addressed to this degree. Finding these alleles nonetheless required genotyping ∼63,000 individuals over the course of three studies, each explaining <4% of the variance in height. Recent analysis of ∼300,000 SNPs, without regard to the significance of their association, demonstrated that a total of 45% of the variance in height could potentially be explained, with most effects too small to pass significance tests (Yang et al. 2010).
However, the polygenic nature of a disease may also offer potential opportunities to more efficiently discover new and relevant genes. In particular, we might expect that the genes associated with a disease will often organize into pathways or functional groupings linked to the disease formation and progression. Thus, knowing some disease genes in advance, it may occasionally be possible to apply guilt-by-association (GBA) in gene networks (for review, see Ideker and Sharan 2008). In particular, it is now possible to construct large gene network models, as has been done, e.g., for yeast, worms, plants, mice, and humans, summarizing thousands of functional associations among genes (for review, see Christensen et al. 2007; Lee et al. 2007b; Bonneau 2008; Feist et al. 2009). Gene pairs are coupled in these networks if they are inferred to participate in the same biological process (Marcotte et al. 1999) and may have corresponding measures of confidence (Troyanskaya et al. 2003; Lee et al. 2004; Rhodes et al. 2005; Alexeyenko and Sonnhammer 2009; Jensen et al. 2009). GBA in such networks has been shown to correctly identify disease and phenotype-linked genes based on their network connections to previously known genes (e.g., Fraser and Plotkin 2007; Lage et al. 2007; McGary et al. 2007; Lee et al. 2008, 2010a; Chen et al. 2009b; Huttenhower et al. 2009; Linghu et al. 2009), based on the observation that genes involved in a common biological process often tend be associated with similar mutational phenotypes (as seen, e.g., in Fraser and Plotkin 2007; Hart et al. 2007; Lage et al. 2007; Lee et al. 2008).
In principle, the GWAS-based association of genetic loci with a disease and the functional association of genes into pathways represent independent sets of observations that can be logically combined to improve identification of relevant disease genes. For example, networks have been applied to search for interacting loci in human GWAS data (Emily et al. 2009; Rossin et al. 2011) and in yeast (Hannum et al. 2009), to identify GWAS- and cancer genome-enriched pathways (Baranzini et al. 2009; Wu et al. 2010), and to rank genes in implicated chromosomal intervals (Franke et al. 2006; Pico et al. 2009; Wu et al. 2009). Other studies have looked at previously studied pathways for a disease, and tried to improve the ranking of the candidate genes using this information (e.g., Chang et al. 2008; Saccone et al. 2008; for review, see Wang et al. 2010b). Here, we have tested and expanded the general validity of the approach of using functional networks for prioritizing candidate disease genes. We propose a theoretical framework for combining the large-scale, unbiased pathway and association information encoded by functional gene networks and GWAS studies, respectively, showing improvements in performance as judged by data from GWAS meta-analyses.
First, we describe the construction of a functional network for human genes. This network spans 87% of validated protein coding genes, and provides strong predictive power for a majority of currently known genetic diseases. We evaluate six alternate approaches for prioritizing candidate disease genes using this network, and demonstrate the strongest overall performance with algorithms related to Google's PageRank. We then show that this network, in conjunction with genome-wide association data for Type 2 diabetes and Crohn's disease, boosts the identification of disease-associated genes that were discovered in later meta-analyses. This work suggests both a specific strategy and a general path to future improvements for the interpretation of GWAS data. Taken together, our work demonstrates that a high-quality functional network for human genes can provide a powerful resource for identifying causal genes in human disease.
Results
HumanNet: an extended functional gene network for H. sapiens
To test the ability of functional networks to improve gene association studies, we first constructed a genome-scale functional network for human genes. Diverse distinct lines of evidence, spanning human mRNA coexpression, protein–protein interactions, protein complex, and comparative genomics data sets, in combination with similar lines of evidence from orthologs in yeast, fly, and worm, were analyzed using an approach previously developed and validated for yeast (Lee et al. 2004, 2007a), C. elegans (Lee et al. 2008, 2010b), and Arabidopsis (Lee et al. 2010a). In total, 21 large-scale genomics and proteomics data sets from the four species (see Methods; Supplemental Tables S1, S2) were integrated into a functional gene network spanning 476,399 scored functional couplings between 16,243 (87%) of validated human protein encoding genes (Fig. 1A).
Figure 1.

Construction and evaluation of a genome-scale human gene network, HumanNet. (A) 21 diverse functional genomic and proteomic data sets (Supplemental Table S1) were evaluated for their tendencies to link genes in the same biological processes. Pairwise gene linkages derived from the individual data sets were then integrated into a composite network of higher accuracy and genome coverage than any individual data set. The integrated network (HumanNet) contains 476,399 functional linkages among 16,243 (86.7%) of the 18,714 genes encoding validated human proteins. The x-axis indicates the log-scale percentage of the 18,714 genes covered by functional linkages derived from the indicated data sets (curves); the y-axis indicates the predictive quality of the data sets, measured as the cumulative log likelihood of linked genes to share Gene Ontology (GO) biological process annotations, tested using 0.632 bootstrapping and plotted for successive bins of 1000 linkages each (symbols). Data sets are named as XX-YY, where XX indicates species of data origin (CE, C. elegans; DM, D. melanogaster; HS, H. sapiens; SC, S. cerevisiae) and YY indicates data type (CC, co-citation; CX, mRNA coexpression; DC, domain co-occurrence; GN, gene neighbor; GT, genetic interaction; LC, literature-curated protein interactions; MS, affinity purification/mass spectrometry; PG, phylogenetic profiles; PI, fly protein interactions; TS, tertiary structure; and YH, yeast two-hybrid). Detailed descriptions are listed in Supplemental Table S1. (B) Essential genes were highly interconnected in HumanNet, and thus predictable from the network, as shown by ROC analysis. Genes were ranked by their sum of network edge weights to the known essential genes, measuring recovery of known essential genes (true positives) and other genes (false positives) using leave-one-out cross-validation. (C) Genes involved in more specific cellular phenotypes—host factors required for HIV infection (HDF) (Brass et al. 2008), modulators of OCT4 (also known as POU5F1) expression (Oct4-GI) (Ding et al. 2009), and synthetic lethal partners of activated KRAS alleles (KRAS-SL) (Luo et al. 2009)—were also well predicted by their interconnectivity in HumanNet, calculated as for B. (D) Finally, network-linked gene pairs were substantially more likely to show similar tissue specificity in their expression patterns, measured as the likelihood of co-occurrence of transcripts of pairs of genes in the same tissues across 30 different human tissues from the TiGER database of tissue-specific gene expression and regulation (Liu et al. 2008).
Table 1.
Selected top-ranked Crohn's disease and type 2 diabetes genes for which network data added support to GWAS evidence, measured as an increase in odds (prior = −1.7 for each)

HumanNet predicts cellular loss-of-function phenotypes
To evaluate the predictive power of the new network, we first examined cellular-level phenotypes. Many human diseases reflect failures of core cellular machinery, e.g., failures of metabolism, DNA repair, replication, etc. For example, hereditary nonpolyposis colorectal carcinoma arises from mutations in DNA mismatch repair (Fishel et al. 1993; Miyaki et al. 1997), Zellweger syndrome arises from mutations in peroxisome biogenesis (Moser et al. 1995), and leukoencephalopathy with vanishing white matter arises from mutations in any of the subunits of translation initiation factor EIF2B1 (Leegwater et al. 2001; van der Knaap et al. 2002). A network for even a single eukaryotic cell will capture many of these basic processes and has the potential to prove predictive for genes for diverse human diseases. We therefore investigated whether the human gene network was predictive of cellular-level mutational phenotypes, focusing on cell survival and proliferation phenotypes from loss-of-function studies in cell culture.
