


Abstract
Of the rules used by the splicing machinery to precisely determine intron–exon boundaries only a fraction is known. Recent evidence suggests that specific short sequences within exons help in defining these boundaries. Such sequences are known as exonic splicing enhancers (ESE). A possible bioinformatical approach to studying ESE sequences is to compare genes that harbor introns with genes that do not. For this purpose two non-redundant samples of 719 intron-containing and 63 intron-lacking human genes were created. We performed a statistical analysis on these datasets of intron-containing and intron-lacking human coding sequences and found a statistically significant difference (P = 0.01) between these samples in terms of 5–6mer oligonucleotide distributions. The difference is not created by a few strong signals present in the majority of exons, but rather by the accumulation of multiple weak signals through small variations in codon frequencies, codon biases and context-dependent codon biases between the samples. A list of putative novel human splicing regulation sequences has been elucidated by our analysis.
INTRODUCTION
Recent findings have shown that inside animal exons there are motifs that are used by the splicing machinery for recognizing exon–intron junctions (1–5). These motifs have been named exonic splicing enhancers (ESEs). A large number of nuclear proteins (SR-proteins) specifically bind to these motifs during the initial steps of splicing to initiate the assembly of the spliceosomal complex (5–7). Even a single point mutation inside an ESE can dramatically change the pattern of gene splicing (8). Thus, knowledge of the ESEs is important for revealing the locations of the exon–intron junctions and for the prediction of novel genes from genomic sequences. Thus far, there have been a limited number of experimental studies of ESEs and as of yet these motifs are poorly understood. The main aims of this paper are to analyze properties of the ESE distribution via a statistical analysis of human coding sequences (CDSs) and to uncover and characterize some ESEs.
It is well known that the order of nucleotides inside a CDS is non-random. This is believed to be for the following reasons: first, the CDS contains the information for the construction of the corresponding protein product by means of a genetic code; secondly, the CDS has codon bias and context-dependent codon bias (a preference of particular nucleotides at the third position of a codon triplet). It is widely accepted that codon bias serves for higher efficiency and accuracy of protein synthesis (9–12) and, possibly, for other biological reasons as well (13). It is reasonable to hypothesize that the necessity of efficient splicing could create additional non-randomness in CDSs through the selection for ESEs inside exons. Another possibility is that the splicing machinery already uses the existing non-randomness of CDSs. ESEs would then be those DNA motifs that are in excess inside CDSs compared to the surrounding intronic sequences. A possible approach to supporting one of these hypotheses is to compare two CDS samples: one composed of intron-containing (IC) genes and another composed of intron-lacking (IL) genes. If there is a difference between the IC and the IL samples, one could view that as evidence of additional splicing signals embedded within CDSs and the difference itself would represent such signals.
In this paper we present results of a statistical analysis performed on a set of IC and IL samples of human genes. We find a statistically significant (P = 0.01) difference between the gene sets in terms of the distributions of 5–6 nt sequences inside CDSs. The difference is due to the accumulation of multiple weak signals through small variations in codon frequencies, codon biases and context-dependent codon biases. Also, lists of putative novel human splicing regulation sequences have been elucidated by our analysis; these putative sequences may be suitable for experimental testing.
MATERIALS AND METHODS
Construction of the IC sample
We extracted coding sequences of human genes from the experimentally confirmed subset of our Exon–Intron Database (EID) (14), based on GenBank 112 (15). Only the genes with all introns conforming to the GT/AG boundary rule were used. From this initial database we removed genes with multiple duplications (if an amino acid 4mer was repeated four or more times, the gene was discarded). The set of human genes was then purged using the program gbpurge (http://www.fallingrain.com/publicserver) at the 20% amino acid similarity level to remove homologous sequences. All the genes were then checked for reading frame validity. We further pruned the set to remove viral and immunoglobulin genes. In the end, the IC sample was composed of 719 coding sequences (1 × 106 nt) with a GC content of 53.2%.
Construction of the IL sample
We extracted all human genes from GenBank 112 (15) without any indication of an exon–intron structure. For this purpose, as the first step, all the genes whose headings included ‘CDS join’, ‘intron’ or ‘exon’ annotations were omitted. Then, by visual inspection of the individual entries, pseudogenes and genes that were likely to be cDNAs were removed. Furthermore, we removed genes with multiple duplications and purged this IL set at the 20% amino acid similarity level in exactly the same manner as was done for the IC sample. All the genes were then checked for validity of the reading frame. Finally, viral and transposon genes were removed from the IL sample. In the end, the IL sample contained 63 coding sequences (6 × 104 nt) with a GC content of 53.9%. Both samples (IC and IL) and related data are available on the web (http://www.mcb.havard.edu/gilbert/exonsplicing).
