

Abstract
Plasticity of gene regulatory encryption can permit DNA sequence divergence without loss of function. Functional information is preserved through conservation of the composition of transcription factor binding sites (TFBS) in a regulatory element. We have developed a method that can accurately identify pairs of functional noncoding orthologs at evolutionarily diverged loci by searching for conserved TFBS arrangements. With an estimated 5% false-positive rate (FPR) in approximately 3000 human and zebrafish syntenic loci, we detected approximately 300 pairs of diverged elements that are likely to share common ancestry and have similar regulatory activity. By analyzing a pool of experimentally validated human enhancers, we demonstrated that 7/8 (88%) of their predicted functional orthologs retained in vivo regulatory control. Moreover, in 5/7 (71%) of assayed enhancer pairs, we observed concordant expression patterns. We argue that TFBS composition is often necessary to retain and sufficient to predict regulatory function in the absence of overt sequence conservation, revealing an entire class of functionally conserved, evolutionarily diverged regulatory elements that we term “covert.”
In recent years, sequence constraint has been widely used as a powerful filter to identify regulatory sequences (Hardison 2000; Bejerano et al. 2004; Pennacchio et al. 2006; Visel et al. 2007a). However, the divergence of regulatory pathways and networks is also predicted to play a major role in the diversification and adaptation of species (King and Wilson 1975). Recent studies indicate that the cis-regulatory sequences constitute the primary substrate of evolutionary divergence, while the remaining components of the transcriptional machinery, such as transcription factors (TFs) and the coding genes they modulate, are predominantly conserved (ENCODE Project Consortium et al. 2007; Wilson et al. 2008). Furthermore, UTRs, introns, and intergenic DNA show unexpectedly high levels of divergence (Andolfatto 2005; Bird et al. 2006). Consequently, only ∼3.5% of noncoding sequence are highly conserved among mammals (Waterston et al. 2002; Siepel et al. 2005), and <1% are conserved with more distant vertebrates, such as teleosts (Thomas et al. 2003).
Regulatory elements (enhancers, silencers, insulators, etc.) display heterogeneous levels of conservation. Sequences that are critical for organism development and homeostasis frequently display evidence of strong selective constraint and are thus conserved among distant lineages (Nobrega et al. 2003; Woolfe et al. 2005; Visel et al. 2009). For instance, the majority of assayed conserved noncoding elements (CNEs) in human and fish genomes have been shown to act as tissue-specific enhancers in the developing brain and neuronal systems (Loots et al. 2002). However, most of the regulatory landscape in vertebrate genomes shows evidence of rapid modification and differs even between closely related species (Dermitzakis and Clark 2002; Kasowski et al. 2010) as well as between individuals within the same population (Borneman et al. 2007; Stranger et al. 2007). Although a substantial fraction of these differences likely corresponds to lineage-specific elements, recent work suggests that lineage-specific TF binding site (TFBS) turnover has resulted in a group of regulatory elements with evolutionarily conserved function but little evidence of sequence constraint (Blow et al. 2010; Kunarso et al. 2010; Schmidt et al. 2010; Xie et al. 2010). In these elements, which we term “covert” elements, the regulatory encryption is conserved, but embedded within a divergent sequence background. Individual instances of covert regulatory elements have been previously reported in Drosophila (Dermitzakis et al. 2003; Ludwig et al. 2005; Wittkopp 2006; Hare et al. 2008).
The difficulty of reliably aligning noncoding sequences of distant species was recognized early by the research community, which has developed several models to assess inference errors (Pollard et al. 2006; Huang et al. 2007; Kim and Sinha 2010). Extensive work has been done to provide further insight into the functional constraints on TFBS organization, concluding that clustered and/or overlapping TFBSs are common requirements for enhancer activity (e.g., Hu et al. 2007; Gotea et al. 2010; Lusk and Eisen 2010). Many state-of-the-art enhancer predictors search for clusters of TFBS, facilitating the discovery of regulatory regions in an “alignment-free” manner (Philippakis et al. 2005; Blanchette et al. 2006; Sinha et al. 2006; Narlikar et al. 2010), even in the scenario where relevant TFs or binding affinities are unknown (Kantorovitz et al. 2009; Arunachalam et al. 2010).
Here, we introduce a computational framework to identify covert regulatory elements in genomes of distantly related species. We demonstrate that, using iterative pairwise alignments among trios of vertebrate species, we can establish orthology relationships between diverged noncoding sequences and identify specific patterns describing these sequences. Furthermore, we show that these patterns are appropriately modeled as arrangements of TFBS. Using this data set of diverged sequences wherein orthology is known, we developed and trained an alignment model capable of accurately identifying regulatory orthologs genome-wide on sequences where no overt alignment is provided by standard metrics. Using alignments of TFBSs instead of nucleotides, we predicted orthology relationships of 300 human/fish noncoding sequence pairs with an estimated false-positive rate (FPR) of 5%. Putative human/zebrafish orthologs were tested in transgenic zebrafish assays, confirming enhancer activity of 7/8 (88%) of the zebrafish sequences for which the human counterpart also showed enhancer activity. Furthermore, 5/7 (71%) of the zebrafish enhancers displayed consistent overlapping function with their human counterparts, despite diverged sequences indicating a high degree of functional conservation during enhancer evolution. These results validate the accuracy of our predictions.
Results
Conservation tunneling can reveal diverged regulatory elements in the human genome
For any given genomic region, both mutation rates and selection pressure fluctuate over time and between species. Accordingly, iterative pairwise comparisons among sequences of multiple species evolving over different divergence times and rates can provide evidence of orthology where standard sequence alignment methods (such as BLASTZ) (Schwartz et al. 2003) might fail. We hypothesized that iterative pairwise comparisons can be used to identify covert regulatory elements, i.e., sequence orthologs that are overtly diverged yet retain a core TFBS composition due to their function.
Using pairwise comparisons among three distantly related species, we cataloged noncoding sequence pairs for which we could detect homology in only two of the three possible comparisons. From these data, we generated a library of sequence pairs that display extensive divergence but are both homologous to a third sequence, and thus, likely to share a common ancestor (Fig. 1). We compared human/frog, human/zebrafish, and frog/zebrafish conserved noncoding elements (CNEs; 70% identity across at least 100 bp) and identified approximately 1500 pairs of human and zebrafish sequences that show similarity to the same sequences in the frog genome, but are not alignable to each other (for the phylogenetic relationships among the involved species, see Supplemental Fig. 1). In this case, the frog sequence is likely to be the most similar to the ancestral sequence, thus serving as an orthology “tunnel” between human and zebrafish.
Figure 1.

Conservation tunneling. (A) Phylogenetic tree constructed for three orthologous sequences in human (hg18: chr18:53271349–53271555), frog (xenTro2: scaffold_97:133388–133595), and zebrafish (danRer5: chr24:28243171–28243307). Only the human and the frog sequences and the frog and the zebrafish sequences can be aligned (with at least 70% identity across at least 100 bp). The frog sequence has evolved more slowly relative to the human and zebrafish sequences, and thus, can be used to establish the orthology of the diverged human and the zebrafish sequences. (B) Pairwise sequence comparisons. Eighty-seven percent of frog nucleotides are conserved in either human or fish (gray squares), while only 42% are conserved in both human and fish (red triangles).
