

Summary
Background
Activated protein C (APC) is a vitamin K-dependent plasma serine protease that down-regulates clotting and inflammatory pathways. It is known that APC exerts a cardioprotective effect by decreasing apoptosis of cardiomyocytes and inhibiting expression of inflammatory mediators after myocardial ischemia.
Objectives
The objective of this study was to understand the mechanism of the APC-mediated cardioprotection against ischemic injury.
Methods
Cardioprotective activities of wild-type APC and two derivatives having either dramatically reduced anticoagulant activity or lacking signaling activity were monitored in an acute ischemia/reperfusion injury model in which the left anterior descending coronary artery (LAD) was occluded.
Results
APC reduced the myocardial infarct size by a mechanism that was largely independent of its anticoagulant activity. Thus, the non-anticoagulant APC-2Cys mutant, but not the non-signaling APC-E170A mutant attenuated myocardial infarct size by EPCR and PAR-1-dependent mechanisms. Further studies revealed that APC acts directly on cardiomyocytes to stimulate the AMP-activated protein kinase (AMPK) signaling pathway. The activation of AMPK by APC ameliorated the post-ischemic cardiac dysfunction in isolated perfused mouse hearts. Moreover, both APC and APC-2Cys inhibited production of TNFα and IL-6 in vivo by attenuating the ischemia/reperfusion-induced JNK and NF-κB signaling pathways.
Conclusions
APC exerts a cardioprotective function in ischemic/reperfusion injury through modulation of AMPK, NF-κB and JNK signaling pathways.
Keywords: APC, Ischemia/reperfusion, Cardioprotection, AMPK signaling, EPCR, PAR-1
Introduction
Protein C is a vitamin K-dependent serine protease zymogen in plasma which upon activation by thrombin in complex with thrombomodulin cleaves factors Va and VIIIa by limited proteolysis to down-regulate the blood clotting cascade [1,2]. In addition to its anticoagulant activity, activated protein C (APC) also exhibits potent cytoprotective and antiinflammatory properties [3–6]. It has been demonstrated that APC exerts its antiinflammatory effect by suppressing expression of proinflammatory cytokines and inhibiting interaction and migration of leukocytes across the endothelium by inhibiting the NF-κB signaling pathway [6–10]. APC also inhibits apoptosis and reverses the hyperpermeability effect of inflammatory mediators on the endothelium [6,8,11]. Moreover, APC exerts a neuroprotective effect in ischemic brain injury by reducing brain infarction and inhibiting cell injury and apoptosis triggered by hypoxia [12,13]. Because of its antiinflammatory properties, the FDA has approved recombinant APC for treating severe sepsis in adult patients [14]. Overwhelming data from several groups indicate that protective activities of APC require its binding to endothelial protein C receptor (EPCR) and subsequent activation of protease-activator receptor 1 (PAR-1) [5,6]. This mechanism of co-receptor signaling by APC is believed to contribute, at least in part, to its beneficial effect in reducing the mortality of severe sepsis [6]. Several in vivo studies, using either function-blocking antibodies or mutant mice with deficiency for either receptor, in different inflammatory and severe sepsis model systems have provided further support for this hypothesis [15–18].
In two recent studies, it has been found that human recombinant APC also decreases cardiomyocyte apoptosis in rat and mouse models of myocardial ischemia/reperfusion injury [19,20]. In support for a cardioprotective role for the protein C pathway, a clinical study monitoring EPCR polymorphism showed that two haplotypes, A1 and A3, in the EPCR gene lower the risk of premature myocardial infarction due to increased levels of APC (A1 haplotype) and soluble EPCR (A3 haplotype) in plasma [21]. Moreover, in an ischemic animal model, treatment with APC during ischemia/reperfusion restored the mean arterial pressure after a short time occlusion and reduced the mortality rate [19,20]. The mechanism by which APC exerts a cardioprotective effect is not fully understood. In this study we investigated this question by using human recombinant APC and its various derivatives having only cytoprotective activity, or dramatically reduced anticoagulant activity in a mouse model of acute ischemia/reperfusion injury. The effect of APC derivatives was assessed on myocardial infarction size, post-ischemic cardiac function recovery and modulation of inflammatory responses on cardiomyocytes. We show that APC attenuates acute ischemic injury in the heart via stimulating the AMP-activated protein kinase signaling and inhibition of NF-κB and JNK signaling pathways by a mechanism(s) that is largely independent of its anticoagulant activity.
