

Abstract
Selenium (Se) is thought to confer cardioprotective effects through the actions of antioxidant selenoprotein enzymes that directly limit levels of ROS such as hydrogen peroxide (H2O2) or that reverse oxidative damage to lipids and proteins. To determine how the selenoproteome responds to myocardial hypertrophy, two mouse models were employed: triidothyronine (T3)- or isoproterenol (ISO)-treatment. After 7 days of T3- and ISO-treatment, cardiac stress was demonstrated by increased H2O2 and caspase-3 activity. Neither treatment produced significant increases in phospholipid peroxidation or TUNEL-positive cells, suggesting that antioxidant systems were protecting the cardiomyocytes from damage. Many selenoprotein mRNAs were induced by T3- and ISO-treatment, with levels of methionine sulfoxide reductase 1 (MsrB1, also called SelR) mRNA showing the largest increases. MsrB enzymatic activity was also elevated in both models of cardiac stress, while glutathione peroxidase (GPx) activity and thioredoxin reductase (Trxrd) activity were moderately and nonsignificantly increased, respectively. Western blot assays revealed a marked increase in MsrB1 and moderate increases in GPx3, GPx4, and Trxrd1, particularly in T3-treated hearts. Thus, the main response of the selenoproteome during hypertrophy does not involve increased GPx1, but increased GPx3 for reducing extracellular H2O2 and increased GPx4, Trxrd1, and MsrB1 for minimizing intracellular oxidative damage.
Keywords: Selenoproteins, cardiac stress, hypertrophy, oxidative stress, antioxidant
1 Introduction
Oxidative stress is induced in the heart by elevated levels of reactive oxygen species (ROS) that are generated in many cardiovascular disorders [1, 2]. Two useful mouse models of cardiac stress that induce increased contractility and cardiomyocyte hypertrophy are treatment with thyroid hormone, triidothyronine (T3), or the β-adrenergic agonist, isoproterenol (ISO). Both treatments exert profound effects on the heart and cardiovascular system. Hyperthyroidism produces a well-recognized spectrum of cardiovascular perturbations that include increased heart rate and contractility, elevated cardiac output, and eventual cardiac hypertrophy [3, 4]. ISO-treatment also causes an increase in heart rate and contractility without increasing systemic blood pressure [5]. Both treatments cause increased myocardial oxygen consumption. Over long periods, these hemodynamic changes can ultimately lead to “high output” heart failure and some data suggest that increased oxidative stress contributes to cardiac pathology caused by hyperthyroidism or chronic catecholamine exposure [6–10]. Excessive or sustained ROS may damage cellular constituents including lipids, proteins, and nucleic acids. In addition, evidence is emerging for a direct role of ROS in mediating the hypertrophic process through specific cell signaling pathways [11]. However, it remains unclear what role ROS or the key enzymes that regulate their levels play in hypertrophy.
Dietary selenium (Se) is an essential micronutrient that has been suggested to play an important role in protecting the heart from oxidative stress [12–14], although mechanisms by which this may occur are not well understood. Se exerts its biological effects mainly through its direct incorporation into selenoproteins as the amino acid, selenocysteine. Twenty-five selenoproteins have been identified in humans, all but one of which also exist as selenocysteine-containing proteins in rodents [15]. Functions have been identified for just over half of the selenoproteins, and these include regulation of thyroid hormone metabolism, intra- and extra-cellular antioxidation, redox regulation, protein retrotranslocation from the ER to cytoplasm, and sperm maturation/protection. Important antioxidant, cardioprotective roles have been described for selenoproteins including glutathione peroxidase (GPx) 1, 3 and 4 and thioredoxin reductase (Txnrd) 1 and 2 [16]. Interestingly, the thioredoxin(Txn)/Txnrd system has been implicated not only in protecting against oxidative damage, but in regulating ROS-dependent hypertrophic signaling in cardiac myocytes by reversing oxidative modification of free reactive thiols (S-thiolation) on the small G protein Ras and regulating Ras activity [17].
Methionine sulfoxide reductase B1 (MsrB1, described initially as SelR or SelX) is one of four methionine sulfoxide reductase (Msr) proteins in mammals, and this group of proteins also includes MsrA, MsrB2, and MsrB3 [18]. ROS can oxidize methionine residues in proteins to produce a mixture of S- and R-forms of methionine sulfoxide, which can be reduced by MsrA and MsrB enzymes, respectively [19]. Among Msr enzymes, MsrB1 is the only selenoprotein. MsrB1 is ubiquitously expressed in all tissues, with highest levels found in liver and kidney, and a MsrB1 knockout mouse model was recently described in which these tissues were the most susceptible to oxidative damage of proteins [20]. Whether MsrB1 protects the heart from oxidative stress under normal or pathological conditions has not been previously examined.
In the present study, we analyzed heart tissue from mice subjected to T3- and ISO-treatment to identify selenoproteins or factors involved in their synthesis that are induced during cardiac stress. Induced expression of MsrB1 mRNA was highest of all proteins examined and was accompanied by increases in protein levels and enzymatic activity. GPx activity correlating with increased GPx3 and GPx4 protein expression was also increased with T3-treatment. Our results define the selenoproteomic response that helps to protect the heart against stress-induced apoptosis.
2 Materials and methods
2.1 Mice and Se status determination
C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Male mice were used at 8–10 weeks of age. All animal protocols were approved by the University of Hawaii Institutional Animal Care and Use Committee. Serum was analyzed for levels of Se by inductively coupled plasma mass spectrometry (West Coast Analytical Services/Exova; Santa Fe Springs, CA, USA).
2.2 T3- and ISO-treatments and tissue collection
An established protocol for inducing hyperthyroidism was followed that involved daily i.p. injections with 1 mg/kg 3,3’,5-triido-L-thyronine sodium salt (T3; Sigma) or PBS as a negative control [21]. For the ISO model, isoproterenol bitartrate was delivered by osmotic minipumps (Alzet Corp., Cupertino, CA) at 20 mg/kg/day, again with PBS as negative controls. After 4 or 8 days of treatment, the mice were weighed and sacrificed. The hearts were immediately removed, washed in PBS, blotted dry, weighed, and frozen in liquid nitrogen. The entire heart was pulverized and divided into separate tubes for RNA or protein extraction. For histology, whole hearts were dissected and were frozen in Tissue-Tek O.C.T. compound (Ted Pella, Inc., Redding, CA) and stored at −80°C until sections were prepared.