We first asked whether genes essential to cell viability could be accurately identified using the gene network. Schlabach and colleagues identified about 600 genes that affect the viability and proliferation of normal human mammary epithelial cells (HMEC) by using multiplex short hairpin RNA (shRNA) screening (Schlabach et al. 2008). Although assayed largely for proliferation defects, these genes are highly likely to be essential for HMEC cell growth, given the incompletely penetrant phenotype induced by shRNA knockdown (Chang et al. 2006).
We found that the essential HMEC genes were, indeed, highly connected in HumanNet (Fig. 1B), as assessed by cross-validated receiver operating characteristic (ROC) analysis (see the Methods section). For example, ∼18% of all known essential genes, but only 2% of all genes not known to be essential, are connected to known essential genes in HumanNet, a ninefold enrichment. From these results we conclude that essential genes can be predicted on the basis of their connectivity to other essential genes in HumanNet.
This general level of predictability was also observed for more specific cellular phenotypes. We tested whether genes known to be required for HIV infection, as measured by large-scale RNAi knockdown (Brass et al. 2008), were predictable by guilt-by-association in HumanNet. Indeed, they showed a moderate degree of predictivity, at a level significantly higher than random chance (Fig. 1C).
The essentiality and viral infectivity phenotypes described above are single gene phenotypes, but yeast and worm gene networks have also proven generally predictive for bigenic phenotypes, such as synthetic genetic interactions (e.g., Lee et al. 2010b). We therefore next asked whether the human gene network could predict genetic interactions, focusing on two large-scale RNAi screens performed in mammalian cell culture. The first screen identified genes modulating expression of a core stemness regulator OCT4 (also known as POU5F1) in mouse embryonic stem cells (Ding et al. 2009). The second found genes acting as synthetic lethal interaction partners with oncogenic KRAS mutants expressed in a colorectal cancer cell line, screening for genes whose knockdown in the activated KRAS background resulted in cellular lethality (Luo et al. 2009). In both cases, genes identified by the screens were well predicted by guilt-by-association in HumanNet at rates significantly higher than random expectation (Fig. 1C). The high predictive strength (AUC = 0.81) for KRAS interactors is particularly notable, as such genes might be useful as cancer cell-specific drug targets (Luo et al. 2009). More generally, these tests confirm that the human gene network is predictive of a variety of cellular level loss-of-function phenotypes, including specific bigenic traits.
Genes linked to specific mouse mutational phenotypes and human diseases are predictable by guilt-by-association in the network
The cellular-level results demonstrate that genes for cell viability and proliferation phenotypes can be identified based on network connectivity in HumanNet. A further trend for genes linked in the network to share tissue-specific expression patterns (Fig. 1D) implies that the network could potentially predict more specific organism-level mutational phenotypes as well. This notion has previously been explored for human diseases by considering network connections among known disease genes, prioritizing the genes most highly connected to the known causal genes as being likely new candidate genes for that disease (Fraser and Plotkin 2007; Lage et al. 2007; McGary et al. 2007; Linghu et al. 2009) as illustrated in Figure 2A. Such approaches primarily consider direct network connections to known disease genes, but related work on predicting gene function from networks (for review, see Sharan et al. 2007; Stolovitzky et al. 2007) has shown wide benefits of also appropriately considering indirect network connections (e.g., as in Mostafavi et al. 2008), and tests have confirmed the utility of these so-called network diffusion algorithms for predicting RNAi phenotypes in worms and loss-of-function phenotypes in yeast cells (Wang and Marcotte 2010). Here, we implemented a representative set of both types of algorithms, collectively termed label propagation algorithms and chosen by their successful application in yeast and worm networks (Wang and Marcotte 2010) for inferring disease genes based on network connectivity, evaluating them for their overall predictive ability using cross-validation and ROC analysis.
Figure 2.

Network-guided prediction of genes for transgenic mouse phenotypes and human diseases. (A) A schematic figure of network-guided prioritization of candidate disease genes. Given some known disease genes (black nodes), additional genes can be predicted by their (weighted) associations in the network, with more strongly connected genes being prioritized more highly (node shading). (B) Known genes associated with several human diseases are well predicted by the Iterative Ranking method for propagating disease labels across HumanNet, as measured using cross-validated ROC analysis. In this and later GBA analyses, we used leave-one-out cross-validation for phenotype sets of 3–10 genes and 10-fold cross-validation for all other sets. The performance can be summarized as the area under the ROC curve (AUC), ranging from 0.5 (random) to 1.0 (perfect). (C) Network GBA predictability of genes associated with 3374 transgenic mouse phenotypes. Bar-and-whiskers plots summarize the predictive performance (measured as cross-validated AUC) for each of six algorithms for using HumanNet to prioritize candidate disease genes. The Iterative Ranking and Gaussian smoothing approaches outperform the others by a significant margin, and show generally high predictability for more than three-quarters of the phenotypes tested. In bar-and-whiskers plots, the central horizontal line in the box indicates the median AUC, and the boundaries of the box indicate the first and third quartiles of the AUC distribution, whiskers indicate the 10th and 90th percentiles, and plus signs indicate individual outliers. The mean AUC is plotted as a dashed blue horizontal line. (D) A related analysis of human disease genes, assembled for 263 diseases from the OMIM database (http://omim.org/), shows similarly strong prediction strengths and the same relative ranking of algorithm performance.
Specifically, we considered six methods of network label propagation. The first are two methods that consider only direct network neighbors: (1) neighbor counting (Schwikowski et al. 2000), in which the genes with the most neighbors already linked to the disease are most highly scored, and (2) naive Bayes label propagation, in which the sum of the HumanNet linkages to implicated neighbors is used rather than their count (Lee et al. 2007a), corresponding to the naive Bayes estimate for a gene to participate in the same process as the known disease genes. We further considered four methods that “diffuse” disease associations across the network, considering both direct and indirect connections, similar to the methods considered in Chen et al. (2009a). Two of these are mathematically related to Google's PageRank algorithm: (3) the Iterative Ranking method, in which a gene's score is calculated from an initial score and the normalized scores of its neighbors, which, when updated over successive iterations, “smear” across the network linkages (Ramakrishnan et al. 2009; Wang and Marcotte 2010), and (4) Gaussian field label propagation (Gaussian smoothing, for short), in which the difference between a gene's initial and final scores and the weighted score difference between a gene and its neighbors are simultaneously minimized (Mostafavi et al. 2008). Finally, we considered (5) a clustering approach, using Markov clustering of genes based on simulation of stochastic flow in the network (Enright et al. 1999), followed by ranking of each gene within a cluster for relevance by considering the sum of the gene's edge-weights within the cluster relative to all of its edge-weights (Wang and Marcotte 2010), and (6) a model based on electrical circuits (Suthram et al. 2008), in which network edge weights are considered to be analogous to electrical conductance and disease-implicated proteins are considered as ground nodes; candidate nodes are identified by modeling the application of current to the resulting circuit and measuring which nodes have the highest modeled current flow.
Figure 2B shows examples of ROC curves associating genes with several human diseases using the Iterative Ranking approach, showing high predictability for these cases. In order to systematically test whether such predictability was common, and in order to judge the relative merits of the network diffusion approaches, we next evaluated a more comprehensive set of mouse phenotypes and human diseases.
We first evaluated the predictive power of HumanNet for genes associated (via orthology) with mouse mutational phenotypes, drawing upon the over 3000 well-annotated gene–phenotype associations identified in gene knock-out, gene trapping, and chemical mutagenesis experiments, and cataloged in the Mouse Genome Database (MGD) database (Eppig et al. 2005). In order to minimize the risk of circular predictions, we performed the tests using a version of the network lacking human literature-based linkages (i.e., no linkages by HS-CC or HS-LC). For each of the six approaches, we measured the network predictability for these mouse phenotypes using cross-validated ROC curve analysis, plotting the distributions of AUC (area under the ROC curve) scores for 3374 gene sets associated with mouse phenotypes in Figure 2C. HumanNet shows broad predictive ability of genes associated with specific mouse phenotypes and is significantly better than expected by chance using each of the six algorithms. However, the closely related Gaussian smoothing and Iterative Ranking approaches perform comparably to each other and significantly better than the other four approaches, indicating that there is a clear benefit to considering indirect connections as well as direct network connections.