Materials and methods
In vivo regional ischemia and myocardial infarct size measurement
Mice (FVB/NJ) were anesthetized, intubated and ventilated with a respirator (Harvard apparatus, Holliston, MA) as described [22,23]. After a left lateral thoracotomy, the left anterior descending coronary artery (LAD) was occluded for 20 min with an 8-0 nylon suture and polyethylene tubing to prevent arterial injury, and reperfused for up to 3h. The myocardial infarct size was calculated as the ratio of the percentage of myocardial necrosis to the ischemic area at risk (AAR). APC derivatives (0.2 μg/g) or saline was administered via the tail vein 5 min before reperfusion.
Supplemental Methodology
For a detailed explanation of methods relative to AMPK [23] and CaMKKβ activity analysis [24], heart perfusion and cardiac functions [23], immunoblotting [22], real-time reverse-transcriptase polymerase chain reaction (RT-PCR), high-energy phosphate analysis [25], measurement of isolated cardiomyocytes contractile function [26], see Methods’ section of the online-only Supplemental Material.
Statistical analysis
Data were expressed as means ± S.E. A variety of statistical tests employing SAS software (version 9.2; SAS Institute Inc, Cary, NC) was used based on designs and specific question asked. This meant employing t-tests, repeated and non-repeated measures 1-way and 2-way ANOVA. For single- and multi-factorial analyses, appropriate post-hoc test(s) were performed to measure individual group differences of interest. A P value of less than 0.05 was considered statistically significant.
Results
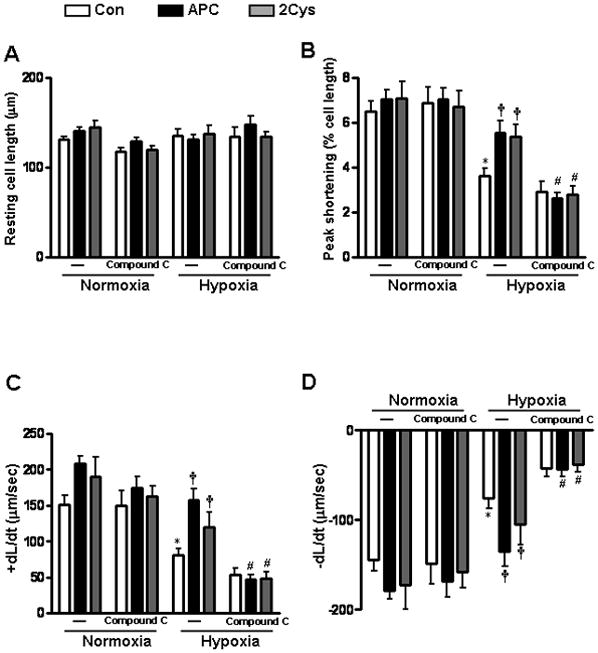
APC reduces myocardial infarction during ischemia/reperfusion
To investigate whether APC contributes to cardiac protection against ischemia/ reperfusion injury in mice, myocardial infarction was measured after 20 min of regional ischemia followed by 3h of reperfusion. Representative pictures of dual staining with TTC and Evan’s blue dye for assessing the infarct size are shown in Figure 1. With a fairly equal ratio of area at risk (AAR) to whole myocardial area (Figure 1B), the administration of APC (0.2 μg/g) 5 min before reperfusion significantly reduced the myocardial infarct size. To determine whether anticoagulant or direct cellular effect of APC are responsible for this function, experiments were conducted with two APC mutants which have either dramatically reduced anticoagulant activity (APC-2Cys) [27] or devoid of intracellular signaling activity (APC-E170A) [28]. As demonstrated in Figure 1, APC-2Cys decreased the size of myocardial infarction but E170A did not show any cardioprotective effect in this model. These results indicate that APC protects against ischemia/reperfusion injury in the heart and that this effect is primarily associated with its signaling activity.