Tunneled elements (TEs) encompass 267 kb in the human genome and are widely but not uniformly distributed across all chromosomes, often residing in clusters—37% lie within 25 kb of another one (in contrast with the 3% expectation, see Supplemental Fig. 2). Nearly all human TEs (TEHs) also exhibit extremely high levels of conservation in other vertebrates, with an average phastCons score (Siepel et al. 2005) of 1.4 (as compared to the average 1.7 for human/zebrafish CNEs). Similarly, 64% of zebrafish counterparts of TEs (TEZ) are conserved in Fugu (at least 70% identity across 100 bp). Collectively, these data establish that TEs are well conserved within vertebrates, supporting their functionality, albeit that this conservation is circumscribed to particular phylogenetic clades.
In addition, as further evidence of their potential association with regulatory function, TEs demonstrate a highly significant overlap with sites of ChIP-seq enrichment for the transcriptional coactivator p300 (Visel et al. 2009) in forebrain (34%), midbrain (32%), and limb (28%) tissue (all P-values <2.2 × 10−16 according to a one-tailed Fisher's exact test). Gene Ontology (GO) categories (Ashburner et al. 2000) enriched among TEHs include regulation of transcription, organ development, and morphogenesis (P-values <0.05 after multiple testing correction, computed with the binomial test, accounting for locus length differences) (Taher and Ovcharenko 2009).
These results provide support for the idea that human/zebrafish TEs predate the tetrapod split and comprise functional regulatory sequences that have diverged beyond the point where standard sequence comparison can reliably detect homology. Thus, TEs constitute an ideal data set to study patterns of regulatory sequence evolution at the extremes of sequence divergence.
TFBS constraint characterizes TEs
Although the most evolutionarily distant counterparts of TEs do not satisfy an empirically optimized similarity threshold (70% identity across 100 bp) (Loots et al. 2000; Ovcharenko et al. 2004), some sequences display lower levels of conservation. To quantify this, we aligned the human counterpart of each TE to the zebrafish genome using BLAT (Kent 2002). We successfully identified the zebrafish orthologous region in only 7% of cases, completely failing to align 33% of the sequences and identifying nonorthologous alignments for the remaining 60%. In whole-genome comparisons, alignments that fall below standard sequence conservation thresholds are likely to reveal spurious alignments, and are considered false positives. By tunneling the sequence similarity through an additional species—in this instance, frog—we minimize this risk, providing strong evidence of common ancestry for all pairs of human and zebrafish sequences.
To better assess the impact of sequence divergence on function, we then analyzed the TFBS composition and variation among TEs. Using the TF binding specificities in TRANSFAC (Matys et al. 2006) with a conservative threshold of less than one occurrence for a given TFBS every 10 kb of random sequence, we identified an average of 70 and 73 different TFBSs in the human and zebrafish counterparts of TEs, respectively. Then, we relaxed the definition of conservation, calling a TFBS conserved if it simply occurs in all sequences being considered. We found that, on average, 22% of the TFBSs predicted in the human counterpart of TEs are conserved in the frog counterpart, and from these, only 7% of the total are conserved in the zebrafish counterpart. This exceeds the expected 4% observed for unrelated human and zebrafish noncoding sequences (P-value <2.2 × 10−16, Wilcoxon rank-sum test). Also, conserved TFBSs display a high level of sequence identity, with an average of 73% (while the average sequence identity between TFBSs in unrelated sequences is 61% [P-value <2.2 × 10−16, Wilcoxon rank-sum test]). Although we should be cautious in interpreting these findings, since we ignore which TFBSs are functional, these observations suggest that TFBSs within these TEs are subject to evolutionary constraint.
TFBS composition can be used to describe covert regulatory elements
With the collection of TEs as foundation, we hypothesized that we could formulate a robust mathematical framework describing the regulatory function encrypted in the arrangements of TFBS of covert elements. To this end, we developed an alignment model that compares sets of conserved TFBSs. In our model, TFBSs were considered conserved if they could be identified in all sequences involved, regardless of the sequence alignment. The main assumptions behind our alignment model are that the order of the TFBSs is conserved among orthologous regulatory elements and that the distance between pairs of functional TFBSs can only vary within a set range. As a first step, we independently searched the sequences with a set of position weight matrices (PWMs), generating a list of TFBS occurrences. Then, for a given PWM, we compared all occurrences on the reference sequence with the occurrences on the corresponding target sequence, producing a list of pairs of occurrences. Lastly, we scored all possible combinations of pairs of TFBS occurrences that would establish consistent alignments, in the sense they do not violate the assumption of order preservation between any two TFBSs. The score of an alignment is a function of the number of conserved TFBSs and their relative position shift (for details, see Methods; for an example, see Supplemental Fig. 3).
Our model was trained and tested on the set of TEs for which we could ascertain true orthologs. To define the search space in the zebrafish genome, we first identified syntenic human and zebrafish loci containing the TEs. These loci were defined using pairs of human/zebrafish CNEs separated by ≤50 kb and encompassing the TEs. We scanned the resulting set of 308 syntenic zebrafish loci using a sliding window approach, looking for the counterparts of the corresponding TEHs. In aggregate, we analyzed more than 3 million windows and selected the highest-scoring window for each of the 308 TEHs (<0.2% of all windows) as predicted zebrafish orthologs. Fifty-one percent of predicted orthologs correctly revealed the location of the corresponding TEZs. Moreover, the center of the majority (88%) of windows is shifted by <100 bp with respect to the center of the corresponding TEZs, indicating that our alignment method recognizes functional orthologs accurately.
A few similar approaches have been proposed in the past (Berezikov et al. 2004; Blanco et al. 2006; Hallikas et al. 2006), focusing on the comparison of mammalian regulatory regions that do not show discernible sequence conservation. In particular, the method by Blanco et al. (2006), which is targeted to promoters, succeeded in retrieving only 7% of the zebrafish orthologs. Similarly, EEL (Hallikas et al. 2006), a tool designed to locate enhancers in mammalian genomes by comparing conserved clusters of TFBSs, succeeded in 18% of the cases. Despite relying on similar models, the optimal parameter configuration of these methods depends on the exact issue to be addressed, explaining the remarkable differences in performance. Additionally, we assessed the ability to recover orthologs in distant species of EMMA (He et al. 2009), a state-of-the-art computational method for cis-regulatory module prediction that performs alignment and binding site prediction simultaneously, based on an evolutionary model. EMMA recognized 13% of TEs as regulatory sequences. We used Cluster-Buster (Frith et al. 2003), a software that finds clusters of TFBSs in DNA sequences, and retrieved 5% of TEs. The best performance among tested previously developed tools was demonstrated by a multiple alignment program, MUSCLE (Edgar 2004), which correctly identified zebrafish orthologs of human TEs in 47% of the instances. Although our method performed better than these alternatives, we would like to emphasize that these tools have been designed to achieve different goals. Thus, whereas EMMA and Cluster-Buster produce robust analysis of well-characterized regulatory regions and exploration of long sequences with strong clusters of TFBSs, respectively, our tool is specifically suited to the alignment and comparison of TFBS profiles. On the other hand, whereas MUSCLE's performance is comparable to our method, it provides little information about the underlying regulatory architecture of the sequences.