Unlike mouse phenotypes, annotations for human disease genes are still extremely limited, spanning ∼3000 gene-disease linkages in human versus nearly 100,000 in mouse (McGary et al. 2010). From annotations available at The Mendelian Inheritance in Man (OMIM) database (http://omim.org/), we selected 263 diseases with at least three associated genes. We tested the network's ability to associate genes with each of the 263 diseases using cross-validated ROC analysis, testing each of the six approaches, just as we did for mouse phenotypes (and again, using the version of the network lacking human literature-based linkages in order to avoid any potential circularity). We observed strong predictability for the human genetic diseases, with many disease gene sets predicted to high accuracy based upon gene–gene associations in the network (Fig. 2D). Again, the Iterative Ranking and Gaussian smoothing approaches performed similarly well and significantly better than the other four approaches, confirming the general applicability of network label propagation for associating genes with human diseases and organism-level phenotypes.
Data from diverse sources is used to predict disease genes
We further investigated how the various data sets derived from high-throughput experiments and model organisms contribute to the mouse and human phenotype predictions. We examined predictions made by direct network connections using the naive Bayes analysis and excluding the human literature-derived HS-LC and HS-CC data sets as for analysis in Figure 2, C and D. These contributions are visualized for the 20 most predictable mouse phenotypes and human diseases in Figure 3. Notably, data sets from worm and fly were strong contributors to the prediction of mouse phenotypes, as were data from human mRNA coexpression patterns (Fig. 3A). Likewise, diverse data sets from yeast were strong contributors to a variety of well-predicted human diseases (Fig. 3B). This demonstrates that most data sets contribute to the predictions, supporting the importance of data integration for effective disease gene identification.
Figure 3.

The predictive power for loss-of-function phenotypes stems from a wide variety of data types integrated into HumanNet. Prediction both of (A) genes associated with mouse phenotypes and (B) of genes associated with human diseases are supported by diverse lines of evidence, including, for example, fly and worm data contributing strongly to mouse phenotypes, and yeast data contributing to human diseases. Grayscale indicates percentage contribution of a given data set's naive Bayes score to a phenotype's total AUC.
Combining evidence from network guilt-by-association and genome-wide association studies
Given that network GBA is strongly predictive of human disease genes, a potentially powerful application of this approach is to combine the network GBA with the data from GWAS for direct discovery of human disease genes from patient and control populations. In order to use the information encoded by HumanNet, our method takes a slightly different approach from the SNP level tests used in the statistical analysis of GWAS today. Instead of focusing on single SNPs, we try to identify which genes and pathways might be involved in the disease. There are a number of reasons for this. First, even the SNPs that are identified in the traditional analysis are rarely thought to be the causal variants underlying the disease. This is due to the fact that the polymorphisms measured by GWAS have been chosen not for their biological significance, but for being the most informative of the surrounding region of the genome. Second, only a very small fraction of the genetic heredity of most diseases studied so far can be explained by the SNPs identified by GWAS (Park et al. 2010). This might be because a very large number of genes are involved in the diseases, or it might be because rarer variants cause a greater fraction of the heredity than previously thought (Dickson et al. 2010; Wang et al. 2010a). If it is due to the latter, we need to identify the regions of the genome where these rare mutations are located so that our search for such variants can be as efficient as possible. Our goal then is to identify genes and pathways of genes involved in the disease, not the marker SNPs most strongly correlated with the disease. Third, by taking a gene-centric approach, we can use the information encoded by HumanNet to improve our predictions. Finally, by working on the level of genes instead of SNPs, the method generalizes to future sequencing data, as long as the genetic variation can be associated with nearby genes.
If a GWAS finds a highly significant gene, it makes sense to attempt to identify the causal mechanism by which this gene influences the disease by looking at which pathways proteins encoded by this gene are active in. For example, this strategy leads to β-catenin expression and WNT signaling as a likely mechanism by which TCF7L2 influences type 2 diabetes (for review, see Jin and Liu 2008). By performing this type of pathway analysis automatically, it might also be possible to uncover genes that would not otherwise easily be found. This is especially true for genes that fall just under the threshold of significance for the GWAS study, but which might be “rescued” by considering their interactions with the confident genes. Recent evidence for the case of human height shows that such minor contributions are common from polymorphisms falling below the significance threshold for association, but nonetheless contributing to total variation (Yang et al. 2010).
Unlike the GBA analyses considered above, for GWAS data, definite seed genes can rarely be found, particularly for the case where the only evidence for disease association comes from the GWAS itself. In order to make use of the information from the genes that are on the verge of being statistically significant, we implemented a “soft category assignment” for the GBA, where only genes that show a very strong signal are given full weight in the GBA. Notably, the performance of guilt-by-association in HumanNet is independent of the number of genes linked to the phenotype (Supplemental Fig. 1), which means that by varying the parameter that assigns weight in the GBA, we can include successively more genes that are increasingly less likely to truly be involved in the disease. We chose to base our method on the naive Bayes GBA rather than the Iterative Ranking or Gaussian smoothing methods, since naive Bayes gave superior recall in the highest precision regime (Supplemental Fig. 2), and the log odds output of the naive Bayes can be combined with the log odds from the GWAS in a natural way.
We developed the following scoring scheme: Let Si denote the total GBA score for a gene i, and denote by pj the probability that some other gene j is involved in the disease. Suppose that j is connected in the functional network to i by a link of strength lij . It would then be natural to assign a “soft” GBA contribution from gene j to gene i by
which gives the total “soft” score Si of gene i as
This gives very poor results in practice, most likely because the network is only built on positive evidence. However, by only keeping positive contributions, we observe good empirical results. Our “soft” GBA score is therefore summed only over those j where 2pj-1 > 0. Note that another natural way to take into account the fact that the network is built only on positive evidence would be to use pj as a weight instead of 2pj−1. In practice, however, this does not work well (data not shown).
If we assume that the data from the GWAS and the data for the network are conditionally independent given the set of genes that are involved in the disease, we can again integrate them in a naive Bayes framework. The posterior log odds that gene i is involved in the disease are then
where 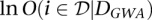 is the log odds of association calculated from the GWAS data, corresponding to the log Bayes factor for association with the disease plus the prior log odds for association. In general, the prior log odds of association can be thought of as a proxy for the number of genes believed to be involved in the disease; in practice, we see significant boosts in performance over a wide range of values for the prior log odds.
is the log odds of association calculated from the GWAS data, corresponding to the log Bayes factor for association with the disease plus the prior log odds for association. In general, the prior log odds of association can be thought of as a proxy for the number of genes believed to be involved in the disease; in practice, we see significant boosts in performance over a wide range of values for the prior log odds.
Considering network linkages increases the power of genome-wide association studies
To evaluate whether the genes highlighted by this method actually are genes that are biologically relevant to diseases, we used ROC analysis to compare how highly the combined GWAS/GBA method ranks the top candidates from meta-analyses for type 2 diabetes and Crohn's disease (Barrett et al. 2008; Zeggini et al. 2008), versus how highly those same genes are ranked by the Wellcome Trust study by itself (Wellcome Trust Case Control Consortium 2007). These meta-analyses contain the Wellcome Trust data used for the predictions, but also incorporate data from a number of similar size studies, and have higher statistical power. For both type 2 diabetes and Crohn's, the Wellcome Trust study considered about 2000 cases and 3000 controls. For Crohn's, the meta-analysis considered 3230 cases and 4829 controls; for diabetes, 4549 cases and 5579 controls. To confirm that it really is the incorporation of the information encoded by the network that improves our predictions, we also compared these results with 200 randomly shuffled networks. As shown in Figure 4A and Figure 5A, the combined GWAS/GBA method clearly improves the ranking of the genes for both diseases, and does so over a wide range of parameter settings for the prior parameter.
Figure 4.