Figure 1.
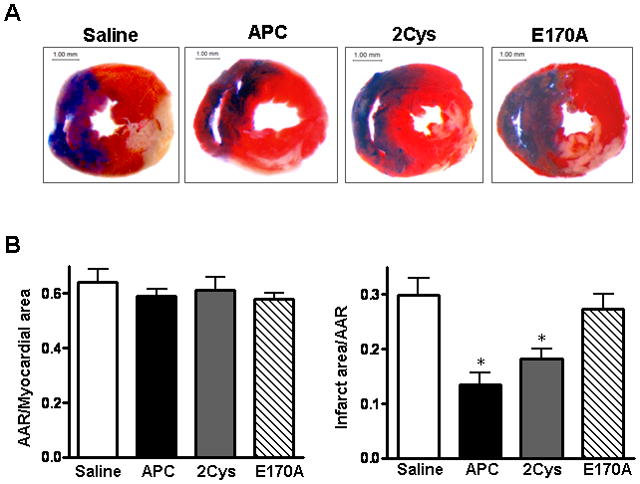
APC reduces myocardial infarct size after ischemia/reperfusion. Hearts were subjected to 20 min ischemia followed by 3h reperfusion. APC derivatives (0.2 μg/g) or saline (control) were administered via the tail vein 5 min before reperfusion. The extent of myocardial necrosis was assessed as described under “Methods”. (A) Representative sections of myocardial infarction. (B) The ratio of area at risk (AAR) to myocardial area (left panel) and the ratio of infarct area to AAR (right panel) in mouse hearts of differe treatment groups. Values are means ± S.E. for 4 independent experiments. *p<0.01 vs. saline, respectively.
APC stimulates AMPK phosphorylation during ischemia/reperfusion
AMP-activated protein kinase (AMPK) is a stress sensitive kinase that is known to be activated in response to ATP depletion [29]. Activated AMPK exerts a cardioprotective effect during ischemia by mediating substrate metabolism in order to compensate for the energy depletion [29]. Mouse hearts subjected to LAD occlusion exhibited an increase of AMPK phosphorylation at Thr-172 of the α catalytic subunit in the left ventricular area early after 2 min of ischemia, and the phosphorylation level of AMPK reached its peak level after 10 min of ischemia (Figure 2A). However, AMPK was quickly dephosphorylated once reperfusion started (Figure 2A). In mice hearts subjected to 20 min of ischemia, administration of APC or APC-2Cys maintained phosphorylation levels of both AMPK and the downstream acetyl-CoA carboxylase (ACC) but APC-E170A did not show this effect (Figure 2B). Moreover, in sham-operated hearts, an elevation of both AMPK and ACC phosphorylation was detected in APC and APC-2Cys, but not in E170A treated groups (Figure 2B). Furthermore, immunoprecipitated AMPKα1 and AMPKα2 activity assay demonstrated that APC and APC-2Cys increase activation of both AMPK isoforms, while E170A has no effect (Figure 2C and 2D). These results suggest that both APC and APC-2Cys stimulate the activation of AMPK in vivo, possibly accounting for their cytoprotective effects in this model.
Figure 2.