Finally, we investigated how alignments between orthologous sequences could be distinguished from incorrect alignments, occurring by chance between unrelated sequences. The ability of our algorithm to recognize the zebrafish counterpart does not depend on the length of the human (average 170 bp) and zebrafish (average 161 bp) counterparts of the element or the GC content of the sequences (averages 45% and 46%, respectively; for details, see Supplemental Fig. 4). Differences in the level of conservation do not have a major effect either: BLAT (Kent 2002) found no evident sequence alignment for neither orthologous (88% of cases) nor incorrect (95%) TFBS-based alignments. However, orthologous alignments have significantly higher scores than incorrect alignments (averages 9.3 and 5.9, respectively; P-value = 1.8 × 10−8, Wilcoxon rank-sum test). Furthermore, the alignment score is correlated with the number of TFBS occurrences in the predicted zebrafish ortholog (R-squared = 0.3). The correlation, however, differs for orthologous versus incorrect alignments, in that, for a given alignment score, incorrect alignments contained a disproportionally small number of TFBSs. Thus, by using the number of TFBSs in addition to the alignment scores, we can perfectly separate 28% of orthologous alignments from incorrect alignments (Fig. 2), a 75% improvement over the separation using alignment scores only. In addition, 74% of orthologous alignments scored higher than control alignments computed with their flanking loci, whereas only 38% of incorrect alignments did (Supplemental Fig. 5). Based on these findings, we integrated the alignment scores of orthologous and control alignments with the number of TFBSs in the corresponding predicted orthologs and used a Support Vector Machine (SVM) to separate reliable from unreliable alignments. We examined the performance of the method by repeating a 10-fold cross-validation 100 times with random partitions of the data and obtained an average sensitivity of 50% for a FPR of 5% (Fig. 2, insert). This strategy provided a theoretical framework to detect covert elements genome-wide.
Figure 2.

Alignment scores as a function of the number of TFBSs in the target window. Trend lines for correct (solid green line) and incorrect (solid red line) alignments; (black dotted line) perfectly separates correct from incorrect alignments. (Inset) The receiver operating characteristic (ROC) curve for the linear SVM classifier separating orthologous from control alignments; the curve profiles the performance in terms of the number of orthologous sequences that are correctly identified among all orthologous sequences (TPR), and the number of control sequences that are incorrectly identified as orthologs among all control sequences (FPR). Gray dotted lines show the standard deviation. The red dotted line displays the ROC curve for a random classifier. The solid red lines indicate the selected operating point (FPR = 0.5).
TFBS-based alignments can discover covert regulatory elements genome-wide
Finally, we set out to determine whether the TFBS-based alignment model trained on TEs could be helpful to discover other regulatory sequences that lie below the radar of sequence conservation, de novo. To ensure that we apply our model to only well-diverged sequences, we filtered out weakly conserved sequences (50% identity across at least 100 bp), as well as successfully tunneled elements, and used our model to align approximately 3000 human/frog CNEs to the zebrafish genome. The corresponding syntenic loci in zebrafish were defined as previously described requiring human/zebrafish CNEs to demarcate locus boundaries (Fig. 3).
Figure 3.

Overview of the detection of covert regulatory elements. We look for functional orthologs of conserved human/frog CNEs in the zebrafish sequence by computing alignments for syntenic and control loci and using a SVM to distinguish significant from random alignments.
Evidently, not all of these human/frog CNEs are expected to have a functional ortholog in the zebrafish genome, as many might have been lost due to lineage specialization or constitute lineage-specific innovation. Using the set of human/frog CNEs we identified approximately 300 high-confidence predictions of human/zebrafish covert elements (with a FPR of 5%). These elements are widely distributed in 236 loci of UCSC Known and RefSeq (Hsu et al. 2006; Pruitt et al. 2007) genes. The human counterparts of 3% of these elements overlap a UTR of a known human protein-coding gene, while 71% are located in introns of known genes and the rest are intergenic. Compared to the complete set of human/frog CNEs, our predictions are significantly enriched in the neighborhood of genes related to somatic muscle development (200-fold enrichment, P-value = 0 after multiple testing correction, binomial test), suggesting that they are biologically meaningful. Other attributes, such as regulation of transcription, are almost twofold enriched with regard to human/frog CNEs. This suggests that covert elements may be specifically associated with developmentally relevant regulatory functions. Moreover, 11 elements (Table 1) are contained in enhancers that have been shown to drive expression in limb, heart, and brain tissues (Visel et al. 2007b). The regulatory function of our predictions is further supported by their overlap with ChIP-seq and histone monomethylation patterns that characterize enhancer activity (H3K4me1) (Barski et al. 2007; Heintzman et al. 2007). The predicted approximately 300 human/zebrafish predicted covert elements significantly overlap with sites of p300 enrichment (ChIP-seq) for p300 in forebrain (71%), midbrain (71%), and limb (66%), with an increase >1.6-fold over human/frog CNEs (all P-values <2.2 × 10−16 according to a Fisher's exact test). Overall, 87% of the predicted sequences overlap with p300 peaks, demonstrating overwhelming support for their regulatory role in forebrain, midbrain, and limb development. Furthermore, ∼26% of the elements have H3K4me1 signatures, emphasizing that a large fraction of the reported elements could exhibit enhancer activity.
Table 1.
Human counterparts of human/zebrafish covert regulatory elements with known enhancer activity

In vivo analyses of putative enhancers reveal concordant tissue-specific expression
The ultimate test for the ability of our method to accurately identify functional orthologs that are diverged at the sequence level is to experimentally demonstrate, in vivo, their regulatory activity. Toward that end, we randomly selected a set of 18 putative human/zebrafish orthologs discovered by the TFBS-based alignments (Table 2), and undertook in vivo analyses of the sequences' ability to drive tissue-specific activity in developing zebrafish embryos. These human and zebrafish sequences are neither overtly conserved at the sequence level nor can they be identified using the conservation tunneling approach. To build up a comprehensive picture of the accuracy of our model, we assayed sequences with a wide range of scores.
Table 2.
In vivo testing of putative regulatory human and zebrafish orthologs in transgenic zebrafish assays

As expected for deeply conserved elements (Pennacchio et al. 2006), 8/18 (44%) of the assayed human sequences displayed enhancer activity in vivo. To assess if the predicted zebrafish sequences correspond to the functional orthologs of the human sequences, we similarly assayed the zebrafish counterparts of all identified human enhancers. Remarkably, 7/8 (88%) of the assayed zebrafish sequences also directed tissue-specific expression. Moreover, the orthologous sequences displayed notable similarity in enhancer activity (Fig. 4; Supplemental Fig. 6), in that they directed expression in concordant anatomical discrete units. For example, the human sequence E (Table 2) directed expression in the forebrain, notochord, and somites; the predicted orthologous zebrafish sequence directed expression in the same tissues. In two cases, however, we observed expression in different tissues (Fig. 4, sequences B and G; Supplemental Fig. 6). For instance, human sequence G weakly drives expression in the forebrain and in spinal cord neurons, while its zebrafish counterpart shows stronger expression in the notochord (Fig. 4). Unless we have failed to recognize a related element in the zebrafish locus, the observed divergence in function likely corresponds to adaptive changes in the function of these enhancers.