Counting 4–6mer oligonucleotides and codon with contexts in IL and IC samples
We counted 4–6 nt sequences inside CDSs in two different ways. First, the sequences were counted in all of the three possible reading frames, a case where we called them 4–6mer oligonucleotides. And second, the sequences were counted in only one reading frame, the one where the first three nucleotides of the sequence represented a real codon. In this case they were named codons with context N1 (for 4 nt sequences), codons with context N1N2 (for 5 nt sequences) and dicodons (for 6 nt sequences). Context Ni stands for the next i-nucleotide after the codon, according to the notation of Berg and Silva (16).
Details of the calculations
To compare samples of genes with regard to their nucleotide, amino acid, codon and oligonucleotide compositions we employed the χ2 test. Since the samples to be compared were of different sizes (R and S), the following standard formula was used (17):
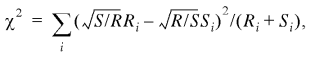 1
1
where

and
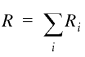 ;
;
i goes over all the 4 nt, 20 amino acids, 61 codons, etc., depending on the nature of the comparison. Because of the limited size of the IL sample, the 6mer sequences are the longest ones for which the χ2 test is applicable. Rare 6mers were pooled so that all the bins used in equation 1 had at least five members.
Monte Carlo simulations
Since the CDS nucleotide composition varies from one gene to another, we used a Monte Carlo approach to estimate the IL/IC sample difference. For this purpose 200 random IC subsets of the same size as the IL sample were created. Each random IC subset was obtained by the following procedure: we randomly chose a gene from the IC sample and placed it in the random IC-subset until the number of codons in the subset became the same as in the IL sample. The rest of the genes in the IC sample comprised the so-called IC complement set (IC sample = IC subset + IC complement). We performed the most rigid estimation of the IL/IC sample difference by comparison of the χ2i(IC subset – IC complement) value for the ICi subset and its ICi complement (i varies from 1 to 200) with the X2i(IL – IC complement) value for the IL and the corresponding ICi complement. We then counted how many times (k) the difference between the ICi subset and its ICi complement was greater than the difference between the IL and the corresponding ICi complement (χ2i(IC subset – IC complement) ≥ χ2i(IL – IC complement)). Finally the P value for the IL/IC difference was calculated using the formula: P = k/200.
All the calculations were performed with computer programs written in PERL.
RESULTS
Statistical comparison of IL and IC samples
The χ2 test revealed a very significant difference in oligonucleotide occurrence between the IC and IL samples. For example, for the 4mer oligonucleotide distributions, the statistical values were the following: χ2 = 1598, degrees of freedom = 254, P = 2.6 × 10–193. However, because our samples are of genes, not oligonucleotides, and because each gene has its own unique nucleotide composition, the P values obtained from the χ2 test are not reliable. For that reason, while using the χ2 calculation for measuring the oligonucleotide difference, we resorted to Monte Carlo simulations to estimate the significance of the IC/IL difference (see Materials and Methods). Using this approach we studied the differences between the samples with regard to the various types of compositions [nucleotide, amino acid, codon, codon with contexts and 4–6mer oligonucleotide (Table 1)].
Table 1. Comparison of results for the IC and IL samples: χ2 tests for IL set and 200 random IC subsets.
| Type of comparison |
IC subsets versus IC complements rangea (mean) |
IL-sample versus IC-complements rangeb (mean) |
Pc |
| χ2 (nucleotide) | 1–590 (73) | 36–51 (43) | 0.45 |
| χ2 (amino acid) | 18–309 (72) | 118–136 (127) | 0.065 |
| χ2 (codon) | 62–824 (217) | 403–438 (418) | 0.09 |
| χ2 (codon_N1) | 249–1204 (449) | 883–948 (910) | 0.02 |
| χ2 (codon_N1N2) | 918–2066 (1217) | 1820–1906 (1858) | 0.01 |
| χ2 (dicodon) | 2431–3943 (2830) | 3771–3889 (3825) | 0.01 |
| χ2 (4mer oligo)d | 267–3032 (674) | 1545–1720 (1612) | 0.035 |
| χ2 (5mer oligo)d | 1031–4497 (1576) | 2952–3183 (3043) | 0.025 |
| χ2 (6mer oligo)d | 3707–8044 (4517) | 6488–6817 (6620) | 0.01 |
aThe results from comparing 200 randomly chosen IC subsets with their IC complements.