2.3 RNA extraction and real-time PCR
Tissue samples were thawed and RNA extracted using RNeasy Mini kit and RNase-free DNase I (both from Qiagen). Concentration and purity of extracted RNA was determined using A260/A280 measured on an ND1000 Spectrophotometer (NanoDrop Technologies). Synthesis of cDNA was carried out using Superscript III (Invitrogen) and oligo dT primer, with 2 µg of total RNA per 50 µl reaction. For real-time PCR, 1 µl of this cDNA was used in 10 µl reactions with Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen). Reactions were carried out in a 9700HT thermal cycler (Applied Biosystems, Foster City, CA). Oligonucleotides used for murine selenoproteins and selenoprotein synthesis factors have been previously described [22]. In addition, for real-time PCR amplification of BNP we used: fwd: 5’-ATC TCC TGA AGG TGC TGT CCC AG-3’; rev: 5’-GGT CTT CCT ACA ACA ACT TCA GTG CGT TAC-3’. Cycling conditions included 45 cycles with a hybridization temperature of 55°C. For relative quantitation, pilot studies were performed using several housekeeping genes, and hypoxanthine phosphoribosyltransferase (hprt) and ubiquitin c (ubc) were found to be relatively stable comparing PBS-treated to T3-treated conditions. To normalize target mRNA of high abundance such as BNP, the high abundance housekeeping mRNA, ubc, was chosen. In examining the entire selenoprotein transcriptome that contain several low abundance mRNAs, the lower abundance hprt was utilized.
2.4 Protein extraction, western blots, and enzymatic assays
Protein was extracted from whole hearts by homogenizing powdered tissue on ice in 10 ml of CellLytic MT buffer (Sigma) containing 1 mM DTT, 1X protease inhibitor cocktail (Calbiochem), and 5 mM EDTA. Homogenate was centrifuged at 12,000 rpm (13,000 g; Beckman Coulter microcentrifuge) for 10 min and supernatant removed and stored at −70°C. Bradford assay was carried out using Bradford Reagent (Bio-Rad) and 30 µg total protein was combined with reduced Laemmli buffer, boiled at 95°C for 10 min, cooled on ice, and loaded into wells of 10–14.5% polyacrylamide gradient gels (Bio-Rad). Protein was transferred to PVDF membranes (Millipore), blocked for 1 h with 5% BSA, and then probed for 1 h with primary antibodies, including polyclonal anti-GPx1 and anti-GPx4 (Lab Frontier, Seoul, Korea), anti-GPx3 (Imgenex), and mouse monoclonal anti-α-tubulin (Novus Biologicals). Also used in western blots were rabbit polyclonal antibodies raised against bacterially expressed, Cys-mutant versions of MsrB1, Txnrd1 and 2, and proteins MsrB2 and MsrA. Appropriate HRP-conjugated secondary antibodies from Jackson Immunolabs were incubated with the membranes for 45 min and detected using ECL Plus (GE Healthcare). For densitometry, digital images of autoradiographic film were captured using Gel Logic 200 and Kodak MI software (Kodak Scientific Imaging Systems). This software was used to measure mean intensity from regions of interest (ROI) that corresponded to the bands to be measured. The intensity of the target bands (e.g. GPx4) was normalized to that of the loading control band (e.g. α-tubulin) to obtain normalized levels of target proteins. Txnrd activity was measured using an activity assay kit (Sigma) based on the reduction of 5,5'-dithiobis(2-nitrobenzoic) acid (DTNB) with NADPH to 5-thio-2-nitrobenzoic acid (TNB), which produces a strong yellow color that is spectrophotometrically measured at 412 nm. Glutathione peroxidase activity (GPx) was measured using the GPx-340 kit (OxisResearch), which is based on GPx generating GSSG, which is then reduced by gltathione reductase. The latter leads to the oxidation of NADPH to NADP+ that is detected by a decreased A340. MsrB activity was assayed as previously described [20], using a substrate specific for MsrB that is not recognized by MsrA. Hydrogen peroxide was measured using a fluorescence-based assay involving an iron and xylenol orange reagent (Thermo Fisher) and caspase-3 activity was assayed using a Caspase 3 Fluorescence Assay kit (R&D Systems). The LPO-586 kit (OxisResearch) was used to measure malonaldehyde (MDA) levels as an indicator of lipid peroxidation.
2.5 Measurement of anatomical structures and TUNEL Assay
Left ventricular mass was measured in unsedated mice with transthoracic echocardiography (VEVO 2100, VisualSonics, Canada) using a 38MHz transducer. Left ventricular parasternal short-axis views were obtained in M-mode, imaging at the papillary muscle level. Three consecutive beats in two M-mode images (6 beats total) were used for measurements of diastolic values for left ventricular interventricular septum (IVS;d), left ventricular interior diameter (LVID;d), and left ventricle posterior wall (LVPW;d). The corrected LV Mass was calculated as follows: (M-Mode LV Mass (1.053 *((LVID;d + LVPW;d + IVS;d)3 − LVID;d3)) * 0.8). Measurements were taken at baseline and on T3- and ISO-treated mice prior to sacrifice by an operator blinded to treatment. Frozen heart tissue sections were analyzed for TUNEL-positive cells using the In Situ Cell Death Detection kit (Roche). Negative controls from healthy mice were included as well as positive controls using DNAse I per manufacturer’s instructions. Slides were examined using a Zeiss Axioscop 2 plus equipped with Axio Cam MRc.
2.6 Statistical Analyses
All statistical tests were performed using GraphPad Prism version 4.0 for Windows (GraphPad Software, San Diego, CA). Means of two groups were compared using a Student’s t test and significance was defined as p < 0.05. For constructing standard curves, non-linear regression analysis and curve fitting were performed using Prism 4.0.
3 Results
3.1 T3- and ISO-treatment result in hypertrophic hearts
T3- and ISO-treatments both lead to myocardial hypertrophy, but the signaling pathways, redox effects, and alterations in systemic Se status may differ for each disease state. Thus, we used both treatments to induce myocardial hypertrophy in C57BL/6 mice and their effectiveness was analyzed by determining the heart:body mass ratio. No changes in body mass were observed during T3- or ISO-treatment, so detectable changes in the ratios were attributable to changes in heart mass. Results showed that heart:body mass ratios were significantly increased in both T3- and ISO-treated mice compared to controls (Figure 1A). Brain natriuretic peptide (BNP) is a cardiac neurohormone [23] and its expression increases to counteract ventricular wall stress [24]. T3- and ISO-treatments resulted in approximately 2-fold and 1.5-fold increases in mRNA levels for BNP, respectively (Figure 1B), consistent with other mouse models of hypertrophy [25]. Increased left ventricular mass was confirmed using ultrasound at baseline and after 7 d T3- or ISO-treatment to determine the change in mass per mouse (Fig. 1C). As expected, both treatments resulted in increased left ventricular mass, confirming adaptive hypertrophic remodeling. Importantly, there were no differences in serum Se concentration with either T3- or ISO-treatment (Table I).
Figure 1.