Figure 4.

Putative human and zebrafish enhancer pairs direct similar tissue-specific expression (covert regulatory elements D, E, F, G) (Table 2). (A) Composite overviews of in vivo GFP expression data from 16–20 individual zebrafish embryos per construct. The keys for the marked expression are provided next to each image, followed by the number of fish in the set with that specific expression. (B) One representative GFP live image from each enhancer set is displayed. All zebrafish are 24 hpf oriented with anterior to the left and dorsal to the top. The dotted box demarcates the forebrain. The stacked structures of the notochord are between the dotted lines. Arrows refer to the somites. The solid line box contains the spinal cord. The pronephric duct-consistent expression is marked by the solid line ovals.
In general, despite the great evolutionary distance and the absence of sequence conservation, both human and zebrafish counterparts of the tested covert elements displayed strong similarity in enhancer activity in vivo (zebrafish). Three of the four pairs that demonstrated enhancer activity had highly overlapping expression patterns, and the fourth pair was similar in that both had expression in neural tissues, suggesting functional specialization as compared to the ancestral sequence.
Taken collectively, our experimental data confirm that our computational approach captures essential functional information despite lack of sequence similarity, and therefore, constitutes an important step toward understanding the encryption and evolution of the regulatory code.
Discussion
Changes in transcriptional regulation are frequently assumed to constitute key players in the recent evolution of humans (King and Wilson 1975). However, despite immense efforts in the field, we still have a very limited knowledge of the regulatory architecture of vertebrate genomes. In particular, we know that transcriptional regulatory sequences appear to be relatively flexible, allowing considerable sequence mutation while retaining functional equivalence (Ludwig 2002; Elgar 2006; Polavarapu et al. 2008). Several examples also demonstrate the existence of covert regulatory elements, i.e., elements that have maintained their function despite extensive function divergence (Fisher et al. 2006a; Hare et al. 2008; McGaughey et al. 2008). Yet, most predictions of functional noncoding sequences are still achieved through the analysis of evolutionary conservation, suggesting that many functional sequences may remain undetected.
To improve our understanding of the language of transcriptional regulation, we have established a strategy to ascertain the ancestral identity of diverged noncoding sequences. In the comparison of two distantly related species, e.g., human and zebrafish, the addition of a third species that is also a descendant of the last common ancestor of the original two, e.g., frog, often serves as a tunnel to establish a relationship between them. The inclusion of the frog sequence often provides us with a better estimation of the sequence of interest in the last common ancestor of human and frog, and this sequence is likely to share more similarity with its hypothetical zebrafish ortholog than the original human sequence. Thereby, incorporating a third species into the genomic pairwise comparison of two distant species facilitates the detection of ancestral sequence identity. We have applied this principle to establish orthology relationships between 1500 noncoding elements in human and zebrafish that fail to align under standard pairwise sequence comparison, increasing the number of predicted functional elements in approximately 5%. Human, frog, and zebrafish are certainly not the only species to which the conservation tunneling principle can be applied, but only examples to illustrate our approach. Likewise, we have inferred the orthology of 3600 human/frog (tunneled through chicken) and 6400 human/chicken (tunneled though mouse) elements that fail to align under standard pairwise sequence comparison methods.
A significant fraction of diverged noncoding sequences defy detection based on sequence similarity, even after including more species into the analysis. For those cases, we have designed an alignment model based solely on the distribution of TFBSs. The main limitation of the method resides in the need of ensuring the orthology of the search locus, which we addressed by requiring conserved elements on both sides of the putative diverged element. Also, our model assumes that the set of transcription factors binding to each particular cis-regulatory module is very similar in the species compared and will fail to identify orthologous elements if extensive changes in the transcriptional machinery have taken place. Evidence suggests that weakly advantageous (or deleterious) mutations at different positions within a binding site are likely to be strongly selected for (or against). Consistently, our model does not require the TFBSs to be identical on both sequences and permits variation as long as the signal represented by the shared TFBSs is not drowned out by the noise of matches to unrelated sequences.
We evaluated 3000 zebrafish, 8000 frog, and 290,000 chicken loci, looking for functional orthologs of human/frog, human/chicken, and human/mouse CNEs, respectively, and found conclusive evidence of the existence of covert regulatory elements in 1% to 10% of them. Predicted covert elements are particularly enriched in loci of genes displaying transcriptional and developmental functions, and sequence divergence of these elements could be explained by extensive sequence changes in nonbinding site regions. An in vivo screen for enhancer activity of 18 human counterparts of putative human/zebrafish functional orthologs yielded eight positive enhancers with roles in mesoderm and nervous system development. The zebrafish counterpart of seven of these eight elements also exhibited enhancer activity. Moreover, 5/7 (71%) of the human/zebrafish pairs of sequences directed gene expression in overlapping sets of discrete anatomical units, with 40% driving transcription in identical structures. This demonstrates that enhancers can maintain their function despite sequence divergence. Two zebrafish sequences, however, exhibited divergent activities as compared with their human counterparts. Indeed, known examples show that extensive expression pattern changes may result from mutations in only a few nucleotides (Wittkopp 2006). Our experimental data are consistent with these interpretations and confirm the theoretical possibility of predicting regulatory function using comparisons that rely on the TFBS structure rather than nucleotide composition of the sequences.
In summary, although the analysis of regulatory elements that control gene expression has placed much emphasis on the conservation of noncoding regions at the sequence level, recent studies demand a rethink of this approach. Here, we showed how iterative pairwise sequence comparisons among multiple species can be applied to detect orthology relationships between noncoding regions that have diverged at the sequence level. We also modeled the evolution of regulatory elements based on arrangements of TFBSs identified therein, detecting orthology relationships where conventional strategies failed. With an approach based on this model, we then searched the zebrafish genome for orthologs of functional human sequences (as supported by both functional analysis and post hoc evaluation of ChIP-identified sequences) previously undetected/undetectable using common metrics of constraint, confirming their broader existence.
Few available examples of well-characterized sets of enhancers that have diverged at the sequence level but preserved their function limit our understanding of covert regulatory sequences. To address this, we have proposed a systematic approach for detecting further instances and capturing general properties of covert sequences. In turn, additional experimental investigation will allow us to develop more robust models and gain greater insight into the evolution of regulatory sequences.
Methods
Conserved elements
Our method relies on the identification of short sequences conserved between two or three species. Using UCSC single-coverage pairwise alignments (axtNet) (Kent et al. 2003) produced from BLASTZ (Schwartz et al. 2003), we looked for sequences that are at least 100 bp long and show at least 70% identity.