bThe comparisons of the IL sample with the IC complements.
cReflects how frequently the difference between the IC subset and its IC complement was greater than the difference between the IL sample with the same IC complement.
dn-mer oligonucleotides counted in all possible reading frames. On the other hand, a codon with context is an oligonucleotide counted in the first reading frame only (see Materials and Methods).
The obtained results show that the nucleotide composition of the IL sample is very similar to that of the IC sample (P = 0.45; Table 1). Also, these two samples have similar GC content (the IL and IC samples have GC contents of 53.9 and 53.2%, respectively; Table 2). The amino acid and codon composition difference between the IC and IL samples is more prominent but still not significant (P = 0.065 and 0.09, respectively; Table 1). It is clear from Table 1 that, as expected, the IC sample is not homogeneous. If the IC sample were homogeneous then, according to statistical theory, in 99.9% of all cases the value of the χ2 test between the random IC subset and its complement would not exceed 16 for the nucleotide, 44 for the amino acid or 100 for the codon comparisons. Nevertheless, despite the sample in-homogeneity, the difference between the IL and IC samples is significant for the 5mer oligonucleotide analysis within the 97% confidence interval (P = 0.03), and for the 6mer oligonucleotide analysis within the 99% confidence interval. Similar or even more pronounced figures were obtained for the distributions of dicodons and codons with contexts N1 and N1N2 (Table 1). With respect to 6mer oligonucleotide, dicodon and codon with N1N2 context comparisons, for only two out the 200 IC subsets did the χ2 values exceed those obtained from the corresponding IL sample comparisons. These two particular subsets were the ones with the highest divergence from the IC sample in GC content (50.2 and 48.5%), nucleotide (χ2 = 234 and 590), codon (χ2 = 506 and 824) and amino acid (χ2 = 309 and 244) composition. So the difference in the oligonucleotide distributions of these two particular random IC subsets and their IC complements was presumably due to the anomaly in their nucleotide composition. Therefore, we can state with confidence that the 5–6mer composition of the IL sample differs significantly from the random IC samples.
Table 2. Characteristics of the IL sample and the 200 random IC subsets and their IC complements.
| Type of comparison |
IC subsets range (mean) |
IC complements range (mean) |
IL sample |
| GC composition (%) | 48.5–56.8 (53.2) | 52.9–53.5 (53.2) | 53.9 |
| Gene length mean (nt) | 1126–2186 (1445) | 1173–1203 (1183) | 963.0 |
| Gene length median (nt) | 879–1563 (1181) | 1403–1460 (1436) | 873.0 |
The gene length median is the length of the gene in the middle of the sample that was sorted by length.
In short, we found a statistically significant difference between sets of IC and IL human genes. Because the two samples were constructed on the basis of intron presence, it is immediately suggestive that our result can be directly explained by the fact that one sample contains introns while the other does not. We performed several tests to discount other possibilities, which could have caused the IC/IL difference.
Statistical analysis of controls
The average length of the IL genes (mean = 963 nt, median = 873 nt) was found to be considerably lower than that of the IC genes (mean = 1310 nt, median = 1197 nt; Table 2). We generated an IC subset composed of the shortest genes (mean = 516, median = 522) of the IC sample and compared this subset to its IC complement. The resulting χ2 values for the ‘short’ IC subset (Table 3) fell in the middle of the range of χ2 values for the 200 randomly constructed IC subsets (Table 1). Thus, it is extremely unlikely that the dicodon IC/IL difference arose from a discrepancy in gene length distributions. The ‘longest’ IC subset differed much more strongly from its complement (presumably, due to the anomaly of 51.0% GC content), but again, this difference did not exceed the IL/IC difference.