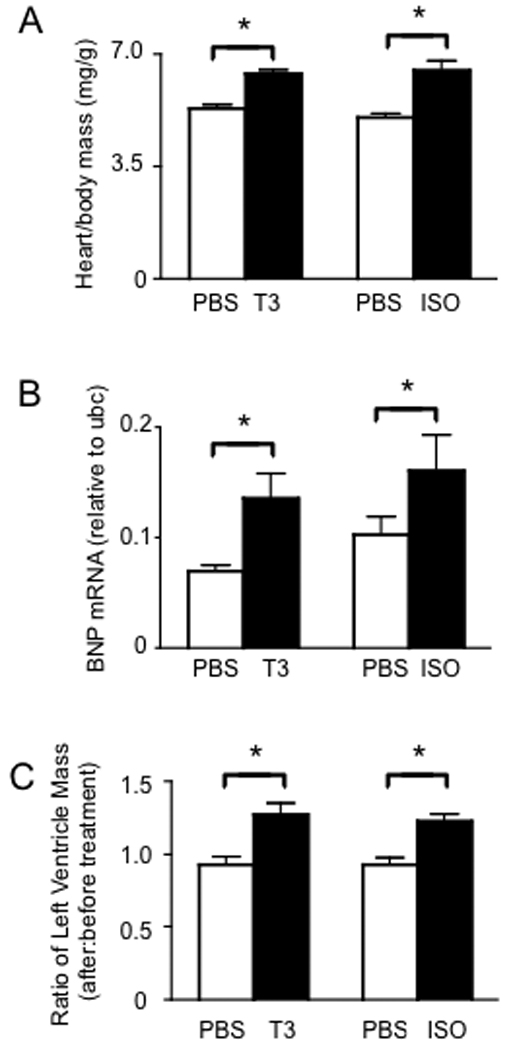
Treatment with T3 or ISO results in myocardial hypertrophy. Male C57BL/6 mice were treated with 1 mg/kg T3 or 20 mg/kg/day ISO for 7 d, with PBS serving as a negative control for both treatments. (A) Heart to body mass ratios demonstrate hypertrophy (N = 10 mice per group). (B) Real-time PCR measuring BNP mRNA levels relative to the high-abundance, stable housekeeping mRNA, ubiqutin-c (ubc). Each real-time reaction was performed in triplicate (N = 4 mice per group). C) Anatomical features were measured before and after T3- or ISO-treatment using ultrasound and left ventricular mass calculated as described in the Methods section (N = 3 for PBS groups and N = 5 for T3- and ISO-treatment groups). For all graphs, results represent mean + SE with means compared using a student's t test, *p < 0.05.
Table I.
Serum selenium concentrations
| T3 Experiment | PBS | 384 ± 18 |
| T3 | 384 ± 25 | |
| ISO Experiment | PBS | 385 ± 8.2 |
| ISO | 368 ± 21 |
Numbers represent mean ± S.D. (N = 5)
3.2 T3- and ISO-treatments increase cardiac stress but do not damage heart tissue
Heart tissues were first evaluated for stress by measuring levels of caspase-3 activity. Both T3- and ISO-treatment led to increased caspase-3 activity in the heart (Fig. 2A). However, extensive evaluation of heart tissue by TUNEL assay revealed no detectable apoptosis in hearts of T3- and ISO-treated mice (data not shown). Both treatments increased levels of H2O2 in heart tissue (Fig. 2B). Increases in lipid peroxidation, if not corrected by enzymes such as GPx4, can lead to cellular injury. Therefore, we analyzed lipid peroxidation by measuring the lipid peroxide break-down product, malonaldehyde (MDA), which was not significantly increased in hearts from either treatment. Collectively, these results suggest that hypertrophy induced by T3 and ISO is accompanied by oxidative stress, but this does not lead to detectable levels of cellular damage or death at the doses administered.
Figure 2.
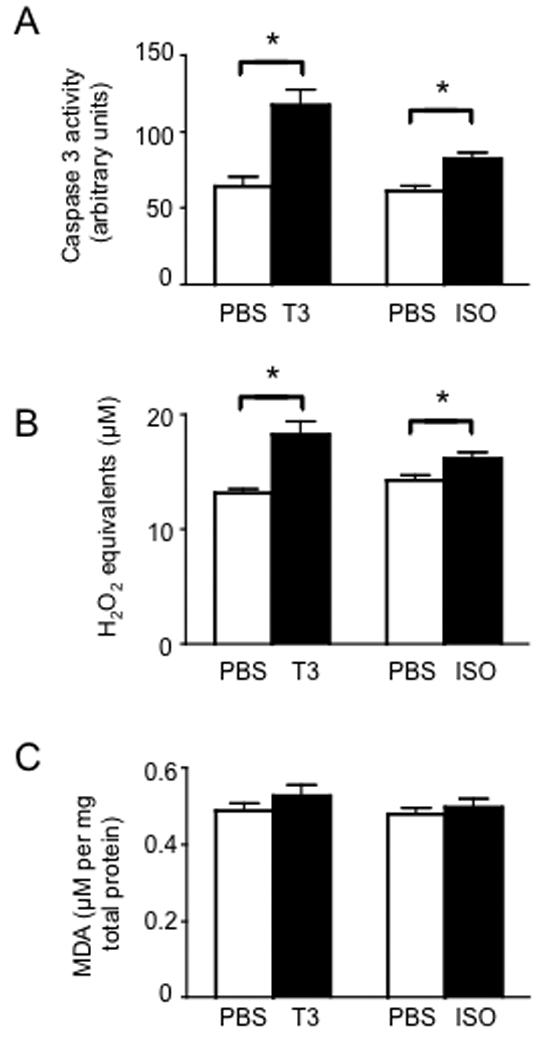
Myocardial hypertrophy is accompanied by increased cardiac stress. (A) Caspase-3 activity was measured in hearts from mice treated for 7 d with either T3 or ISO compared to PBS as negative controls, with fluorescence generated by caspase-3 activity (arbitrary fluorescence units). (B) Hydrogen peroxide was measured using a fluorescence-based assay. (C) Levels of malonaldehyde (MDA) as an indicator of lipid peroxidation were measured by spectrophotometry. For all graphs, results represent mean + SE (N = 5 mice per group) with means compared using a student's t test, *p < 0.05.
3.3 Selenoprotein mRNA levels are increased in heart tissue during hypertrophy
We next used real-time PCR to analyze the relative abundance of mRNA for all 24 murine selenoproteins and 3 factors involved in their synthesis. Consistent with results from our previous study [22], five mRNAs (DIO 1, 2, and 3, Sel V and GPx2) were found to be at levels below reliable detection in heart tissue and this was found to be the case in hearts from hypertrophic mice and controls (data not shown). Therefore, we focused our analyses on the remaining members of the selenoprotein transcriptome and results demonstrated that mRNA abundance of several selenoproteins was increased in hypertrophic hearts (Fig. 3). Selenoprotein mRNAs were particularly elevated in hearts of T3-treated mice, with highest levels exhibited for MsrB1 mRNA. MsrB1 mRNA was increased in hypertrophic hearts above the housekeeping mRNA included as comparison transcripts, including actin, gapdh and ubc. For the ISO-treated mice, selenoprotein mRNAs showing the largest increases compared to PBS controls at both days 4 and 8 included GPx1 and MsrB1. The T3-treatment increased these mRNAs more robustly than the ISO-treatment, consistent with data in Fig. 2 demonstrating higher levels of oxidative stress induced by T3 compared to ISO. The 4 and 8 day time-points to evaluate early and late levels, respectively, of mRNA during the hypertrophic response. Interestingly, T3 showed increases of most selenoprotein mRNAs at both time-points, while increases for selenoprotein mRNAs was mostly induced by ISO-treatment after 8 days of treatment. This suggests the higher ROS found with T3-treatment may have a broader, more sustained impact across the entire selenoprotein transcriptome compared to ISO-treatment.