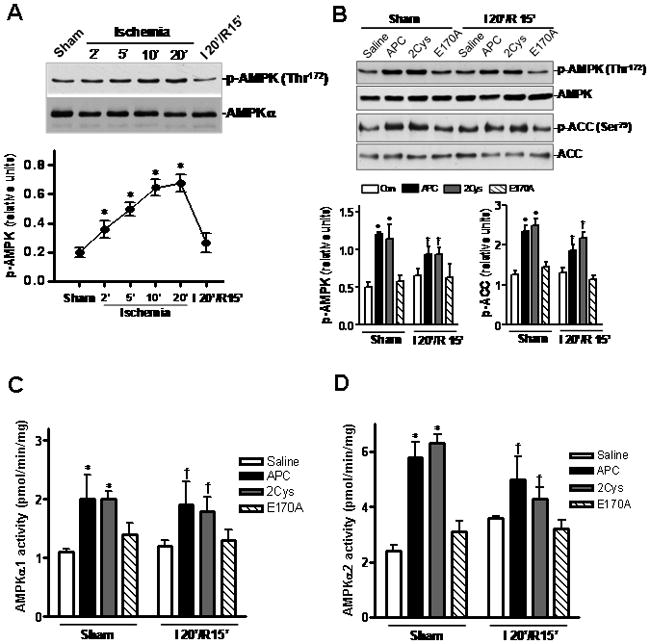
APC stimulates AMPK activation. (A) In vivo regional ischemia stimulates phosphorylation of AMPK as shown by immunoblots. Values are means ± S.E., n=3. *p<0.05 vs. Sham. (B) Both APC and 2Cys but not E170A trigger AMPK and ACC phosphorylation during reperfusion. (C) and (D) Activation of AMPKα1 and AMPKα2 in the same heart samples as assessed by a kinase assay. Values are means ± S.E., n=4–6. *p<0.05 vs. Sham saline; †p<0.05 vs. I/R saline.
APC mediates phosphorylation of AMPK in mouse cardiomyocytes
To determine whether APC can increase AMPK phosphorylation in heart tissues, we isolated cardiomyocytes (major heart cells) from experimental animals and examined them for the induction of AMPK activation. We first demonstrated that cardiomyocytes express EPCR (Figure 3A). We then studied the dose- and time-dependence of AMPK phosphorylation by APC. The data presented in Figures 3B and 3C indicated that 20 nmol/L APC for 20 min optimally induces AMPK phosphorylation in cardiomyocytes. Under these conditions, APC-2Cys activated AMPK (both AMPKα1 and AMPKα2) to the similar level as APC (Figures 3D-F), but E170A did not exhibit any activity, suggesting that the signaling effect of APC is primarily responsible for its protective activity. Function-blocking antibodies to EPCR and PAR-1 blocked both APC and 2Cys-mediated activation of AMPK in cardiomyocytes (Figure 3G). These results indicate that APC triggers phosphorylation of AMPK in cardiomyocytes by EPCR and PAR-1-dependent mechanisms.
Figure 3.
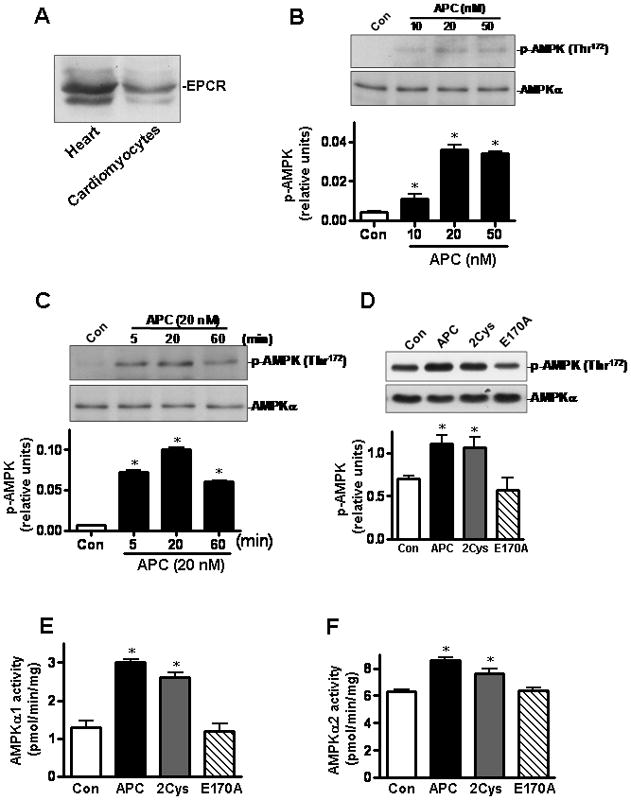
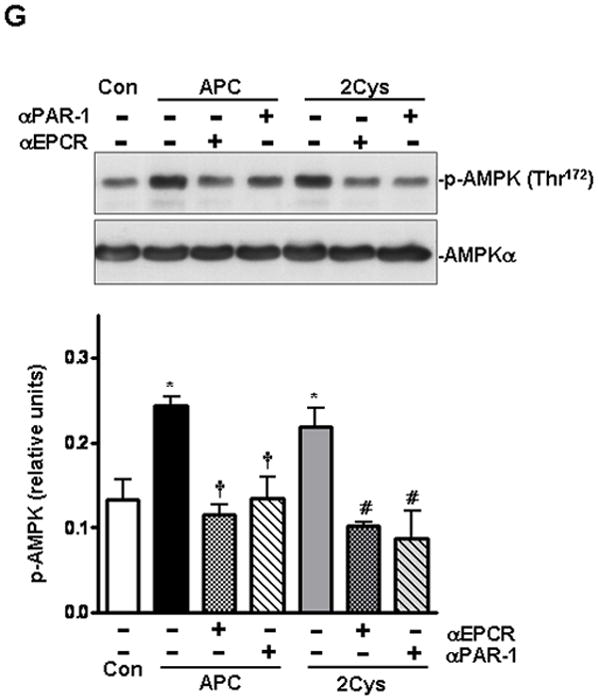
APC activates AMPK phosphorylation in isolated mice cardiomyocytes. (A) Immunoblotting for EPCR in mice heart or cardiomyocytes. (B) APC stimulates the phosphorylation of AMPK in cardiomyocytes by a concentration- and (C) time-dependent manner. (D) The phosphorylation of AMPK in isolated mouse cardiomyocytes treated with different APC derivatives. Activation of AMPKα1 (E) and AMPKα2 (F) by different APC derivatives in isolated mouse cardiomyocytes. Values