Pairwise alignments of the human genome (hg18) with mouse (mm9), chicken (galGal3), frog (xenTro2), and zebrafish (danRer5) genomes, as well as between mouse and chicken, chicken and frog, and frog and zebrafish (danRer4), were obtained from the UCSC Genome Browser (Karolchik et al. 2009). The danRer4 coordinates were converted to danRer5 coordinates using the UCSC liftOver tool.
We used annotation from RefSeq (Pruitt et al. 2007) and UCSC Known Genes (Hsu et al. 2006) to identify protein-coding regions.
Syntenic search loci
Syntenic loci are defined by two deeply conserved (i.e., conserved in all species of interest) elements separated on the reference sequence (in our case, human) by a minimum length of 1 kb and a maximum length of 50 kb. We require consistency on the chromosome where the flanking elements are located. For example, for each human/frog CNE that appears to have diverged in zebrafish, we used human/frog/zebrafish CNEs to delimit the corresponding syntenic loci in human and zebrafish; the human/frog/zebrafish CNEs are required to flank the human and frog counterpart of the diverged CNE. For the genome-wide prediction of diverged regulatory elements, we did not enforce any constraint on the frog sequence other than existence of a conserved element, but required a minimum length of 1 kb and a maximum length of 50 kb for the zebrafish sequence.
Sequence representation
Each nucleotide sequence was masked for annotated repeat regions (Smit et al. 1996-2010) and translated into a map of TF binding sites using a set of position weight matrices (PWMs) that represent TF binding specificities.
Position weight matrices (PWMs)
We used a set of 701 PWMs for vertebrate TFs from TRANSFAC 11.4 (Matys et al. 2003) for the analysis. These PWMs include 5 to 30 nucleotide positions. TF binding sites were mapped using tfSearch (Ovcharenko et al. 2005). tfSearch scores each position in the sequence for each PWM and reports positions with a score above a given threshold. We optimized the threshold for each PWM on a 10-Mb random nucleotide sequence (consisting of multiple short pieces randomly extracted from the human genome) to obtain at most k binding sites every 10 kb. We tested different values of k, which result in different overall sequence densities of TF binding sites predictions (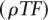 ) (Table 3). PWMs that produce more than the desired number of binding sites predictions for any possible well-defined threshold are excluded from the final set, resulting in slightly different sets of PWMs for each value of k. Many PWMs represent the same transcription factor; we only exclude sites starting at the same position in the sequence during the alignment procedure.
) (Table 3). PWMs that produce more than the desired number of binding sites predictions for any possible well-defined threshold are excluded from the final set, resulting in slightly different sets of PWMs for each value of k. Many PWMs represent the same transcription factor; we only exclude sites starting at the same position in the sequence during the alignment procedure.
Table 3.
Thresholds imposed on the number of occurrences for each TFBS and resulting overall density of TF binding site predictions (for both strands combined, 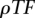 ) computed on a 10-Mb random sequence
) computed on a 10-Mb random sequence

Alignment model
To identify functional orthologs of diverged noncoding elements, we applied a loose definition of TF binding site conservation, comparing pairs of sequences on the basis of the collections of TF binding sites that are shared among them. Our method uses a sliding window approach to calculate an alignment score between the query sequence 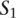 and each window
and each window 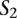 with length
with length 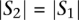 in the target locus, with
in the target locus, with 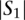 being the length of
being the length of 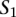 .
.
Let us define a set of labels for different TF binding sites, 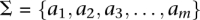 . We break each nucleotide sequence
. We break each nucleotide sequence 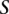 of length
of length 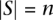 , into a set of ordered pairs
, into a set of ordered pairs 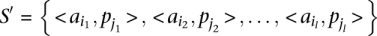 , with
, with 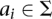 and
and 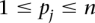 , the starting position of the site.
, the starting position of the site.
Let us now consider two nucleotide sequences, 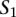 and
and 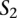 , and their sets of ordered pairs
, and their sets of ordered pairs 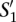 and
and 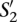 , respectively. Then, we define a TF binding site match between
, respectively. Then, we define a TF binding site match between 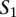 and
and 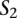 as a triplet,
as a triplet, 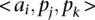 , such that
, such that 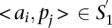 and
and 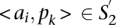 . The score for a match is given by
. The score for a match is given by
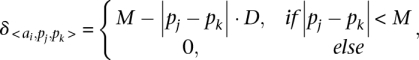 |
where 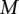 and D are two parameters, heuristically determined, punishing shifts in the relative positions of the TF binding sites in both sequences.
and D are two parameters, heuristically determined, punishing shifts in the relative positions of the TF binding sites in both sequences.
Next, we call a set of TF binding site matches between two sequences 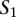 and
and 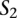 consistent if any two triplets
consistent if any two triplets 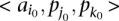 and
and 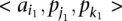 satisfy
satisfy 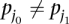 ,
, 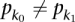 , and
, and 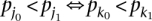 .
.
A TF binding site-based alignment of sequence 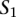 to sequence
to sequence 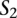 is a mapping from
is a mapping from 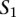 and
and 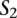 that identifies a consistent set of TF binding site matches. Finding an optimal alignment is equivalent to identifying an alignment with maximum score, which is given by the sum of the scores of involved TF binding site matches. Let
that identifies a consistent set of TF binding site matches. Finding an optimal alignment is equivalent to identifying an alignment with maximum score, which is given by the sum of the scores of involved TF binding site matches. Let 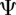 be a consistent set of TF binding site matches, then the score of an optimal alignment is simply:
be a consistent set of TF binding site matches, then the score of an optimal alignment is simply:
TF binding sites are predicted on both forward and reverse strands, which are treated independently for alignment purposes. Regions with no predicted TF binding sites will remain unaligned.
Given a query sequence 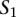 and a search locus
and a search locus 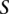 , we compute the optimal alignment between the entire query sequence and each of the possible
, we compute the optimal alignment between the entire query sequence and each of the possible  −
− 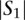 +1 windows of length
+1 windows of length 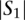 base pairs. We report the location of the window in the search locus with the maximum optimal alignment score.
base pairs. We report the location of the window in the search locus with the maximum optimal alignment score.
Alignment model parameterization
First, we optimized the number of predictions for each PWM in the database. This was done by setting a maximum number of overall TF binding site predictions per base pair (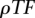 ) in the training data.
) in the training data.
The alignment model has two parameters, 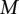 and
and 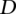 , which concern differences in relative positions of the TF binding sites in the sequences that are being aligned. A TF binding site-match
, which concern differences in relative positions of the TF binding sites in the sequences that are being aligned. A TF binding site-match 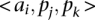 between two sequences
between two sequences 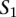 and
and 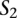 will not contribute to the alignment score (i.e.,
will not contribute to the alignment score (i.e., 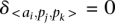 ) if
) if 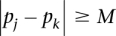 .
. 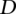 is a penalty for the shift in the location of a given TF binding site. We considered only positive contributions to the scoring function (
is a penalty for the shift in the location of a given TF binding site. We considered only positive contributions to the scoring function (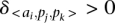 ), assessing only
), assessing only 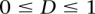 . The optimal values for
. The optimal values for 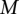 and
and 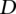 were also empirically optimized on the training data.
were also empirically optimized on the training data.