Figure 3.
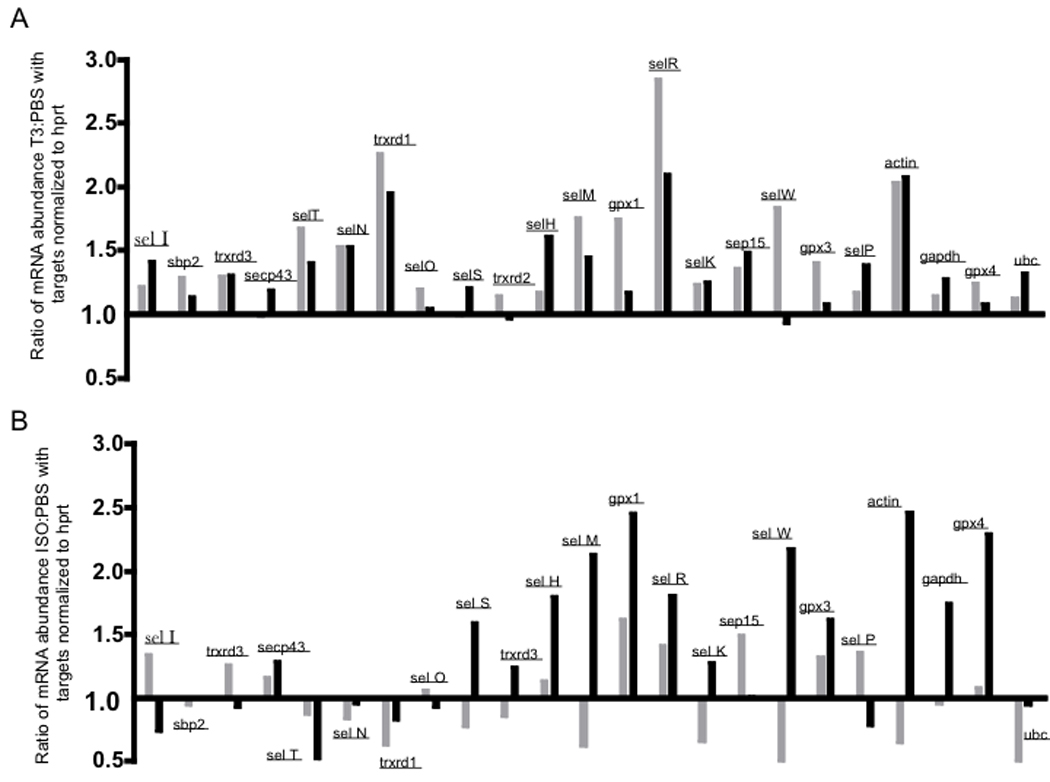
Abundance of mRNA from hearts obtained on days 4 (grey bars) or 8 (black bars), was compared to the low-abundance, stable housekeeping mRNA, hypoxanthine phosphoribosyltransferase (hprt). The relative levels of each target mRNA in T3-treated (A) or ISO-treated (B) mice were compared to relative levels in PBS-treated controls, with three mice included per group at each end-point. Results for five selenoproteins (dio-1, -2, and -3, and sel V and GPx2) were excluded from analyses due to negligible detection of mRNA in either treated mice or controls.
3.4 Protein levels for certain selenoproteins were increased in hypertrophic hearts
Selenoprotein mRNA abundance is regulated at both levels of transcription and mRNA stability [26, 27], which may change levels of selenoprotein mRNA during stress. However, this may not necessarily result in altered protein levels. To evaluate protein expression, we focused on representative antioxidant selenoproteins for which enzymatic assays are available for measuring biological activity. First, GPx activity was increased in hearts from T3-treated mice compared to controls (Fig. 4A). Although a slight increase was also found with ISO-treatment, the difference compared to controls was not statistically significant. To determine which GPx enzymes may contribute to the increased GPx activity in these cells, we performed western blot analyses and found GPx3 and GPx4, but not GPx1, were increased in hearts from T3-treated mice (Fig. 4B–C).
Figure 4.
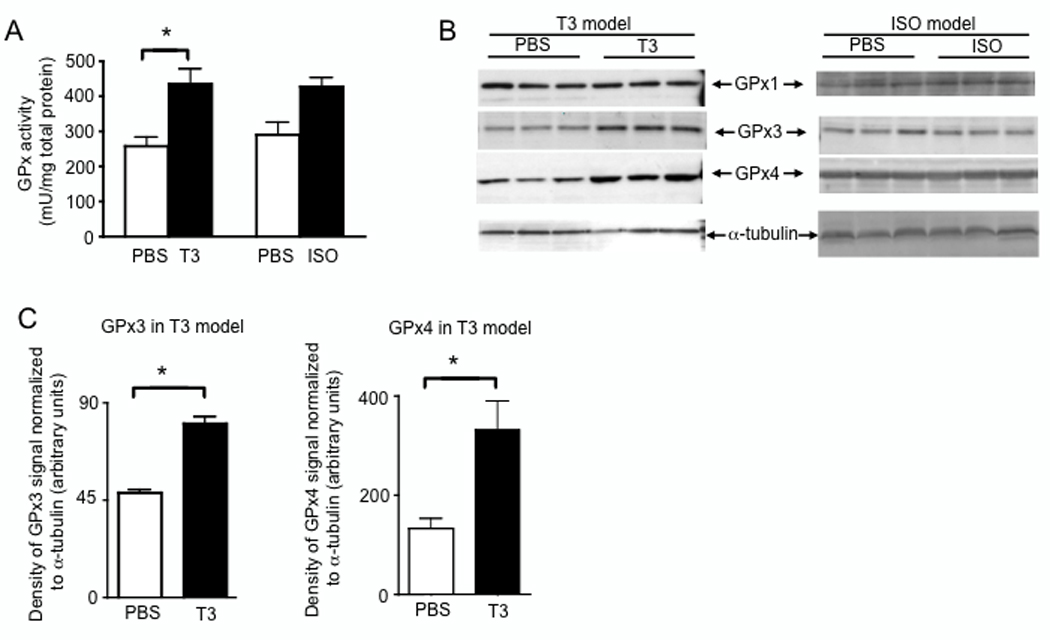
Protein levels of GPx3 and GPx4, but not GPx1, are increased in hearts during T3-induced myocardial hypertrophy. (A) After 7 d of T3- or ISO-treatment, protein extracted from heart tissue was analyzed for total GPx activity. Enzymatic activity was normalized to total protein as determined by Bradford assay. (B) Levels of GPx1 and GPx4 were analyzed by western blot. For each lane, 30 µg total protein was loaded and equivalent loading was verified using anti-α-tubulin. (C) Densitometry was used to estimate quantities of GPx protein detected by western blot as described in the Methods section. For (A) and (C) results represent mean ± SE (N = 3 per group) with means compared using a student's t test, *p < 0.05.