Consideration of the human gene network boosts recovery of validated Crohn's disease genes from GWAS analysis of 2000 cases and 3000 controls. (A) The performance improvement achieved by network-boosted GWAS relative to GWAS alone (Wellcome Trust Baseline, [Wellcome Trust Case Control Consortium 2007]), measuring performance as the area under a ROC curve up to 5% false positive rate (AUC, <5% FPR) for recovering the top 22 Crohn's disease genes identified in a larger meta-analysis of 4549 cases and 5579 controls (Barrett et al. 2008). For the AUC (<5% FPR) measure of performance, a perfect predictor achieves a score of 0.05, while random predictors score near 0.00125. The network boosted approach (colored red line) outperforms the GWAS alone (straight dashed blue line) over a wide range of parameter values. For comparison we also show the results of network boosting when randomized networks are used, plotting the mean (dotted line) and range of performance (2 SD) for 1000 random trials. B plots the network of candidate genes (rounded rectangles) identified from the combination of HumanNet and GWAS data, visualized using Cytoscape (Cline et al. 2007). The node size corresponds to the strength of the combined evidence from the Wellcome Trust Case Control Consortium (WTCCC) data and the network, and the intensity of the red color indicates how much the gene was boosted by the HumanNet GBA. HumanNet linkages are drawn as directed arrows connecting genes, with edge weight scaled by strength of boost contributed by the source to the sink. All genes are drawn with positive posterior log-odds when the prior log-odds of association are −1.7, except for network singletons, and the 50 highest scoring nonsingleton genes are shown. Note the strong boost given to GRB2 and SHC1, which are known to be involved in healing gastric ulcers (Pai et al. 1999), and to JAK2 and STAT3, which were also identified in later meta-analyses (Van Limbergen et al. 2009).
Figure 5.

Consideration of the human gene network boosts recovery of validated type 2 diabetes genes from GWAS analysis of 2000 patients and 3000 controls. (A,B) Plotted using the same conventions as in Figure 4, analyzing WTCCC GWAS data (Wellcome Trust Case Control Consortium 2007) for type 2 diabetes alone and in combination with HumanNet and measuring performance as AUC (<5% FPR) for recovering the top 20 genes from a type 2 diabetes meta-analysis of 4549 cases and 5579 controls (Zeggini et al. 2008). As for Crohn's disease, consideration of the network boosts performance across a wide range of parameter values. Notably, consideration of the network strongly implicates the genes CTNNB1 and BACH2 in type 2 diabetes; CTNNB1 is well studied in connection with type 2 diabetes and BACH2 has been previously implicated in type 1 diabetes and celiac disease (e.g., Cooper et al. 2008; Madu et al. 2009), but not type 2 diabetes.
Genes boosted in Crohn's
Prior to the Wellcome Trust study, strong association signals for Crohn's disease had been observed in NOD2, IL23R, ATG16L1, ZNF365, and in 5q31 and the gene desert, 5p13.1. Furthermore, the Wellcome Trust study identified four more strong associations that were replicated in follow up studies. These were IRGM; a locus on chromosome 3 containing BSN, MST1, MST1R, TRAIP, and some other genes; NKX2-3; and finally, PTPN2. Moderate association was also seen in the regions 1q24, 5q23, 6p22, 6p21, 6q23, 7q36, 10p15, and 19q13, which contain a number of plausible candidate genes, such as STAT3 and TNFAIP3.
Using the evaluation method described above, we saw a distinct increase in the top portion of the ROC curve for a wide range of values for the prior parameter from −3.5 to −1.5 (Fig. 4A). Using −1.7 as our value for the prior parameter, we then surveyed the gene groups that had strong network support. Interestingly, many of the gene clusters that emerged in this analysis showed strong connections with TNF signaling, which suggests multiple points of failure for the TNF pathway in Crohn's disease. We note that one of the most successful drugs against Crohn's disease is the TNF antibody Infliximab.
IL23R, STAT3, IL12RB2, and JAK2 have all been indicated as candidate genes for Crohn's disease, probably affecting the disease through their involvement in the differentiation of Th17 cells (Van Limbergen et al. 2009). These are strongly connected in our network, and therefore boost each other's rankings. For our choice of the free prior parameter, STAT3 gets bumped from rank 17 to 8, and JAK2 from rank 3139 to 38 (Table 1). Many of these are functionally connected in our network to both the gene coding for adaptor protein GRB2 (rank 99 to 7) and to its interaction partner SHC1 (6125 to 17). GRB2 and SHC1 are also involved in gastric ulcer healing (Pai et al. 1999). GRB2 and SHC1 are furthermore supported by their functional interactions with PTPN2 and MST1, which probably affect Crohn's disease via their roles in the orchestration of the secondary immune response (Van Limbergen et al. 2009). Lastly, GRB2 is a binding partner to TNFRISF1A, TNF receptor type I, which can mediate a majority of TNF-dependent activities (Hildt and Oess 1999). All of this, taken together, indicates that GRB2-SHC1 warrants further study as disease candidate genes for Crohn's disease.
The cluster containing CYLD, TRAIP, and TRAF1 could also show a mechanism of action for Crohn's disease candidate genes. CYLD is located next to NOD2 on chromosome 16. However, Cyld−/− knockout mice have an irritable bowel disorder phenotype (Reiley et al. 2007), and CYLD has been shown to interact with TRAIP (TRAF interacting protein) by yeast two-hybrid screens (Regamey et al. 2003). TRAIP is located in the 3p21 locus, which contains multiple independent signals for association with Crohn's disease (Beckly et al. 2008). Both of these genes are connected in HumanNet to TRAF1, TNF receptor-associated factor 1, which is involved in TNF signaling and NF-kappaB signaling.
We also see encouraging support of already known loci—TNFRSF6B and TNFSF15 are both known to be involved in Crohn's disease, and they are connected in HumanNet.
Another interesting gene association is given by ATG16L1 and CAPN9, which boost each other. ATG16L1 is involved in autophagy and has been implicated in multiple GWAS. CAPN9 is a stomach-specific calpain, and mouse Capn9−/− knockouts are sensitive to gastric mucosal injury induced by ethanol administration (Hata et al. 2010). This, together with the connection to ATG16L1, indicates that this is another plausible candidate gene for Crohn's disease.
Genes boosted in type 2 diabetes
Before the Wellcome Trust study, PPARG, KCNJ11, and TCF7L2 had all been identified as genes involved in type 2 diabetes through genome-wide association studies and replicated in follow-up studies (for review, see Bonnefond et al. 2010). The strongest candidate gene for type 2 diabetes, TCF7L2, was also the strongest signal seen in the Wellcome trust study, although the others were not so strong. However, the exact mechanism by which TCF7L2 acts was not entirely clear. In our analysis (Fig. 5), we find it directly connected to the β-catenin/WNT signaling pathway by its functional connection to CTNNB1, as well as to BACH2, a gene that has been repeatedly implicated in type 1 diabetes (e.g., Cooper et al. 2008; Madu et al. 2009), but which has not yet been linked to type 2 diabetes. BACH2 is among the genes most strongly boosted by network linkages, deriving additional signal from CREB5 and PARD3B, which both score highly in the GWAS data. PARD6G, PARD3B, and CDC42 are also emphasized by the method. Notably, these genes form a complex with PRKCZ (Koh et al. 2008), a variant of which correlates with type 2 diabetes in Han Chinese (Qin et al. 2008). EBF1, a known regulator of adipocyte differentiation (Akerblad et al. 2005) is also strongly boosted by the network, supporting a possible role in type 2 diabetes.
Thus, for both Crohn's disease and type 2 diabetes, the combined GWAS/GBA approach both boosts genes that have support in other populations and that have been replicated in later meta-analyses and highlights new connections between functionally connected genes among the genes that show moderate association to the disease.