Table 3. χ2 test results for four non-random IC subsets versus their IC complements.
| Type of comparison |
Receptor subset versus IC complement |
Kinase subset versus IC complement |
Short_gene subset versus
IC complement |
Long_gene subset versus IC complement |
| χ2 (nucleotide) | 88 | 12 | 22 | 46 |
| χ2 (amino acid) | 115 | 59 | 140 | 160 |
| χ2 (codon) | 406 | 115 | 236 | 363 |
| χ2 (codon_N1) | 781 | 320 | 508 | 698 |
| χ2 (codon_N1N2) | 1821 | 1040 | 1313 | 1656 |
| χ2 (dicodon) | 3635 | 2538 | 2911 | 3473 |
| χ2 (4mer oligo)a | 806 | 352 | 467 | 903 |
| χ2 (5mer oligo)a | 1839 | 1167 | 1340 | 2038 |
| χ2 (6mer oligo)a | 5013 | 3979 | 4219 | 5487 |
an-mer oligonucleotides counted in all possible reading frames. A codon with context, however, is an oligonucleotide counted in the first reading frame only (see Materials and Methods).
For further investigation of the difference between the IC and the IL samples we studied the sequence composition of several non-random IC subsets of the same size as the IL sample. Since our gene samples were purged with the 20% homology threshold, most of the genes represent unrelated proteins. Nevertheless, examination of the annotations associated with genes in the IC sample showed that ∼10% of its genes are various types of receptors. The next most abundant group inside the IC sample was composed of kinases. Based on these two distinct functional classes, the two IC subsets were then constructed and investigated independently. The set of receptors was the largest functional subset within the IC sample, and showed the greatest deviation in 4–6mer sequence composition from its IC complement (Table 3). However, the difference was clearly due to the large biases in amino acid (χ2 = 247) and nucleotide composition (GC content = 51.4%) of the receptor genes. Despite these nucleotide and amino acid biases, the 5 and 6mer difference between the receptor subset and its IC complement was still below that of the IL comparison (Tables 1 and 3). All calculated χ2 values for kinase IC subset were in the middle range of the corresponding values for the 200 random IC subsets.
To ensure that the known splicing motifs at exon–intron junctions could not have produced the IC/IL dicodon difference, we performed another comparison with a modified IC sample. The modification involved treating each exon separately, while removing terminal bases from both 5′- and 3′-ends of the exon (for the computational convenience we removed the first codon and the last two nucleotides of every exon). In every other respect the new comparison was identical to the one presented above. The results (http://www.mcb.harvard.edu/gilbert/exonsplicing) were very similar to the ones presented in Table 1.
Characterization of sequences over- and under-represented in the IL sample
The frequencies of the 4mer and 5mer oligonucleotides and codons with N1 and N1N2 contexts in the IL sample and 200 random IC subsets were compared with one another. Those oligonucleotides that were over- or under-represented in the IL sample compared with every one of the 200 random IC subsets are presented in Table 4. Interestingly, the number of IL over-represented oligonucleotides (13 for 4mers and 42 for 5mers) is considerably larger than the IL under-represented oligonucleotides (5 for 4mers and 18 for 5mers). The same type of comparison of oligonucleotide frequencies in 200 random IC-subsets revealed that on average only one 4mer and four 5mers of a random IC subset were over-represented compared to the other 199 IC subsets. The presence of over-represented oligonucleotides in the IC subsets was due to statistical fluctuations engendered by limited size of these subsets (which were the size of the IL sample).
Table 4. Over- and under-represented sequences in the IL sample.