Another important subgroup of selenoenzymes in the heart includes Txnrd1 and 2. Txnrd activity was not significantly increased in hearts of T3- or ISO-treated mice compared to controls (Fig. 5A). Consistent with this finding, no significant differences were found in Txnrd 1 or 2 protein levels for hearts from T3- or ISO-treated mice compared to controls (Fig. 5B). Overall, the oxidative stress induced by T3- or ISO-treatment does not appear to affect expression of either Txnrd1 or 2.
Figure 5.
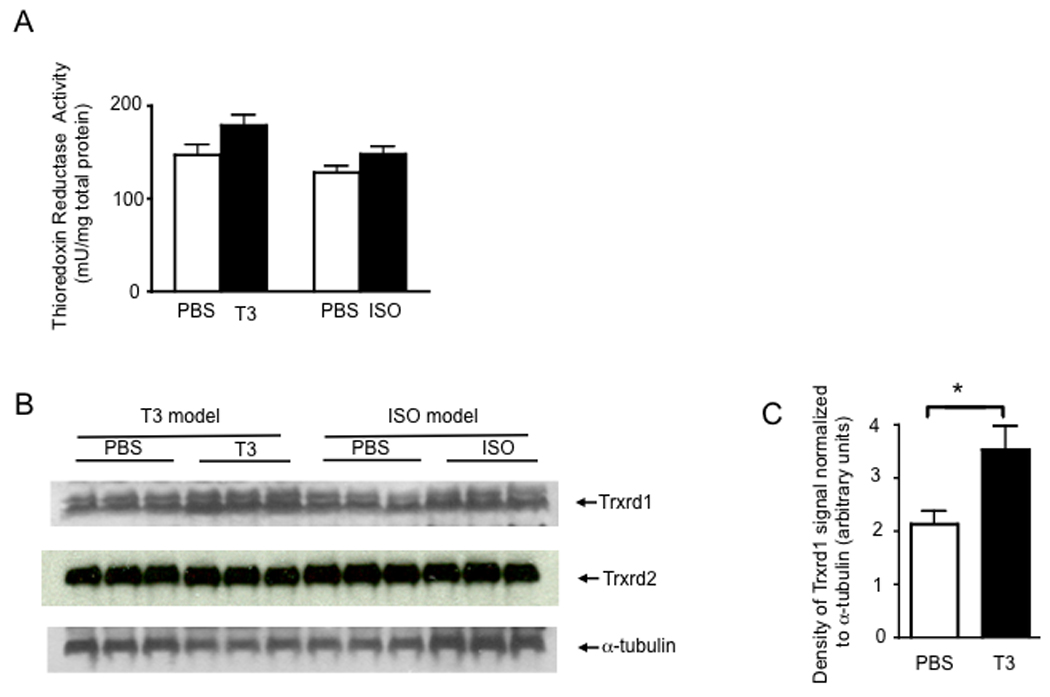
Thioredoxin reductase 1 is significantly increased with T3-treatment. (A) After 7 d of T3- or ISO-treatment, protein extracted from heart tissue was analyzed for total Trxrd activity. Enzymatic activity was normalized to total protein as determined by Bradford assay. Results represent mean ± SE (N = 4 per group) with means compared using a student's t test, T3-treatment vs. PBS p = 0.09; T3-treatment vs. PBS p = 0.13. (B) Western blot analysis of lysates showed increased Txnrd1, but not Txnrd2. (C) Densitometry results for Txnrd1 with results representing mean ± SE (N = 3 per group) with means compared using a student's t test, *p < 0.05.
Another antioxidant selenoprotein evaluated was MsrB1, which together with non-selenoproteins MsrB2, MsrB3, and MsrA constitute an enzymatic defense system responsible for reversing sulfoxidation of methionine residues in proteins, thereby preserving the functional integrity of these proteins [28]. Specific MsrB activity was significantly increased in response to both T3- and ISO-treatment (Fig. 6A). Western blot analyses revealed that, of the Msr enzymes expressed in heart (MsrA, MsrB1 and 2), only MsrB1 expression was significantly induced in hearts with both treatments, with particularly strong MsrB1 expression induced by T3-treatment (Fig. 6B–C). These results are consistent with the relatively high levels of MsrB1 mRNA detected in both cardiac stress models as shown in Fig. 2.
Figure 6.
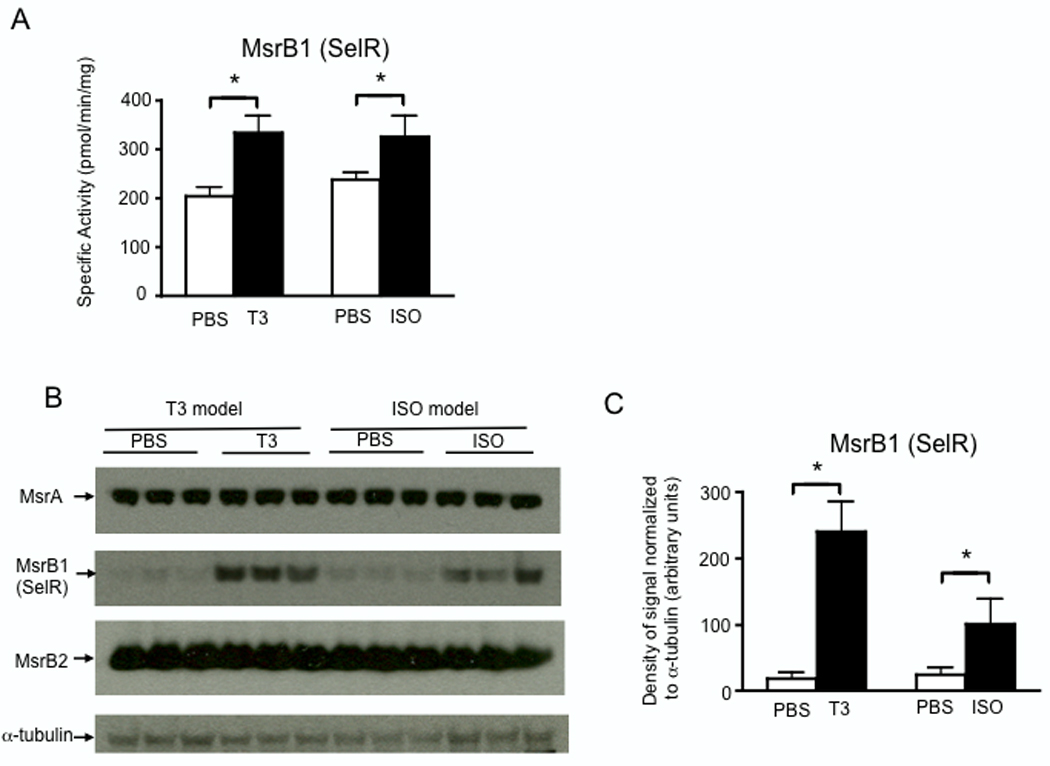
Protein levels of MsrB1 (SelR), but not MsrB2 or MsrA, are increased in hearts during myocardial hypertrophy. (A) After 7 d of T3- or ISO-treatment, protein extracted from heart tissue was analyzed for MsrB activity. Enzymatic activity was normalized to total protein as determined by Bradford assay. (B) Levels of MsrB1, MsrB2, and MsrA were analyzed by western blot. For each lane, 30 µg total protein was loaded and equivalent loading was verified using anti-α-tubulin. (C) Densitometry was used to estimate quantities of MsrB1 protein detected by western blot. For (A) and (C) results represent mean ± SE (N = 3 per group) with means compared using a student's t test, *p < 0.05.