Discussion
A new functional gene network for human genes
In order to test the general ability of a gene network to prioritize disease genes, particularly in conjunction with GWAS studies, we constructed a genome-scale functional network of human genes, incorporating diverse expression, protein interaction, genetic interaction, sequence, literature, and comparative genomics data, including both data collected directly from human genes, as well as that from orthologous genes of yeast, worm, and fly. The resulting HumanNet gene network can be accessed through a web interface (http://www.functionalnet.org/humannet). Using this interface, researchers can easily search the network using a set of “seed” genes of interest. The interface returns a list of genes ranked according to their connections to the seed genes, together with the evidence used to identify each coupling. The interactions and evidence can be downloaded, and a network visualization tool has been incorporated. All linkages can also be downloaded for independent analysis.
Functional networks provide a general strategy for prioritizing disease genes
We demonstrate here that connectivity of human genes in an integrated functional network is a strong predictor of disease genes, both for cellular phenotypes and for diseases at the level of the whole organism. This predictability is strong even when considering only direct network connections, as shown both here and by related previous work (e.g., Fraser and Plotkin 2007; Lage et al. 2007; McGary et al. 2007; Linghu et al. 2009). We further show that algorithms developed originally for predicting gene function using gene networks also perform well at prioritizing candidate disease genes. Importantly, the consideration of indirect connections in diffusion algorithms, such as Iterative Ranking (Ramakrishnan et al. 2009) and Gaussian smoothing (Mostafavi et al. 2008), greatly improves the correct identification of disease genes. Thus, knowing a few genes implicated in a disease, the networks offer a strong tool for prioritizing additional likely candidate genes.
One primary limitation of this approach is that genes must already be affiliated with the disease in order to predict new candidates. Typically, these seed genes would come from prior studies. However, we demonstrate that the approach is still valuable when used in combination with GWAS data, where no genes are definitively associated with the disease.
Recent work also demonstrates that functional networks inworm and yeast can successfully predict genetic modifiers of genes (Lee et al. 2010b) using the same network guilt-by-association approach. The effectiveness of this strategy in yeast and worms strongly supports using a human gene network in the same manner to predict genes of synthetic or epistatic phenotypes. While relatively few such genetic interactions are known currently among human genes (Flint and Mackay 2009) as compared with the cases for yeast (e.g., Tong et al. 2001, 2004; Davierwala et al. 2005; Costanzo et al. 2010) or worm (Lehner et al. 2006; Byrne et al. 2007), functional gene networks offer a potential directed strategy for expanding current sets of human genetic interactions by prioritizing the tested interactions using gene networks, and our preliminary results demonstrating prediction of KRAS and OCT4 modifiers (Fig. 1C) support such an approach.
Tissue specificity profiles are shared by linked genes
One important characteristic of HumanNet is the tendency for linked genes to share specificity of expression in distinct tissues (Fig. 1D). The observation of tissue specificity embedded in networks is consistent with our expectation for colocalization of proteins in the same functional modules (e.g., protein complexes and pathways) in specific cell types. However, this is nonetheless notable, since many of the raw data sets for network construction were not themselves tissue specific. For example, yeast-two-hybrid (Y2H) interactions are tested not in human cells but in yeast cells and, in fact, linkages derived only from Y2H do not show high tissue specificity (data not shown). Similarly, the phylogenetic profiling and gene neighbor comparative genomics approaches are strictly based on analysis of genome sequences and make no reference to tissue expression, nor do, for example, linkages inferred by homology from yeast. This trend for linked proteins in a genome-wide functional gene network to share tissue specificity has also been previously observed for worm and Arabidopsis gene networks (Lee et al. 2008, 2010a), and thus seems to be a result of the training process and integration of multiple data types correctly capturing the sorts of functional relationships reflected by the tissue specificity. A practical consequence is that a single genome-wide network of genes is nonetheless able to successfully implicate genes in tissue- and cell-type-specific disorders as, for example, the case of liver cirrhosis genes, which are well predicted (AUC = 0.88) (Fig. 2B).
Network-aided association studies: A general strategy for prioritizing genome-wide associations in human disease
The success of our approach suggests that analysis of GWAS data sets using gene networks offers a useful strategy for identifying both directly causal genes and even potential modifier loci in human disease, and since neither the pathway information encoded by the network nor the disease-association likelihoods that come out of the GWAS make any prior assumptions about the disease studied, this strategy is free from the study design bias that is inherent in candidate gene or candidate pathway analyses. The altered prioritization offered by the network-based association approach has the effect of shifting attention for follow-up studies to those genes (not SNPs) that are both best supported independently, and most likely to impinge upon the process(es) that are themselves best supported by the GWAS data, as determined from the current state of biological knowledge that has been objectively reconstructed and summarized in the gene network. Since this technique is gene focused and not SNP focused, it can be used with any future sequencing technology as long as the genetic variations can be associated with genes. In our analyses of Crohn's disease and type 2 diabetes, the network boosted identification of correct associations by ∼10% (measured in area under the first 5% of the ROC curve), which translated in practice to one to two genes more for these cases, a statistically significant, but not large effect. However, the organization of the associated genes into processes offered a large practical benefit, such as focusing attention to BACH2, CTNNB1, and EBF1, which were not well-supported by the type 2 diabetes GWAS, but which were prominent network connectors between the well-supported genes. Furthermore, this boost is an effect of using the full network; individual sources of data do not provide nearly the same coverage and accuracy as the integrated network, and the kinds of data that is informative varies for the two different diseases studied (data not shown).
A second overall strategy also presents itself for integrating GBA and GWAS data sets, that of a candidate gene-based approach: It seems quite feasible to use GBA to known causal genes in order to select additional candidates, then to evaluate those candidate genes in a directed fashion, either by interrogating the GWAS data for associations involving these loci, or by directed sequencing of the candidate genes in patient populations. By focusing only on those genes ranked highly by GBA, the multiple testing explosion of typical GWAS is eased considerably, allowing for smaller patient samples to be tested and easier statistical significance thresholds to meet.
Concluding remarks
In summary, the approach outlined here provides a general method for prioritizing human disease genes, both for the case where seed genes associated with the disease are known already, and for the case where no such seed genes are known, but GWAS data for the disease is available. Our results suggest that the network will be useful for a considerable fraction of human diseases with genetic components, and thus provides a general resource for diverse genetic diseases.
Methods
Construction of HumanNet
This study is based on 18,714 human Entrez genes with validated coding proteins (downloaded from NCBI; March 2007). Gene functional associations were trained using a reference set of gene pairs sharing Gene Ontology (GO) biological process annotations (downloaded from NCBI; March 2007). We used only annotations supported by experimental evidence: IDA (inferred from direct assay); IMP (inferred from mutant phenotype); IPI (inferred from protein interaction); IGI (inferred from genetic interaction); and TAS (traceable author statement). To minimize training bias, we excluded highly over-represented annotations: (1) “signal transduction” (GO:0007165) (this term alone would otherwise account for 38% of total positive reference gene pairs); (2) three additional phosphorylation terms that have highly diverse biological roles, “protein amino acid phosphorylation” (GO:0006468), “protein amino acid autophosphorylation” (GO:0046777), and “protein amino acid dephosphorylation” (GO:0006470); and (3) all terms at the first and second levels of the GO hierarchy (assuming the term “biological process” is level zero). The resulting data set of 270,704 reference gene pairs covers 5369 (29%) human genes.
Functional associations were learned (as described in detail in the Supplemental Methods) in a supervised training framework using the log likelihood scoring (LLS) scheme of (Lee et al. 2008, 2010a), monitoring overtraining with 0.632 bootstrapping as in Lee et al. (2008). Gene associations from each separate data set described below were optimized to maximize performance as measured by precision-recall analysis, in accord with the rationales presented in Lee et al. (2008, 2010a). Multiple LLS for each gene pair were integrated using the weighted sum method with linearly decaying weights as in (Lee et al. 2008).