| IL under-represented oligonucleotides | |||||
| 4mer | cagt(0.80) | ctga(0.81) | gatg(0.81) | tcag(0.81) | tgga(0.83) |
| 5mer | acctt(0.61) | agcat(0.70) | cacag(0.72) | cagga(0.78) | cagtg(0.74) |
| ctcag(0.67) | ctgac(0.61) | ctgga(0.80) | gaggt(0.69) | gattg(0.58) | |
| gctga(0.78) | ggagt(0.64) | ggcac(0.66) | gtgga(0.76) | tcagg(0.71) | |
| tgcct(0.65) | tggac(0.74) | ttgcc(0.58) | |||
| IL over-represented oligonucleotides | |||||
| 4mer | cgaa(1.36) | cgac(1.47) | cgag(1.27) | cgca(1.58) | cgcg(2.49) |
| cggc(1.51) | cggt(1.46) | ctcg(1.66) | gccg(1.64) | gcga(1.80) | |
| gcgc(1.94) | ggcg(1.79) | tcgc(1.48) | |||
| 5mer | aaaaa(1.58) | aaaag(1.38) | aagcg(1.49) | accgc(1.67) | acgac(1.60) |
| acgcg(2.22) | actcg(1.77) | agaaa(1.46) | ccgaa(1.63) | ccggt(2.17) | |
| cgacg(2.33) | cgcag(1.65) | cgccg(2.53) | cgcga(2.95) | cgcgc(3.17) | |
| cgcgg(2.20) | cgctc(1.59) | cggcg(2.36) | cggtg(1.76) | ctagc(1.84) | |
| ctcgc(2.31) | ctcgt(1.68) | gaaag(1.44) | gccga(1.70) | gccgg(1.54) | |
| gcgaa(2.13) | gcgag(1.67) | gcgat(1.99) | gcgca(1.95) | gcgcg(3.22) | |
| gcggc(1.85) | gctcg(1.92) | ggccg(1.71) | ggcga(1.69) | ggcgc(2.20) | |
| ggcgg(1.63) | gggcg(1.85) | gtgcg(1.83) | taggg(1.94) | tcgcg(3.86) | |
| tctcg(1.58) | tgcga(1.90) | ||||
| IL under-represented codons with context | |||||
| codon_N1 | aca g(0.74) | gat g(0.76) | tca g(0.66) | tcc a(0.80) | |
| codon_N1N2 | aca gg(0.49) | acc tt(0.65) | cag ga(0.66) | cca ga(0.58) | cct ga(0.62) |
| cgg at(0.26) | ctg ac(0.62) | ctg ca(0.63) | gct ga(0.68) | gct ta(0.27) | |
| ggc ac(0.64) | gtg at(0.56) | gtg ga(0.72) | tac at(0.61) | tcc at(0.54) | |
| IL over-represented codons with context | |||||
| codon_N1 | aga a(1.62) | aga g(1.51) | agg g(1.56) | atg t(1.32) | ccc g(1.59) |
| ccg a(1.95) | cgc a(1.62) | cgc g(2.18) | ctc g(1.57) | gcc g(1.82) | |
| gcg c(2.10) | gcg g(1.91) | ggc g(2.22) | tcg g(1.66) | ||
| codon_N1N2 | acc gc(2.06) | aga aa(1.85) | aga ga(1.83) | cac gc(2.00) | ccg aa(2.46) |
| ccg ag(2.29) | ccg gt(2.90) | cga cg(3.66) | cgc ac(2.12) | cgc ag(1.96) | |
| cgc cg(2.08) | cgc ga(3.19) | cgc gg(2.63) | ctc gt(1.75) | gcc ga(1.73) | |
| gcg aa(2.65) | gcg cc(2.25) | gcg cg(3.51) | gcg gc(2.44) | gcg gt(2.00) | |
| ggc ga(1.78) | ggc gc(3.55) | tca cg(2.21) | tcg cg(4.01) | tcg gg(2.53) | |
| tgc ga(2.45) | tta gg(2.48) | ||||
List of all 4–5mer oligonucleotides and codons with N1 and N1N2 context over- or under-represented in the IL sample compared to all the 200 random IC subsets.
The number in parentheses (k) beside every motif represents the corresponding relative abundance in the IL sample compared to the whole IC sample and was calculated using the formula: k = NIL/NIC × LIC/LIL, where NIL and NIC are the occurrences of the examined sequence in the IL and IC samples, respectively, and LIL and LIC are the sizes of the samples.
It should be noted that putative exonic splicing enhancers are among the IL under-represented sequences and putative exonic splicing silencers are among the IL over-represented.
The largest numbers of over-represented oligonucleotides among the 200 random IC subsets (the maximum number was 15 for 4mers and 32 for 5mers) were found in the IC subsets with the highest deviations of GC content from the average value of 53.2%. Analogous data was obtained for oligonucleotide under-representation. On average only one 4mer and six 5mers of a random IC subset were under-represented with respect to the rest of the 199 IC subsets. Maximal numbers of under-represented sequences among the 200 IC subsets were 24 for 4mers and 43 for 5mers. As above, these maximums were also found in the IC subsets with the highest deviation from average in terms of the GC content. The number of IL over- and under-represented oligonucleotides was considerably higher than the average numbers in the IC subsets. This is especially striking if we take into account the fact that the GC content difference between the IL and the IC samples is small (53.9 and 53.2%, respectively).