4 Discussion
Oxidative stress is increased in many pathological conditions, including excessive thyroid hormone or catecholamines [3, 29, 30]. It follows that T3- or ISO-treatment may result in increased expression and/or activity of antioxidant enzymes induced in cardiomyocytes as a protective response to limit cellular damage. Identification of antioxidant enzymes or molecules involved in mitigating oxidative damage is a key step in understanding compensatory mechanisms that protect the heart. Results presented in this study demonstrate that certain selenoprotein-based enzyme systems are induced during cardiac stress. In particular, expression of MsrB1, a selenoprotein with no previously described role in cardioprotection, is upregulated in heart tissue in response to T3- and ISO-treatment. These data are relevant to human cardiovascular health as genetic variation or other factors that reduce the activity of this protein could increase the damage of an oxidative stress to the heart. Furthermore, MsrB1 may provide a more specific therapeutic target for modulating cardiac stress compared to altering Se intake, which affects expression levels of several selenoproteins and may also alter redox status in cells through small molecular weight selenocompounds.
The development of concentric left ventricular (LV) hypertrophy in pressure overload is considered adaptive because the parallel deposition of new sarcomeres and the corresponding LV wall thickening normalizes wall stress despite the high intracavitary systolic pressure [31]. While a role for MsrB1 in hypertrophic hearts has not been previously described, a protective role for the nonselenoprotein MsrA in the heart has been suggested by others. For example, overexpression of MsrA in primary neonatal rat cardiomyocytes was shown to protect against ischemia/reperfusion-induced cell death [32]. Furthermore, in vivo studies by Erickson et al demonstrated a requirement for MsrA in regulating pro-apoptotic signals during myocardial infarction [33]. MsrA and MsrB1 enzymes may play overlapping roles in reversing oxidative damage to methionine (Met) residues in proteins, and there remains the possibility that they regulate apoptosis through differential reduction of sulfoxidated Met in signaling molecules. Others have noted that the general phenotypes of the MsrB1−/− and MsrA−/− mice appear to differ in vulnerability to oxidative stress in different tissues [20], suggesting that the functions of these two Msr enzymes may not be completely complementary. Investigation is underway to elucidate specific, non-complementary roles played by MsrB1 and MsrA in reversing methionine sulfoxidation induced by different levels or types of ROS under different conditions in heart tissue.
T3 treatment was recently shown to increase serum Se concentrations and affect the expression of several selenoproteins in serum, liver and kidney [34]. However, we found no differences in serum Se concentration with either T3- or ISO-treatment, suggesting that this was not a major factor affecting the selenoprotein transcriptome in the models as used in this study. The discrepancy with the study by Mittag et al could be due to higher Se in our chow, as suggested by higher serum Se in our PBS group compared than untreated mice the previous study, and was actually closer to the T3-treated group in the previous study. Other differences between our study and the Mittag et al study included period of T3-treatment (7 d vs. 12 d) and route of administration (i.p. injection vs. drinking water).
The GPx enzymes are selenoproteins that have been studied extensively in models of cardiovascular disease. Different GPx enzymes use glutathione (GSH) to detoxify hydroperoxides in intracellular and extracellular spaces as well as lipid peroxides in cellular membranes. GPx1 is particularly important for detoxifying intracellular ROS such as H2O2. Loscalzo and colleagues have shown that heterozygous deficiency of GPx1 leads to endothelial dysfunction, which in turn produces significant structural abnormalities in vascular and cardiac tissues [35]. This group has also shown that GPx1 is important for protecting against oxidative damage from ischemia-reperfusion, findings corroborated by others [36]. Interestingly, we did not observe an increase in GPx1 protein, despite the increased H2O2 detected in both T3- and ISO-treated hearts. Elevated levels of H2O2 that accompany the hypertrophic process may instead induce expression of GPx3 to minimize exposure of extracellular matrix to H2O2 and expression of GPx4 to minimize damage to lipids. Lipid peroxidation has been shown to occur from increased ROS in several different models of myocardial hypertrophy, including that induced by hyperthyroidism [30]. Recent work has established a role for GPx4 as a sensor of oxidative stress and a transducer of cell death signals [37]. Thus, the increased levels of GPx4 observed in heart tissue during T3- and ISO-treatment may contribute to increased apoptotic signaling (activated caspase-3) found in our investigation. However, the increased caspase-3 activity induced by T3- or ISO-treatment was not sufficient for inducing apoptosis in the cardiomyocytes.
Our GPx data also show discordance in the translational response to increased mRNA abundance for selenoproteins. In particular, both T3- and ISO-treatment resulted in increased mRNA levels of GPx1 compared to GPx3 or GPx4. Yet we found higher levels of GPx3 and GPx4 proteins, and not GPx1, particularly with T3-treatment. This emphasizes the levels at which levels of these antioxidant selenoproteins may be regulated involve processes in addition to transcription. The lack of increased protein expression for GPx1 is more difficult to reconcile, given the increased ROS in cardiomyocytes that occur during hypertrophy. A possible explanation is that increased cytoplasmic ROS result from the cellular processes that drive the increase in cardiomyocyte size. The cardiomyocytes may maintain levels of GPx1 protein at basal levels so that pro-hypertrophic ROS that act as secondary messengers, such as H2O2, are not enzymatically reduced by GPx1 and are thus available for directing adaptive cellular hypertrophy.