Analysis of tissue-specificity of network linkages
The similar tissue specificity of linked gene pairs was measured as the likelihood of co-occurrence of transcripts of pairs of genes in the same tissues, calculated as likelihood score = 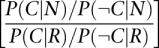 , where
, where 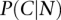 and
and 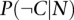 are probabilities that genes connected by the network (N) are coexpressed (C) and not coexpressed
are probabilities that genes connected by the network (N) are coexpressed (C) and not coexpressed 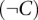 in the same tissue.
in the same tissue. 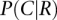 and
and 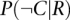 represent similar calculations based on randomized networks (R), repeating calculations for 100 randomized networks. As a reference for tissue-specific expression, we collected 5018 tissue-specific genes and their expression profiles across 30 different human tissues from the TiGER database of tissue-specific gene expression (Liu et al. 2008).
represent similar calculations based on randomized networks (R), repeating calculations for 100 randomized networks. As a reference for tissue-specific expression, we collected 5018 tissue-specific genes and their expression profiles across 30 different human tissues from the TiGER database of tissue-specific gene expression (Liu et al. 2008).
Implementation of network guilt-by-association algorithms
The naive Bayes GBA algorithm was implemented as previously described (Lee et al. 2008). Briefly, a gene score consists of the sum of LLSs to seed genes. For neighbor counting, the LLS sum is simplified to a count of neighboring seed genes. For Markov clustering, MCL software was downloaded from www.micans.org/mcl (van Dongen 2000; Enright et al. 2002). We obtained network clusters using the default granularity settings. The final score for a gene consists of the sum of the gene's maximal coverage scores to clusters containing seeds. The coverage score is an MCL measure, comprised of the sum of edge weights from a node to a cluster, with larger edge weights rewarded. To obtain random scores for a phenotype set, we randomly selected from the genome a set of seeds of the same size, and performed naive Bayes GBA as before.
The following methods were implemented in Matlab: GeneMANIA Gaussian field label propagation (Gaussian smoothing) was implemented as previously described (Mostafavi et al. 2008). Briefly, seeds were assigned initial scores of 1, and all others n/N, where n is the number of seeds and N is the total number of network genes. We then solved the system y = (I + L)f, where y is the set of initial scores, L is the graph Laplacian matrix of the network, and f is the set of final scores. The method for Iterative Ranking is derived in detail elsewhere (Ramakrishnan et al. 2009). However, rather than iteratively computing the final scores, we solved the system y = (I - U)f, where U is the matrix of network edges weighted by the sum of outgoing edges from each node. For the circuit-based method, we followed the electrical model proposed previously (Suthram et al. 2008). Each edge in the network is treated as the conductance between the connecting nodes. The seed nodes are designated as the ground reference, and a current is simultaneously applied to all other nodes in the network. Using Kirchoff Laws, we solved for the voltage for each node. The final score for a node is the flow, or the node's total current multiplied by its voltage.
Integrating the gene network with genome-wide association study data
GWAS data came from the Wellcome Trust Case Control Consortium (Wellcome Trust Case Control Consortium 2007). We selected the additive Bayes factor as a measure of association between SNPs and diseases, and represent each gene by the strongest association signal within 10 kb from the beginning or end of the gene. The same analysis for different cutoffs, varying from 0 to 250 kb, did not significantly change the boosting from the network.
We approximated the probability of a gene being involved in a disease by assuming that the space of possible hypotheses was limited to the null hypothesis and the additive hypothesis used for calculating the Bayes factors, and chose the value for the prior odds by optimizing the area under the first 5% of the area under the ROC curve. In general, we observed an improvement for prior (log10) odds ranging from roughly −2.5 to −1, corresponding to ∼60–1900 associated genes, respectively. Finally, in testing the effect of normalizing for node degree in the gene network, we observed a loss of performance, presumably because node degree does carry information for associating genes with diseases.
Acknowledgments
This work was supported by grants from the National Research Foundation of Korea (NRF), funded by the Korean government (MEST) (No. 2010-0017649), and POSCO TJ Park Science Fellowship to I.L. and from the N.S.F., N.I.H., U.S. Army Research (58343-MA), and Welch (F1515) and Packard Foundations to E.M.M. This study makes use of data generated by the Wellcome Trust Case-Control Consortium. A full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk. Funding for the project was provided by the Wellcome Trust under award 076113 and 085475.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.118992.110.
References
- Akerblad P, Mansson R, Lagergren A, Westerlund S, Basta B, Lind U, Thelin A, Gisler R, Liberg D, Nelander S, et al. 2005. Gene expression analysis suggests that EBF-1 and PPARγ2 induce adipogenesis of NIH-3T3 cells with similar efficiency and kinetics. Physiol Genomics 23: 206–216 [DOI] [PubMed] [Google Scholar]
- Alexeyenko A, Sonnhammer EL 2009. Global networks of functional coupling in eukaryotes from comprehensive data integration. Genome Res 19: 1107–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler D, Daly MJ, Lander ES 2008. Genetic mapping in human disease. Science 322: 881–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranzini SE, Galwey NW, Wang J, Khankhanian P, Lindberg R, Pelletier D, Wu W, Uitdehaag BM, Kappos L, Polman CH, et al. 2009. Pathway and network-based analysis of genome-wide association studies in multiple sclerosis. Hum Mol Genet 18: 2078–2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, et al. 2008. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet 40: 955–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckly JB, Hancock L, Geremia A, Cummings JR, Morris A, Cooney R, Pathan S, Guo C, Jewell DP 2008. Two-stage candidate gene study of chromosome 3p demonstrates an association between nonsynonymous variants in the MST1R gene and Crohn's disease. Inflamm Bowel Dis 14: 500–507 [DOI] [PubMed] [Google Scholar]
- Bonetta L 2008. Getting up close and personal with your genome. Cell 133: 753–756 [DOI] [PubMed] [Google Scholar]
- Bonneau R 2008. Learning biological networks: from modules to dynamics. Nat Chem Biol 4: 658–664 [DOI] [PubMed] [Google Scholar]
- Bonnefond A, Froguel P, Vaxillaire M 2010. The emerging genetics of type 2 diabetes. Trends Mol Med 16: 407–416 [DOI] [PubMed] [Google Scholar]
- Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ 2008. Identification of host proteins required for HIV infection through a functional genomic screen. Science 319: 921–926 [DOI] [PubMed] [Google Scholar]
- Byrne AB, Weirauch MT, Wong V, Koeva M, Dixon SJ, Stuart JM, Roy PJ 2007. A global analysis of genetic interactions in Caenorhabditis elegans. J Biol 6: 8 doi: 10.1186/jbiol58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K, Elledge SJ, Hannon GJ 2006. Lessons from nature: microRNA-based shRNA libraries. Nat Methods 3: 707–714 [DOI] [PubMed] [Google Scholar]
- Chang JS, Yeh RF, Wiencke JK, Wiemels JL, Smirnov I, Pico AR, Tihan T, Patoka J, Miike R, Sison JD, et al. 2008. Pathway analysis of single-nucleotide polymorphisms potentially associated with glioblastoma multiforme susceptibility using random forests. Cancer Epidemiol Biomarkers Prev 17: 1368–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Aronow BJ, Jegga AG 2009a. Disease candidate gene identification and prioritization using protein interaction networks. BMC Bioinformatics 10: 73 doi: 10.1186/gb-200809-s1–s4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Bardes EE, Aronow BJ, Jegga AG 2009b. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 37: (Web Server issue): W305–W311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen C, Thakar J, Albert R 2007. Systems-level insights into cellular regulation: inferring, analysing, and modelling intracellular networks. IET Syst Biol 1: 61–77 [DOI] [PubMed] [Google Scholar]
- Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, et al. 2007. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc 2: 2366–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JD, Smyth DJ, Smiles AM, Plagnol V, Walker NM, Allen JE, Downes K, Barrett JC, Healy BC, Mychaleckyj JC, et al. 2008. Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nat Genet 40: 1399–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, et al. 2010. The genetic landscape of a cell. Science 327: 425–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davierwala AP, Haynes J, Li Z, Brost RL, Robinson MD, Yu L, Mnaimneh S, Ding H, Zhu H, Chen Y, et al. 2005. The synthetic genetic interaction spectrum of essential genes. Nat Genet 37: 1147–1152 [DOI] [PubMed] [Google Scholar]
- Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB 2010. Rare variants create synthetic genome-wide associations. PLoS Biol 8: e1000294 doi: 10.1371/journal.pbio.1000294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Paszkowski-Rogacz M, Nitzsche A, Slabicki MM, Heninger AK, de Vries I, Kittler R, Junqueira M, Shevchenko A, Schulz H, et al. 2009. A genome-scale RNAi screen for Oct4 modulators defines a role of the Paf1 complex for embryonic stem cell identity. Cell Stem Cell 4: 403–415 [DOI] [PubMed] [Google Scholar]
- Emily M, Mailund T, Hein J, Schauser L, Schierup MH 2009. Using biological networks to search for interacting loci in genome-wide association studies. Eur J Hum Genet 17: 1231–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, Iliopoulos I, Kyrpides NC, Ouzounis CA 1999. Protein interaction maps for complete genomes based on gene fusion events. Nature 402: 86–90 [DOI] [PubMed] [Google Scholar]
- Enright AJ, Van Dongen S, Ouzounis CA 2002. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res 30: 1575–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppig JT, Bult CJ, Kadin JA, Richardson JE, Blake JA, Anagnostopoulos A, Baldarelli RM, Baya M, Beal JS, Bello SM, et al. 2005. The Mouse Genome Database (MGD): from genes to mice–a community resource for mouse biology. Nucleic Acids Res 33: D471–D475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feist AM, Herrgard MJ, Thiele I, Reed JL, Palsson BO 2009. Reconstruction of biochemical networks in microorganisms. Nat Rev Microbiol 7: 129–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, Kane M, Kolodner RD 1993. The human mutator gene homologue MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 75: 1027–1038 [DOI] [PubMed] [Google Scholar]
- Flint J, Mackay TF 2009. Genetic architecture of quantitative traits in mice, flies, and humans. Genome Res 19: 723–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke L, Bakel H, Fokkens L, de Jong ED, Egmont-Petersen M, Wijmenga C 2006. Reconstruction of a functional human gene network, with an application for prioritizing positional candidate genes. Am J Hum Genet 78: 1011–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser HB, Plotkin JB 2007. Using protein complexes to predict phenotypic effects of gene mutation. Genome Biol 8: R252 doi: 10.1186/gb-2007-8-11-r252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudbjartsson DF, Walters GB, Thorleifsson G, Stefansson H, Halldorsson BV, Zusmanovich P, Sulem P, Thorlacius S, Gylfason A, Steinberg S, et al. 2008. Many sequence variants affecting diversity of adult human height. Nat Genet 40: 609–615 [DOI] [PubMed] [Google Scholar]
- Hannum G, Srivas R, Guenole A, van Attikum H, Krogan NJ, Karp RM, Ideker T 2009. Genome-wide association data reveal a global map of genetic interactions among protein complexes. PLoS Genet 5: e1000782 doi: 10.1371/journal.pgen.1000782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart GT, Lee I, Marcotte EM 2007. A high-accuracy consensus map of yeast protein complexes reveals modular nature of gene essentiality. BMC Bioinformatics 8: 236 doi: 10.1186/1471-2105-8-236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata S, Abe M, Suzuki H, Kitamura F, Toyama-Sorimachi N, Abe K, Sakimura K, Sorimachi H 2010. Calpain 8/nCL-2 and calpain 9/nCL-4 constitute an active protease complex, G-calpain, involved in gastric mucosal defense. PLoS Genet 6: e1001040 doi: 10.1371/journal.pgen.1001040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildt E, Oess S 1999. Identification of Grb2 as a novel binding partner of tumor necrosis factor (TNF) receptor I. J Exp Med 189: 1707–1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn JN, Daly MJ 2005. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet 6: 95–108 [DOI] [PubMed] [Google Scholar]
- Huttenhower C, Haley EM, Hibbs MA, Dumeaux V, Barrett DR, Coller HA, Troyanskaya OG 2009. Exploring the human genome with functional maps. Genome Res 19: 1093–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T, Sharan R 2008. Protein networks in disease. Genome Res 18: 644–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, et al. 2009. STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res 37: D412–D416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin T, Liu L 2008. The Wnt signaling pathway effector TCF7L2 and type 2 diabetes mellitus. Mol Endocrinol 22: 2383–2392 [DOI] [PubMed] [Google Scholar]
- Koh W, Mahan RD, Davis GE 2008. Cdc42- and Rac1-mediated endothelial lumen formation requires Pak2, Pak4 and Par3, and PKC-dependent signaling. J Cell Sci 121: 989–1001 [DOI] [PubMed] [Google Scholar]
- Lage K, Karlberg EO, Storling ZM, Olason PI, Pedersen AG, Rigina O, Hinsby AM, Tumer Z, Pociot F, Tommerup N, et al. 2007. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat Biotechnol 25: 309–316 [DOI] [PubMed] [Google Scholar]
- Lee I, Date SV, Adai AT, Marcotte EM 2004. A probabilistic functional network of yeast genes. Science 306: 1555–1558 [DOI] [PubMed] [Google Scholar]
- Lee I, Li Z, Marcotte EM 2007a. An improved, bias-reduced probabilistic functional gene network of baker's yeast, Saccharomyces cerevisiae. PLoS ONE 2: e988 doi: 10.1371/journal.pone.0000988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Narayanaswamy R, Marcotte EM 2007b. Bioinformatic prediction of yeast gene function. In Yeast gene analysis (ed. I Stansfield, M Stark). Elsevier, Maryland Heights, MO [Google Scholar]
- Lee I, Lehner B, Crombie C, Wong W, Fraser AG, Marcotte EM 2008. A single gene network accurately predicts phenotypic effects of gene perturbation in Caenorhabditis elegans. Nat Genet 40: 181–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Ambaru B, Thakkar P, Marcotte EM, Rhee SY 2010a. Rational association of genes with traits using a genome-scale gene network for Arabidopsis thaliana. Nat Biotechnol 28: 149–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Lehner B, Vavouri T, Shin J, Fraser AG, Marcotte EM 2010b. Predicting genetic modifier loci using functional gene networks. Genome Res 20: 1143–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leegwater PA, Vermeulen G, Konst AA, Naidu S, Mulders J, Visser A, Kersbergen P, Mobach D, Fonds D, van Berkel CG, et al. 2001. Subunits of the translation initiation factor eIF2B are mutant in leukoencephalopathy with vanishing white matter. Nat Genet 29: 383–388 [DOI] [PubMed] [Google Scholar]
- Lehner B, Crombie C, Tischler J, Fortunato A, Fraser AG 2006. Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat Genet 38: 896–903 [DOI] [PubMed] [Google Scholar]
- Lettre G, Jackson AU, Gieger C, Schumacher FR, Berndt SI, Sanna S, Eyheramendy S, Voight BF, Butler JL, Guiducci C, et al. 2008. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat Genet 40: 584–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew CC, Dzau VJ 2004. Molecular genetics and genomics of heart failure. Nat Rev Genet 5: 811–825 [DOI] [PubMed] [Google Scholar]
- Linghu B, Snitkin ES, Hu Z, Xia Y, Delisi C 2009. Genome-wide prioritization of disease genes and identification of disease-disease associations from an integrated human functional linkage network. Genome Biol 10: R91 doi: 10.1186/gb-2009-10-9-r91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Yu X, Zack DJ, Zhu H, Qian J 2008. TiGER: a database for tissue-specific gene expression and regulation. BMC Bioinformatics 9: 271 doi: 10.1186/1471-2105-9-271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ 2009. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 137: 835–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madu IG, Roth SL, Belouzard S, Whittaker GR 2009. Characterization of a highly conserved domain within the severe acute respiratory syndrome coronavirus spike protein S2 domain with characteristics of a viral fusion peptide. J Virol 83: 7411–7421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte EM, Pellegrini M, Thompson MJ, Yeates TO, Eisenberg D 1999. A combined algorithm for genome-wide prediction of protein function. Nature 402: 83–86 [DOI] [PubMed] [Google Scholar]
- McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JP, Hirschhorn JN 2008. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 9: 356–369 [DOI] [PubMed] [Google Scholar]
- McGary KL, Lee I, Marcotte EM 2007. Broad network-based predictability of Saccharomyces cerevisiae gene loss-of-function phenotypes. Genome Biol 8: R258 doi: 10.1186/gb-2007-8-12-r258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGary KL, Park TJ, Woods JO, Cha HJ, Wallingford JB, Marcotte EM 2010. Systematic discovery of nonobvious human disease models through orthologous phenotypes. Proc Natl Acad Sci 107: 6544–6549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaki M, Konishi M, Tanaka K, Kikuchi-Yanoshita R, Muraoka M, Yasuno M, Igari T, Koike M, Chiba M, Mori T 1997. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Struct Biol 17: 271–272 [DOI] [PubMed] [Google Scholar]
- Moser AB, Rasmussen M, Naidu S, Watkins PA, McGuinness M, Hajra AK, Chen G, Raymond G, Liu A, Gordon D, et al. 1995. Phenotype of patients with peroxisomal disorders subdivided into sixteen complementation groups. J Pediatr 127: 13–22 [DOI] [PubMed] [Google Scholar]
- Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q 2008. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol 9: S1– S4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai R, Jones MK, Tomikawa M, Tarnawski AS 1999. Activation of Raf-1 during experimental gastric ulcer healing is Ras-mediated and protein kinase C-independent. Am J Pathol 155: 1759–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Wacholder S, Gail MH, Peters U, Jacobs KB, Chanock SJ, Chatterjee N 2010. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat Genet 42: 570–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pico AR, Smirnov IV, Chang JS, Yeh RF, Wiemels JL, Wiencke JK, Tihan T, Conklin BR, Wrensch M 2009. SNPLogic: an interactive single nucleotide polymorphism selection, annotation, and prioritization system. Nucleic Acids Res 37: D803–D809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomp D, Allan MF, Wesolowski SR 2004. Quantitative genomics: exploring the genetic architecture of complex trait predisposition. J Anim Sci 82: E300–E312 [DOI] [PubMed] [Google Scholar]
- Qin L, Zhou L, Wu X, Cheng J, Wang J, Du Y, Hu J, Shen M, Zhou J 2008. Genetic variants in protein kinase C zeta gene and type 2 diabetes risk: a case-control study of a Chinese Han population. Diabetes Metab Res Rev 24: 480–485 [DOI] [PubMed] [Google Scholar]
- Ramakrishnan SR, Vogel C, Kwon T, Penalva LO, Marcotte EM, Miranker DP 2009. Mining gene functional networks to improve mass-spectrometry-based protein identification. Bioinformatics 25: 2955–2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regamey A, Hohl D, Liu JW, Roger T, Kogerman P, Toftgard R, Huber M 2003. The tumor suppressor CYLD interacts with TRIP and regulates negatively nuclear factor κB activation by tumor necrosis factor. J Exp Med 198: 1959–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiley WW, Jin W, Lee AJ, Wright A, Wu X, Tewalt EF, Leonard TO, Norbury CC, Fitzpatrick L, Zhang M, et al. 2007. Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J Exp Med 204: 1475–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes DR, Tomlins SA, Varambally S, Mahavisno V, Barrette T, Kalyana-Sundaram S, Ghosh D, Pandey A, Chinnaiyan AM 2005. Probabilistic model of the human protein-protein interaction network. Nat Biotechnol 23: 951–959 [DOI] [PubMed] [Google Scholar]
- Rossin EJ, Lage K, Raychaudhuri S, Xavier RJ, Tatar D, Benita Y, Cotsapas C, Daly MJ 2011. Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet 7: e1001273 10.1371/journal.pgen.1001273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccone SF, Saccone NL, Swan GE, Madden PA, Goate AM, Rice JP, Bierut LJ 2008. Systematic biological prioritization after a genome-wide association study: an application to nicotine dependence. Bioinformatics 24: 1805–1811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlabach MR, Luo J, Solimini NL, Hu G, Xu Q, Li MZ, Zhao Z, Smogorzewska A, Sowa ME, Ang XL, et al. 2008. Cancer proliferation gene discovery through functional genomics. Science 319: 620–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwikowski B, Uetz P, Fields S 2000. A network of protein-protein interactions in yeast. Nat Biotechnol 18: 1257–1261 [DOI] [PubMed] [Google Scholar]
- Sharan R, Ulitsky I, Shamir R 2007. Network-based prediction of protein function. Mol Syst Biol 3: 88 doi: 10.1038/msb4100129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolovitzky G, Monroe D, Califano A 2007. Dialogue on reverse-engineering assessment and methods: the DREAM of high-throughput pathway inference. Ann N Y Acad Sci 1115: 1–22 [DOI] [PubMed] [Google Scholar]
- Suthram S, Beyer A, Karp RM, Eldar Y, Ideker T 2008. eQED: an efficient method for interpreting eQTL associations using protein networks. Mol Syst Biol 4: 162 doi: 10.1038/msb.2008.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Page N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H, et al. 2001. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294: 2364–2368 [DOI] [PubMed] [Google Scholar]
- Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, Young J, Berriz GF, Brost RL, Chang M, et al. 2004. Global mapping of the yeast genetic interaction network. Science 303: 808–813 [DOI] [PubMed] [Google Scholar]
- Troyanskaya OG, Dolinski K, Owen AB, Altman RB, Botstein D 2003. A Bayesian framework for combining heterogeneous data sources for gene function prediction (in Saccharomyces cerevisiae). Proc Natl Acad Sci 100: 8348–8353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Knaap MS, Leegwater PA, Konst AA, Visser A, Naidu S, Oudejans CB, Schutgens RB, Pronk JC 2002. Mutations in each of the five subunits of translation initiation factor eIF2B can cause leukoencephalopathy with vanishing white matter. Ann Neurol 51: 264–270 [DOI] [PubMed] [Google Scholar]
- van Dongen S 2000. A cluster algorithm for graphs. National Research Institute for Mathematics and Computer Science in the Netherlands, Amsterdam [Google Scholar]
- Van Limbergen J, Wilson DC, Satsangi J 2009. The genetics of Crohn's disease. Annu Rev Genomics Hum Genet 10: 89–116 [DOI] [PubMed] [Google Scholar]
- Visscher PM 2008. Sizing up human height variation. Nat Genet 40: 489–490 [DOI] [PubMed] [Google Scholar]
- Wang PI, Marcotte EM 2010. It's the machine that matters: Predicting gene function and phenotype from protein networks. J Proteomics. 73: 2277–2289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Dickson SP, Stolle CA, Krantz ID, Goldstein DB, Hakonarson H 2010a. Interpretation of association signals and identification of causal variants from genome-wide association studies. Am J Hum Genet 86: 730–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H 2010b. Analysing biological pathways in genome-wide association studies. Nat Rev Genet 11: 843–854 [DOI] [PubMed] [Google Scholar]
- Weedon MN, Lango H, Lindgren CM, Wallace C, Evans DM, Mangino M, Freathy RM, Perry JR, Stevens S, Hall AS, et al. 2008. Genome-wide association analysis identifies 20 loci that influence adult height. Nat Genet 40: 575–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium 2007. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447: 661–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Liu Q, Jiang R 2009. Align human interactome with phenome to identify causative genes and networks underlying disease families. Bioinformatics 25: 98–104 [DOI] [PubMed] [Google Scholar]
- Wu G, Feng X, Stein L 2010. A human functional protein interaction network and its application to cancer data analysis. Genome Biol 11: R53 doi: 10.1186/gb-2010-11-5-r53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, Madden PA, Heath AC, Martin NG, Montgomery GW, et al. 2010. Common SNPs explain a large proportion of the heritability for human height. Nat Genet 42: 565–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T, de Bakker PI, Abecasis GR, Almgren P, Andersen G, et al. 2008. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet 40: 638–645 [DOI] [PMC free article] [PubMed] [Google Scholar]