Using the same type of analysis we found that the number of IL over- and under-represented codons with context N1 and N1N2 (Table 4) was considerably higher than the average numbers of codons with corresponding contexts in random IC subsets.
DISCUSSION
IL ↔ IC transition
In the IL sample we found several processed pseudogenes (such as olfactory receptors) that have IC homologs in the human genome. On the other hand, some IL genes from our sample of human genes (histone H2A, H2B, H3) contain intron(s) in the genomes of other species (for example, H2A in Drosophila melanogaster and Arabidopsis thaliana, H2B in Aspergillus nidulans and H2 in Fusarium proliferatum) (14). Similar observations have been known since the early 1980s and have become the main argument in support of the intron-late hypothesis, which makes a claim that introns can move from one gene into another (18,19). The facts support transitions between IC and IL gene forms during evolution. What is important to stress about this transition, however, is that the process occurs infrequently over evolutionary time. The overwhelming majority of orthologous genes in humans and mice have the same number of introns in the corresponding positions; these organisms diverged ∼50–70 million years ago. These data allow us to conclude that, in general, a human intronless gene has existed in the IL form for many millions of years. Due to the infrequency of the transitions, we feel that their effects (residual splicing signals in IL genes, for instance) can be discounted when comparing nucleotide compositions of the IL and IC sets.
Nucleotide composition difference between IL and IC samples
We have found a statistically significant difference between IC and IL samples in terms of the 4–6 nt sequence distributions. The rationale behind the application of the 4–6 nt sequence statistics relies on a simple biological explanation, namely that known ESE elements frequently have the same length (5–6 nt) (3,4). With this in mind, the 4–5mer oligonucleotides with frequencies differing considerably between the IC and IL samples were then characterized. The 6mer oligonucleotides are not presented here because their occurrence in the IL sample was small, and thus statistically unreliable. The numbers of over- and under-represented oligonucleotides in the IL sample (compared to all 200 random IC subsets) were found to be considerably higher than the average numbers of over- and under-represented oligonucleotides in a random IC subset (compared to the rest of the 199 random IC subsets). Therefore, we assumed that between the IL over- and under-represented oligonucleotides, presented in Table 4, there could exist a subgroup of functional sequences involved in the splicing process.
It is of interest to compare our list of IL over- and under-represented oligonucleotides with those that have previously been identified as ESEs based on experimental studies. Comparison of sequences from Table 4 with experimentally determined lists of ESEs essential for in vitro splicing performed in nuclear HeLa extracts, which contained some Drosophila proteins (3,4), shows no strong similarity between these two groups of sequences. However, most strikingly, a recently published functional human ESE sequence corresponds well with the IL under-represented oligonucleotides. This experimentally documented ESE was corrupted by a C→T point mutation in position 6 of the seventh exon of the human SMN gene, which caused a dramatic change in the splicing pattern in vivo (8).
The C→T transition occurred inside the exonic sequence, TCAG, which is identical to one of the five 4mers under-represented in the IL sample. Moreover, CAG is one of the most abundant 3 nt motifs in the whole list of IL under-represented sequences in Table 4. This observation supports the notion of functional splicing properties in some sequences listed in Table 4.
Interestingly, sequences of IL over-represented oligonucleotides from Table 4 are highly enriched by CG dinucleotides (45 occurrences among 42 5mer oligonucleotides) and GC dinucleotides (33 occurrences). So, there exists a possibility that CG and GC dinucleotides were unfavorable for splicing and a fraction of these dinucleotides were then removed from exons by natural selection. We suggest that the sequences from Table 4 are good candidates for experimental testing on splicing enhancer/silencer properties.
Length difference of IC and IL genes
We found that the average length of the IL genes is significantly smaller than the average length of the IC genes (Table 2). The gene length difference between our samples is in agreement with the notion that the average length of prokaryotic genes (which are intronless) is significantly shorter than that of eukaryotic genes, many of which are IC genes (20). The observed gene length difference between the IL and IC samples indirectly supports the exon-early theory of gene evolution, claiming that modern genes were assembled from ‘exon pieces’ by the exon shuffling process (21,22). Exon shuffling is possible only for genes having exon–intron structure and shuffling events will tend to elongate the CDS.
Acknowledgments
ACKNOWLEDGEMENTS
We acknowledge the support and critical comments by Dr Walter Gilbert concerning the merits of this approach. We extend our gratitude to two anonymous referees for their valuable suggestions in improving this manuscript.