In addition to the GPx, Trxrd, and Msr enzymes analyzed in that latter part of our study, mRNAs for several other selenoproteins were altered during T3- and ISO-treatment (e.g. SelH, SelM, and SelW). The changes in SelW are puzzling, given SelW protein is not expressed in rodent hearts, but is detected in primate hearts [22, 38]. The increases in SelH may reflect its suggested role in redox-regulated gene transcription [39, 40]. Similar to SelH, SelM contains a thioredoxin fold motif, although this selenoprotein is localized to the endoplasmic reticulum. Although SelH and SelM both may function as oxidoreductases in nucleus and endoplasmic reticulum, respectively, it remains unclear the specific biological roles these selenoproteins play. Further investigation into the redox functions of these and other selenoproteins during hypertrophy is warranted.
The protective effects of Se supplementation in humans is controversial due to several factors including problems with study design including differences in starting Se status [41]. Conflicting findings in human studies related to cardiovascular disease as well as other endpoints suggest that Se supplementation may benefit only certain populations, such as Se-deficient or genetically predisposed groups [16]. A fuller understanding of how dietary Se levels affect human health requires direct evaluation of the effects of selenoproteins on cellular processes. Overall, our data provide insight into how the selenproteome responds to cardiac stress, and in the heart it appears that MsrB1, GPx3, and GPx4 play an important protective role in circumstances of oxidative stress.
> We model myocardial hypertrophy using thyroid hormone and isoproteranol in mice. > The relationship between oxidative stress and selenoprotein expression in hearts is investigated. > H2O2 and caspase-3 activity are increased, but no apparent tissue damage or apoptosis. > MsrB1(Sel R), GPx3, and GPx4 are increased. > GPx1, Trxrd1, and Trxrd2 expression are not changed in heart.
Acknowledgements
We thank the Histology, Genomics, and Mouse Phenotyping Cores at JABSOM for assistance with this study. This work was supported by NIH grant RR016453.
Abbreviations used
- BNP
Brain natriuretic peptide
- GPx
glutathione peroxidase
- ISO
isoproterenol
- Msr
methionine sulfoxide reductase
- ROS
reactive oxygen species
- Se
selenium
- Trxrd
thioredoxin reductase
- T3
triidothyronine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hori M, Nishida K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc Res. 2009;81:457–464. doi: 10.1093/cvr/cvn335. [DOI] [PubMed] [Google Scholar]
- 2.Berndt C, Lillig CH, Holmgren A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: implications for diseases in the cardiovascular system. Am J Physiol Heart Circ Physiol. 2007;292:H1227–H1236. doi: 10.1152/ajpheart.01162.2006. [DOI] [PubMed] [Google Scholar]
- 3.Klein I, Ojamaa K. Thyroid hormone and the cardiovascular system. N Engl J Med. 2001;344:501–509. doi: 10.1056/NEJM200102153440707. [DOI] [PubMed] [Google Scholar]
- 4.Klein I, Ojamaa K. Thyroid hormone-targeting the heart. Endocrinology. 2001;142:11–12. doi: 10.1210/endo.142.1.7986. [DOI] [PubMed] [Google Scholar]
- 5.Grimm D, Elsner D, Schunkert H, Pfeifer M, Griese D, Bruckschlegel G, Muders F, Riegger GA, Kromer EP. Development of heart failure following isoproterenol administration in the rat: role of the renin-angiotensin system. Cardiovasc Res. 1998;37:91–100. doi: 10.1016/s0008-6363(97)00212-5. [DOI] [PubMed] [Google Scholar]
- 6.Venditti P, Di Meo S. Thyroid hormone-induced oxidative stress. Cell Mol Life Sci. 2006;63:414–434. doi: 10.1007/s00018-005-5457-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asayama K, Dobashi K, Hayashibe H, Megata Y, Kato K. Lipid peroxidation and free radical scavengers in thyroid dysfunction in the rat: a possible mechanism of injury to heart and skeletal muscle in hyperthyroidism. Endocrinology. 1987;121:2112–2118. doi: 10.1210/endo-121-6-2112. [DOI] [PubMed] [Google Scholar]
- 8.Civelek S, Seymen O, Seven A, Yigit G, Hatemi H, Burcak G. Oxidative stress in heart tissue of hyperthyroid and iron supplemented rats. chaJ Toxicol Environ Health A. 2001;64:499–506. doi: 10.1080/152873901753215957. [DOI] [PubMed] [Google Scholar]
- 9.Chatelain P, Gremel M, Brotelle R. Prevention by amiodarone of phospholipid depletion in isoproterenol-induced ischemia in rats. Eur J Pharmacol. 1987;144:83–90. doi: 10.1016/0014-2999(87)90012-4. [DOI] [PubMed] [Google Scholar]
- 10.Blasig IE, Blasig R, Lowe H. Myocardial lipid peroxidation during isoproterenol-induced blood flow reduction in rat myocardium. Biomed Biochim Acta. 1984;43:S171–S174. [PubMed] [Google Scholar]
- 11.Pimentel DR, Adachi T, Ido Y, Heibeck T, Jiang B, Lee Y, Melendez JA, Cohen RA, Colucci WS. Strain-stimulated hypertrophy in cardiac myocytes is mediated by reactive oxygen species-dependent Ras S-glutathiolation. J Mol Cell Cardiol. 2006;41:613–622. doi: 10.1016/j.yjmcc.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 12.de Lorgeril M, Salen P. Selenium and antioxidant defenses as major mediators in the development of chronic heart failure. Heart Fail Rev. 2006;11:13–17. doi: 10.1007/s10741-006-9188-2. [DOI] [PubMed] [Google Scholar]
- 13.Ostadalova I, Vobecky M, Chvojkova Z, Mikova D, Hampl V, Wilhelm J, Ostadal B. Selenium protects the immature rat heart against ischemia/reperfusion injury. Mol Cell Biochem. 2007;300:259–267. doi: 10.1007/s11010-006-9391-4. [DOI] [PubMed] [Google Scholar]
- 14.Venardos KM, Perkins A, Headrick J, Kaye DM. Myocardial ischemia-reperfusion injury, antioxidant enzyme systems, and selenium: a review. Curr Med Chem. 2007;14:1539–1549. doi: 10.2174/092986707780831078. [DOI] [PubMed] [Google Scholar]
- 15.Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, Gladyshev VN. Characterization of mammalian selenoproteomes. Science. 2003;300:1439–1443. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- 16.Reeves MA, Hoffmann PR. The human selenoproteome: recent insights into functions and regulation. Cell Mol Life Sci. 2009 doi: 10.1007/s00018-009-0032-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuster GM, Siwik DA, Pimentel DR, Colucci WS. Role of reversible, thioredoxin-sensitive oxidative protein modifications in cardiac myocytes. Antioxid Redox Signal. 2006;8:2153–2159. doi: 10.1089/ars.2006.8.2153. [DOI] [PubMed] [Google Scholar]