All the parameters of the model were heuristically determined using a 10-fold-cross-validation on the set of approximately 300 human/zebrafish elements obtained through the tunneling conservation strategy. 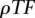 is the most sensitive parameter. The optimal classification rate was obtained for 1.10 TF binding site predictions per base pair (for both strands combined), which corresponds to an average of 22 hits for a given PWM every 10 kb of sequence. Several combinations of
is the most sensitive parameter. The optimal classification rate was obtained for 1.10 TF binding site predictions per base pair (for both strands combined), which corresponds to an average of 22 hits for a given PWM every 10 kb of sequence. Several combinations of 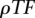 ,
, 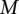 , and
, and 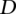 result in a classification rate of 51%. We set
result in a classification rate of 51%. We set 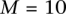 and
and 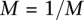 .
.
Significance of the alignment scores
Our alignment algorithm selects the window(s) in the syntenic search locus where the alignment of the probe reaches the maximum score. We discard alignments to multiple windows. To evaluate the significance of the alignments, we analyze the alignments between the probe and two unrelated sequences of similar length and GC content (control loci). In particular, we chose the two loci that flank the syntenic locus and have exactly the same length. Consequently, each human counterpart of a tunneled element is associated with three alignment scores—the score of the alignment to the syntenic locus in the species of interest, e.g., zebrafish, in addition to the scores of the two alignments with the control loci.
In general, the alignment score is (weakly) correlated with the number of TF binding site occurrences in the probe and the number of TF binding site occurrences in the target window (R-squared = 0.3). Because of this, target windows with a higher number of TF binding site occurrences are expected to score higher than windows with a lower number of TF binding site occurrences. In any case, correct alignments tend to have higher alignment scores than random alignments with the same number of TF binding site occurrences (P-value = 1.8 × 10−8, Wilcoxon rank-sum test).
Thus, we used the three alignment scores in addition to the number of PWM occurrences in the reference and target sequence hits to train an SVM with a linear kernel to discriminate orthologous alignments from those found merely by chance. We selected SVMs because they constitute a well-known classification algorithm, suitable for dealing with multidimensional data and able to learn the best features for classification with minimal prior assumptions on the data distribution. To train this classifier, we used our database of tunneled elements; human sequences that are correctly aligned to their zebrafish, frog, and chicken counterparts, respectively, correspond to positive instances, while sequences that are incorrectly aligned constitute negative instances. The average sensitivity of our method at the FPR of 0.05 is 0.5, suggesting that we can use the classifier to make high-confidence predictions of ancestral relationships.
Comparison with alternative methods
We compared our method with four freely available tools. It is worth pointing out that they have been designed with different aims. Each program was run with its default settings and PWM data sets, unless stated otherwise. The method by Blanco et al. (2006) addresses the problem of comparing and characterizing the promoter regions of genes with similar expression patterns and has been optimized by the authors in a collection of human–mouse orthologous gene pairs. As the tool computes global pairwise alignments, we split the zebrafish loci using windows with the same length of TEH, attempted to align TEH to this window, and reported the window with the highest score, similarly to what we do for our own tool. EEL (Hallikas et al. 2006) compares conserved clusters of TFBSs to locate enhancers in mammalian genomes. EMMA and Cluster-Buster (Frith et al. 2003) identify clusters of TFBSs in DNA sequences. EMMA was run using the JASPAR (Bryne et al. 2008) vertebrate collection of PWMs; we pre-computed alignments between the human and zebrafish loci with MUSCLE (Edgar 2004) and then ran EMMA to determine how many of the TEZ were successfully identified as cis-regulatory modules. Free parameters were computed according to the authors' instructions on the training data.
Analysis of the distribution of CNEs in the human genome
To test whether particular elements were randomly distributed, we reallocated each element within its chromosome randomly, following a uniform distribution. We repeated this process 1000 times and computed average cluster sizes. These cluster sizes were then compared to the original cluster sizes.
Functional analysis
To assess whether these elements disproportionally occur near genes with particular functions, we obtained the Gene Ontology (GO) (Ashburner et al. 2000), CVS Version 1.171, GOC Validation Date 11/29/2010 annotations of the closest neighboring UCSC known genes (Hsu et al. 2006) for all noncoding elements and assigned those annotations to each element. Gene-to-GO mapping was achieved by combining the UCSC known gene table and GOA (Barrell et al. 2009) association table using UniProt IDs. All P-values were corrected for multiple hypothesis testing (Bonferroni's method) (Abdi and Salkind 2007). Where applicable, we also corrected for differences in locus length (Taher and Ovcharenko 2009).
Synteny blocks
Synteny blocks were downloaded from the ECRBase database of evolutionarily conserved regions and synteny relationships among vertebrate genomes (Ovcharenko et al. 2005).
ChIP data
As p300 (also known as EP300) binding sites have been mapped to the mouse genome, we first identified their human counterparts using a set of conserved human/mouse elements (Loots and Ovcharenko 2007). Binding events correspond to reads in the Gene Expression Omnibus (GEO) series GSE13845.
In vivo regulatory activity assays
Human and zebrafish sequences were PCR-amplified and subcloned into GFP reporter system constructs (Fisher et al. 2006a,b). These vectors were injected into 200+ zebrafish embryos and were analyzed from 24 to 96 h post-fertilization (hpf) and at 7 d post-fertilization (dpf) for consistent tissue-specific GFP expression. Because expression of enhancers in G0 zebrafish is mosaic, it is problematic to use individual zebrafish for analyses. To provide a better representation of the putative enhancer's regulatory potential, 16 or more zebrafish for each positive set were photographed (Supplemental Figs. 7–9). The expression patterns present in each individual zebrafish embryo were overlaid on a camera lucida zebrafish to create a single composite image (Fig. 4B). The expression was maintained unchanged past 24 hpf in all of the zebrafish sets with positive expression.
Fish care
Zebrafish were raised and bred in accordance with standard conditions (Kimmel et al. 1995; Westerfield 2000). Embryos were raised in embryo medium containing 0.003% phenylthiocarbamide to prevent pigmentation and maintained at 28°C and staged in accordance with standard methods (Kimmel et al. 1995; Westerfield 2000; McGaughey et al. 2008).
Acknowledgments
We gratefully acknowledge three anonymous reviewers for helpful suggestions that greatly improved the manuscript. This research was supported by the Intramural Research Program of the NIH, National Library of Medicine to I.O.; and by the National Institute of Neurological Disease and Stroke (R01 NS062972; NINDS, NIH) to A.S.M. Disclaimer: Certain commercial equipment or materials are identified in this report to specify adequately the experimental procedures. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials or equipment identified are necessarily the best available for the purpose.
Footnotes
[Supplemental material is available for this article. Generated data sets of covert elements are available at http://www.dcode.org/covert/.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.119016.110.