References
- 1.Watakabe A., Tanaka,K. and Shimura,Y. (1993) The role of exon sequences in splice site selection. Genes Dev., 7, 407–418. [DOI] [PubMed] [Google Scholar]
- 2.Tian H. and Kole,R. (1995) Selection of novel exon recognition elements from a pool of random sequences. Mol. Cell. Biol., 15, 6291–6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coulter L.R., Landree,M.A. and Cooper,T.A. (1997) Identification of a new class of exonic splicing enhancers by in vivo selection. Mol. Cell. Biol., 17, 2143–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schaal T.D. and Maniatis,T. (1999) Selection and characterization of pre-mRNA splicing enhancers: identification of novel SR protein-specific enhancer sequences. Mol. Cell. Biol., 19, 1705–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blencowe B.J. (2000) Exonic splicing enhancers: mechanism of action, diversity and role in human genetic diseases. Trends Biochem. Sci., 25, 106–110. [DOI] [PubMed] [Google Scholar]
- 6.Fu X.-D. (1995) The superfamily of arginine/serine-rich splicing factors. RNA, 1, 663–680. [PMC free article] [PubMed] [Google Scholar]
- 7.Manley J. and Tacke,R. (1996) SR proteins and splicing control. Genes Dev., 10, 1569–1579. [DOI] [PubMed] [Google Scholar]
- 8.Lorson C.L. and Androphy,E.J. (2000) An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum. Mol. Genet., 9, 259–265. [DOI] [PubMed] [Google Scholar]
- 9.Grantham R., Gautier,C., Gouy,M., Jacobzone,M. and Mercier,R. (1981) Codon catalog usage is a genome strategy modulated for gene expressivity. Nucleic Acids Res., 8, 49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ikemura T. (1981) Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli translational system. J. Mol. Biol., 151, 389–409. [DOI] [PubMed] [Google Scholar]
- 11.Precup J. and Parker,J. (1987) Missense misreading of asparagine codons as a function of codon identity and context. J. Biol. Chem., 262, 11351–11356. [PubMed] [Google Scholar]
- 12.Eyre-Walker A. (1996) Synonymous codon bias is related to gene length in Escherichia coli: selection for translational accuracy? Mol. Biol. Evol., 13, 864–872. [DOI] [PubMed] [Google Scholar]
- 13.Karlin S. and Mrazek,J. (1996) What drives codon choices in human genes? J. Mol. Biol., 262, 459–472. [DOI] [PubMed] [Google Scholar]
- 14.Saxonov S., Daizadeh,I., Fedorov,A. and Gilbert,W. (2000) EID: the Exon–Intron Database – an exhaustive database of protein-coding intron-containing genes. Nucleic Acids Res., 28, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benson D.A., Boguski,M.S., Lipman,D.J., Ostell,J., Ouellette,B.F., Rapp,B.A. and Wheeler,D.L. (1999) GenBank. Nucleic Acids Res., 27, 12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berg O.G. and Silva,P.J.N. (1997) Codon bias in Escherichia coli: the influence of codon context on mutation and selection. Nucleic Acids Res., 25, 1397–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Press W.H., Teukolsky,S.A., Vetterling,W.T. and Flannery,B.P. (1988) Numerical Recipes In C: The Art of Scientific Computing. Cambridge University Press, Cambridge, UK, pp. 623.
- 18.Rogers J. (1989) How were introns inserted into nuclear genes? Trends Genet., 5, 213–216. [DOI] [PubMed] [Google Scholar]
- 19.Palmer J.D. and Logsdon,J.M. (1991) The recent origins of introns. Curr. Opin. Genet. Dev., 1, 470–477. [DOI] [PubMed] [Google Scholar]
- 20.Moriyama E.N. and Powell,J.R. (1998) Gene length and codon usase bias in Drosophila melanogaster, Saccharomyces cerevisiae and Escherichia coli. Nucleic Acids Res., 26, 3188–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilbert W. (1978) Why gene in pieces? Nature, 271, 501. [DOI] [PubMed] [Google Scholar]
- 22.Long M., DeSouza,S. and Gilbert,W. (1995) Evolution of the intron–exon structure of eukaryotic genes. Curr. Opin. Genet. Dev., 5, 774–778. [DOI] [PubMed] [Google Scholar]