- 18.Kryukov GV, Kumar RA, Koc A, Sun Z, Gladyshev VN. Selenoprotein R is a zinc-containing stereo-specific methionine sulfoxide reductase. Proc Natl Acad Sci U S A. 2002;99:4245–4250. doi: 10.1073/pnas.072603099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim HY, Gladyshev VN. Methionine sulfoxide reduction in mammals: characterization of methionine-R-sulfoxide reductases. Mol Biol Cell. 2004;15:1055–1064. doi: 10.1091/mbc.E03-08-0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fomenko DE, Novoselov SV, Natarajan SK, Lee BC, Koc A, Carlson BA, Lee TH, Kim HY, Hatfield DL, Gladyshev VN. Methionine-R-sulfoxide reductase 1 (MsrB1) knockout mice: Roles of MsrB1 in redox regulation and identification of a novel selenoprotein form. J Biol Chem. 2008 doi: 10.1074/jbc.M805770200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shohet RV, Kisanuki YY, Zhao XS, Siddiquee Z, Franco F, Yanagisawa M. Mice with cardiomyocyte-specific disruption of the endothelin-1 gene are resistant to hyperthyroid cardiac hypertrophy. Proc Natl Acad Sci U S A. 2004;101:2088–2093. doi: 10.1073/pnas.0307159101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoffmann PR, Hoge SC, Li PA, Hoffmann FW, Hashimoto AC, Berry MJ. The selenoproteome exhibits widely varying, tissue-specific dependence on selenoprotein P for selenium supply. Nucleic Acids Res. 2007 doi: 10.1093/nar/gkm355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yasue H, Yoshimura M, Sumida H, Kikuta K, Kugiyama K, Jougasaki M, Ogawa H, Okumura K, Mukoyama M, Nakao K. Localization and mechanism of secretion of B-type natriuretic peptide in comparison with those of A-type natriuretic peptide in normal subjects and patients with heart failure. Circulation. 1994;90:195–203. doi: 10.1161/01.cir.90.1.195. [DOI] [PubMed] [Google Scholar]
- 24.Woods RL. Cardioprotective functions of atrial natriuretic peptide and B-type natriuretic peptide: a brief review. Clin Exp Pharmacol Physiol. 2004;31:791–794. doi: 10.1111/j.0305-1870.2004.04073.x. [DOI] [PubMed] [Google Scholar]
- 25.Frey N, Barrientos T, Shelton JM, Frank D, Rutten H, Gehring D, Kuhn C, Lutz M, Rothermel B, Bassel-Duby R, Richardson JA, Katus HA, Hill JA, Olson EN. Mice lacking calsarcin-1 are sensitized to calcineurin signaling and show accelerated cardiomyopathy in response to pathological biomechanical stress. Nat Med. 2004;10:1336–1343. doi: 10.1038/nm1132. [DOI] [PubMed] [Google Scholar]
- 26.Stoytcheva ZR, Berry MJ. Transcriptional regulation of mammalian selenoprotein expression. Biochim Biophys Acta. 2009;1790:1429–1440. doi: 10.1016/j.bbagen.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Copeland PR. Regulation of gene expression by stop codon recoding: selenocysteine. Gene. 2003;312:17–25. doi: 10.1016/s0378-1119(03)00588-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim HY, Gladyshev VN. Methionine sulfoxide reductases: selenoprotein forms and roles in antioxidant protein repair in mammals. Biochem J. 2007;407:321–329. doi: 10.1042/BJ20070929. [DOI] [PubMed] [Google Scholar]
- 29.Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Araujo AS, Enzveiler AT, Schenkel P, Fernandes TR, Ribeiro MF, Partata WA, Llesuy S, Bello-Klein A. Oxidative stress activates insulin-like growth factor I receptor protein expression, mediating cardiac hypertrophy induced by thyroxine. Mol Cell Biochem. 2007 doi: 10.1007/s11010-007-9459-9. [DOI] [PubMed] [Google Scholar]
- 31.Grossman W, Jones D, McLaurin LP. Wall stress and patterns of hypertrophy in the human left ventricle. J Clin Invest. 1975;56:56–64. doi: 10.1172/JCI108079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prentice HM, Moench IA, Rickaway ZT, Dougherty CJ, Webster KA, Weissbach H. MsrA protects cardiac myocytes against hypoxia/reoxygenation induced cell death. Biochem Biophys Res Commun. 2008;366:775–778. doi: 10.1016/j.bbrc.2007.12.043. [DOI] [PubMed] [Google Scholar]
- 33.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE, Aykin-Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mittag J, Behrends T, Hoefig CS, Vennstrom B, Schomburg L. Thyroid hormones regulate selenoprotein expression and selenium status in mice. PLoS One. 2010;5:e12931. doi: 10.1371/journal.pone.0012931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Forgione MA, Cap A, Liao R, Moldovan NI, Eberhardt RT, Lim CC, Jones J, Goldschmidt-Clermont PJ, Loscalzo J. Heterozygous cellular glutathione peroxidase deficiency in the mouse: abnormalities in vascular and cardiac function and structure. Circulation. 2002;106:1154–1158. doi: 10.1161/01.cir.0000026820.87824.6a. [DOI] [PubMed] [Google Scholar]
- 36.Maulik N, Yoshida T, Das DK. Regulation of cardiomyocyte apoptosis in ischemic reperfused mouse heart by glutathione peroxidase. Mol Cell Biochem. 1999;196:13–21. doi: 10.1007/978-1-4615-5097-6_2. [DOI] [PubMed] [Google Scholar]
- 37.Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Radmark O, Wurst W, Bornkamm GW, Schweizer U, Conrad M. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8:237–248. doi: 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 38.Gu QP, Sun Y, Ream LW, Whanger PD. Selenoprotein W accumulates primarily in primate skeletal muscle, heart, brain and tongue. Mol Cell Biochem. 2000;204:49–56. doi: 10.1023/a:1007065829068. [DOI] [PubMed] [Google Scholar]
- 39.Panee J, Stoytcheva ZR, Liu W, Berry MJ. Selenoprotein H is a redox-sensing high mobility group family DNA-binding protein that up-regulates genes involved in glutathione synthesis and phase II detoxification. J Biol Chem. 2007;282:23759–23765. doi: 10.1074/jbc.M702267200. [DOI] [PubMed] [Google Scholar]
- 40.Novoselov SV, Kryukov GV, Xu XM, Carlson BA, Hatfield DL, Gladyshev VN. Selenoprotein H is a nucleolar thioredoxin-like protein with a unique expression pattern. J Biol Chem. 2007;282:11960–11968. doi: 10.1074/jbc.M701605200. [DOI] [PubMed] [Google Scholar]
- 41.Navas-Acien A, Bleys J, Guallar E. Selenium intake and cardiovascular risk: what is new? Curr Opin Lipidol. 2008;19:43–49. doi: 10.1097/MOL.0b013e3282f2b261. [DOI] [PubMed] [Google Scholar]