References
- Abdi H, Salkind NJ 2007. Bonferroni and Sidak corrections for multiple comparisons. In Encyclopedia of measurement and statistics. Sage, Thousand Oaks, CA [Google Scholar]
- Andolfatto P 2005. Adaptive evolution of non-coding DNA in Drosophila. Nature 437: 1149–1152 [DOI] [PubMed] [Google Scholar]
- Arunachalam M, Jayasurya K, Tomancak P, Ohler U 2010. An alignment-free method to identify candidate orthologous enhancers in multiple Drosophila genomes. Bioinformatics 26: 2109–2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. 2000. Gene Ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrell D, Dimmer E, Huntley RP, Binns D, O'Donovan C, Apweiler R 2009. The GOA database in 2009—an integrated Gene Ontology Annotation resource. Nucleic Acids Res 37: 396–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K 2007. High-resolution profiling of histone methylations in the human genome. Cell 129: 823–837 [DOI] [PubMed] [Google Scholar]
- Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, Haussler D 2004. Ultraconserved elements in the human genome. Science 304: 1321–1325 [DOI] [PubMed] [Google Scholar]
- Berezikov E, Guryev V, Plasterk RH, Cuppen E 2004. CONREAL: Conserved regulatory elements anchored alignment algorithm for identification of transcription factor binding sites by phylogenetic footprinting. Genome Res 14: 170–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird CP, Stranger BE, Dermitzakis ET 2006. Functional variation and evolution of non-coding DNA. Curr Opin Genet Dev 16: 559–564 [DOI] [PubMed] [Google Scholar]
- Blanchette M, Bataille AR, Chen X, Poitras C, Laganiere J, Lefebvre C, Deblois G, Giguere V, Ferretti V, Bergeron D, et al. 2006. Genome-wide computational prediction of transcriptional regulatory modules reveals new insights into human gene expression. Genome Res 16: 656–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco E, Messeguer X, Smith TF, Guigó R 2006. Transcription factor map alignment of promoter regions. PLoS Comput Biol 2: e49 doi: 10.1371/journal.pcbi.0020049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow MJ, McCulley DJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. 2010. ChIP-Seq identification of weakly conserved heart enhancers. Nat Genet 42: 806–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borneman A, Gianoulis T, Zhang Z, Yu H, Rozowsky J, Seringhaus M, Wang L, Gerstein M, Snyder M 2007. Divergence of transcription factor binding sites across related yeast species. Science 317: 815–819 [DOI] [PubMed] [Google Scholar]
- Bryne JC, Valen E, Tang MH, Marstrand T, Winther O, da Piedade I, Krogh A, Lenhard B, Sandelin A 2008. JASPAR, the open access database of transcription factor-binding profiles: new content and tools in the 2008 update. Nucleic Acids Res 36: D102–D106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermitzakis E, Clark A 2002. Evolution of transcription factor binding sites in mammalian gene regulatory regions: Conservation and turnover. Mol Biol Evol 19: 1114–1121 [DOI] [PubMed] [Google Scholar]
- Dermitzakis ET, Bergman CM, Clark AG 2003. Tracing the evolutionary history of Drosophila regulatory regions with models that identify transcription factor binding sites. Mol Biol Evol 20: 703–714 [DOI] [PubMed] [Google Scholar]
- Edgar RC 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgar G 2006. Different words, same meaning: understanding the languages of the genome. Trends Genet 22: 639–641 [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium, Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, et al. 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher S, Grice EA, Vinton RM, Bessling SL, McCallion AS 2006a. Conservation of RET regulatory function from human to zebrafish without sequence similarity. Science 312: 276–279 [DOI] [PubMed] [Google Scholar]
- Fisher S, Grice EA, Vinton RM, Bessling SL, Urasaki A, Kawakami K, McCallion AS 2006b. Evaluating the biological relevance of putative enhancers using Tol2 transposon-mediated transgenesis in zebrafish. Nat Protoc 1: 1297–1305 [DOI] [PubMed] [Google Scholar]
- Frith MC, Li MC, Weng Z 2003. Cluster-Buster: finding dense clusters of motifs in DNA sequences. Nucleic Acids Res 31: 3666–3668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotea V, Visel A, Westlund JM, Nobrega MA, Pennacchio LA, Ovcharenko I 2010. Homotypic clusters of transcription factor binding sites are a key component of human promoters and enhancers. Genome Res 20: 565–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallikas O, Palin K, Sinjushina N, Rautiainen R, Partanen J, Ukkonen E, Taipale J 2006. Genome-wide prediction of mammalian enhancers based on analysis of transcription-factor binding affinity. Cell 124: 47–59 [DOI] [PubMed] [Google Scholar]
- Hardison RC 2000. Conserved noncoding sequences are reliable guides to regulatory elements. Trends Genet 16: 369–372 [DOI] [PubMed] [Google Scholar]
- Hare EE, Peterson BK, Iyer VN, Meier R, Eisen MB 2008. Sepsid even-skipped enhancers are functionally conserved in Drosophila despite lack of sequence conservation. PLoS Genet 4: e1000106 doi: 10.1371/journal.pgen.1000106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Ling X, Sinha S 2009. Alignment and prediction of cis-regulatory modules based on a probabilistic model of evolution. PLoS Comput Biol 5: e1000299 doi: 10.1371/journal.pcbi.1000299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39: 311–318 [DOI] [PubMed] [Google Scholar]
- Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, Haussler D 2006. The UCSC Known Genes. Bioinformatics 22: 1036–1046 [DOI] [PubMed] [Google Scholar]
- Hu Z, Hu B, Collins JF 2007. Prediction of synergistic transcription factors by function conservation. Genome Biol 8: R257 doi: 10.1186/gb-2007-8-12-r257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Nevins JR, Ohler U 2007. Phylogenetic simulation of promoter evolution: estimation and modeling of binding site turnover events and assessment of their impact on alignment tools. Genome Biol 8: R225 doi: 10.1186/gb-2007-8-10-r225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantorovitz MR, Kazemian M, Kinston S, Miranda-Saavedra D, Zhu Q, Robinson GE, Gottgens B, Halfon MS, Sinha S 2009. Motif-blind, genome-wide discovery of cis-regulatory modules in Drosophila and mouse. Dev Cell 17: 568–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, Hinrichs AS, Kent WJ 2009. The UCSC Genome Browser. Curr Protoc Bioinformatics 28: 1.4.1–1.4.26 [DOI] [PubMed] [Google Scholar]
- Kasowski M, Grubert F, Heffelfinger C, Hariharan M, Asabere A, Waszak S, Habegger L, Rozowsky J, Shi M, Urban A, et al. 2010. Variation in transcription factor binding among humans. Science 328: 232–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ 2002. BLAT—the BLAST-like alignment tool. Genome Res 12: 656–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Baertsch R, Hinrichs A, Miller W, Haussler D 2003. Evolution's cauldron: Duplication, deletion, and rearrangement in the mouse and human genomes. Proc Natl Acad Sci 100: 11484–11489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Sinha S 2010. Towards realistic benchmarks for multiple alignments of non-coding sequences. BMC Bioinformatics 11: 54 doi: 10.1186/1471-2105-11-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel C, Ballard W, Kimmel S, Ullmann B, Schilling T 1995. Stages of embryonic development of the zebrafish. Dev Dyn 203: 253–310 [DOI] [PubMed] [Google Scholar]
- King M-C, Wilson AC 1975. Evolution at two levels in humans and chimpanzees. Science 188: 107–116 [DOI] [PubMed] [Google Scholar]
- Kunarso G, Chia N-Y, Jeyakani J, Hwang C, Lu X, Chan Y-S, Ng H-H, Bourque G 2010. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat Genet 42: 631–635 [DOI] [PubMed] [Google Scholar]
- Loots G, Ovcharenko I 2007. ECRbase: database of evolutionary conserved regions, promoters, and transcription factor binding sites in vertebrate genomes. Bioinformatics 23: 122–124 [DOI] [PubMed] [Google Scholar]
- Loots GG, Locksley RM, Blankespoor CM, Wang ZE, Miller W, Rubin EM, Frazer KA 2000. Identification of a coordinate regulator of interleukins 4, 13, and 5 by cross-species sequence comparisons. Science 288: 136–140 [DOI] [PubMed] [Google Scholar]
- Loots G, Ovcharenko I, Pachter L, Dubchak I, Rubin EM 2002. rVISTA for comparative sequence-based discovery of functional transcription factor binding sites. Genome Res 12: 832–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig MZ 2002. Functional evolution of noncoding DNA. Curr Opin Genet Dev 12: 634–639 [DOI] [PubMed] [Google Scholar]
- Ludwig MZ, Palsson A, Alekseeva E, Bergman CM, Nathan J, Kreitman M 2005. Functional evolution of a cis-regulatory module. PLoS Biol 3: e93 doi: 10.1371/journal.pbio.0030093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusk RW, Eisen MB 2010. Evolutionary mirages: Selection on binding site composition creates the illusion of conserved grammars in Drosophila enhancers. PLoS Genet 6: e1000829 doi: 10.1371/journal.pgen.1000829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, et al. 2003. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res 31: 374–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al. 2006. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res 34: D108–D110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaughey DM, Vinton RM, Huynh J, Al-Saif A, Beer MA, McCallion AS 2008. Metrics of sequence constraint overlook regulatory sequences in an exhaustive analysis at phox2b. Genome Res 18: 252–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narlikar L, Sakabe NJ, Blanski AA, Arimura FE, Westlund JM, Nobrega MA, Ovcharenko I 2010. Genome-wide discovery of human heart enhancers. Genome Res 20: 381–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobrega MA, Ovcharenko I, Afzal V, Rubin EM 2003. Scanning human gene deserts for long-range enhancers. Science 302: 413 doi: 10.1126/science.1088328 [DOI] [PubMed] [Google Scholar]
- Ovcharenko I, Loots GG, Hardison RC, Miller W, Stubbs L 2004. zPicture: Dynamic alignment and visualization tool for analyzing conservation profiles. Genome Res 14: 472–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovcharenko I, Loots GG, Nobrega MA, Hardison RC, Miller W, Stubbs L 2005. Evolution and functional classification of vertebrate gene deserts. Genome Res 15: 137–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, Minovitsky S, Dubchak I, Holt A, Lewis KD, et al. 2006. In vivo enhancer analysis of human conserved non-coding sequences. Nature 444: 499–502 [DOI] [PubMed] [Google Scholar]
- Philippakis AA, He FS, Bulyk ML 2005. Modulefinder: A tool for computational discovery of cis regulatory modules. Pac Symp Biocomput 2005: 519–530 [PMC free article] [PubMed] [Google Scholar]
- Polavarapu N, Marino-Ramirez L, Landsman D, McDonald JF, Jordan IK 2008. Evolutionary rates and patterns for human transcription factor binding sites derived from repetitive DNA. BMC Genomics 9: 226 doi: 10.1186/1471-2164-9-226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard DA, Moses AM, Iyer VN, Eisen MB 2006. Detecting the limits of regulatory element conservation and divergence estimation using pairwise and multiple alignments. BMC Bioinformatics 7: 376 doi: 10.1186/1471-2105-7-376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR 2007. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 35: D61–D65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Wilson M, Ballester B, Schwalie P, Brown G, Marshall A, Kutter C, Watt S, Martinez-Jimenez C, Mackay S, et al. 2010. Five-vertebrate ChIP-Seq reveals the evolutionary dynamics of transcription factor binding. Science 328: 1036–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch R, Hardison RC, Haussler D, Miller W 2003. Human–mouse alignments with BLASTZ. Genome Res 13: 103–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Liang Y, Siggia E 2006. Stubb: a program for discovery and analysis of cis-regulatory modules. Nucleic Acids Res 34: W555–W559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit A, Hubley R, Green P 1996–2010. RepeatMasker Open-3.0. http://www.repeatmasker.org
- Stranger B, Forrest M, Dunning M, Ingle C, Beazley C, Thorne N, Redon R, Bird C, de Grassi A, Lee C, et al. 2007. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315: 848–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taher L, Ovcharenko I 2009. Variable locus length in the human genome leads to ascertainment bias in functional inference for non-coding elements. Bioinformatics 25: 578–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JW, Touchman JW, Blakesley RW, Bouffard GG, Beckstrom-Sternberg SM, Margulies EH, Blanchette M, Siepel AC, Thomas PJ, McDowell JC, et al. 2003. Comparative analyses of multi-species sequences from targeted genomic regions. Nature 424: 788–793 [DOI] [PubMed] [Google Scholar]
- Visel A, Bristow J, Pennacchio LA 2007a. Enhancer identification through comparative genomics. Semin Cell Dev Biol 18: 140–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Minovitsky S, Dubchak I, Pennacchio LA 2007b. VISTA Enhancer Browser—a database of tissue-specific human enhancers. Nucleic Acids Res 35: 88–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. 2009. ChIP-Seq accurately predicts tissue-specific activity of enhancers. Nature 457: 854–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562 [DOI] [PubMed] [Google Scholar]
- Westerfield M 2000. The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio), 4th ed. University of Oregon Press, Eugene, OR. [Google Scholar]
- Wilson MD, Barbosa-Morais NL, Schmidt D, Conboy CM, Vanes L, Tybulewicz VLJ, Fisher EMC, Tavare S, Odom DT 2008. Species-specific transcription in mice carrying human chromosome 21. Science 322: 434–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkopp PJ 2006. Evolution of cis-regulatory sequence and function in Diptera. Heredity 97: 139–147 [DOI] [PubMed] [Google Scholar]
- Woolfe A, Goodson M, Goode DK, Snell P, McEwen GK, Vavouri T, Smith SF, North P, Callaway H, Kelly K, et al. 2005. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol 3: e7 doi: 10.1371/journal.pbio.0030007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie D, Chen C-C, Ptaszek LM, Xiao S, Cao X, Fang F, Ng HH, Lewin HA, Cowan C, Zhong S 2010. Rewirable gene regulatory networks in the preimplantation embryonic development of three mammalian species. Genome Res 20: 804–815 [DOI] [PMC free article] [PubMed] [Google Scholar]