are means ± S.E., n=4. *p<0.05 vs. control. (G) AMPK activation by APC and APC-2Cys in isolated cardiomyocytes with or without pre-incubation with function-blocking anti-EPCR (RCR252) and anti-PAR-1 (H-111) antibodies. Values are means ± S.E., n=4. *p<0.01 vs. control; †p<0.05 vs. APC alone; #p<0.05 vs. APC-2Cys alone.
APC activates AMPK via the Ca2+-CaMKKβ pathway
APC initiates intracellular calcium influx in human brain endothelial cells and human smooth muscle cells [30,31]. The mobilization of intracellular calcium might be responsible for elevated AMPK phosphorylation since it is known that the Ca2+/calmodulin-dependent protein kinase kinase β (CaMKKβ) serves as an upstream AMPK kinase [32,33]. To explore whether AMPK activation is due to the APC-mediated intracellular Ca2+ rise in cardiomyocytes, we evaluated intracellular Ca2+ homeostasis by using the fluorescence dye fura-2/AM [34]. Results showed that intracellular Ca2+ was elevated with both APC and 2Cys, however, E170A did not affect this response (Figure 4A). To provide direct evidence that AMPK activation is mediated via the CaMKKβ kinase activity, we first demonstrated that CaMKKβ is expressed in the heart and cardiomyocytes (Figure 4B). The brain tissue was used as a positive control since CaMKKβ is highly expressed in the brain [35]. The immunoprecipitated (IP) CaMKKβ from either APC- or saline-treated heart tissues were incubated with the IP-AMPK obtained from saline-treated heart tissues in a kinase buffer with or without Ca2+/calmodulin (CaM). AMPK was specifically activated by the IP-CaMKKβ derived from the APC-treated heart tissues in the presence of Ca2+/CaM, indicating a higher kinase activity for CaMKKβ in APC-treated heart samples (Figure 4C). Moreover, mouse hearts pre-treated with the CaMKKβ inhibitor, STO-60935, blocked the phosphorylation of AMPK (Figure 4D) and the activation of both AMPKα1 and AMPKα2 (Figures 4E and 4F) triggered by APC. These results suggest that APC activates AMPK in cardiomyocytes through the stimulation of the upstream CaMKKβ kinase activity.
Figure 4.
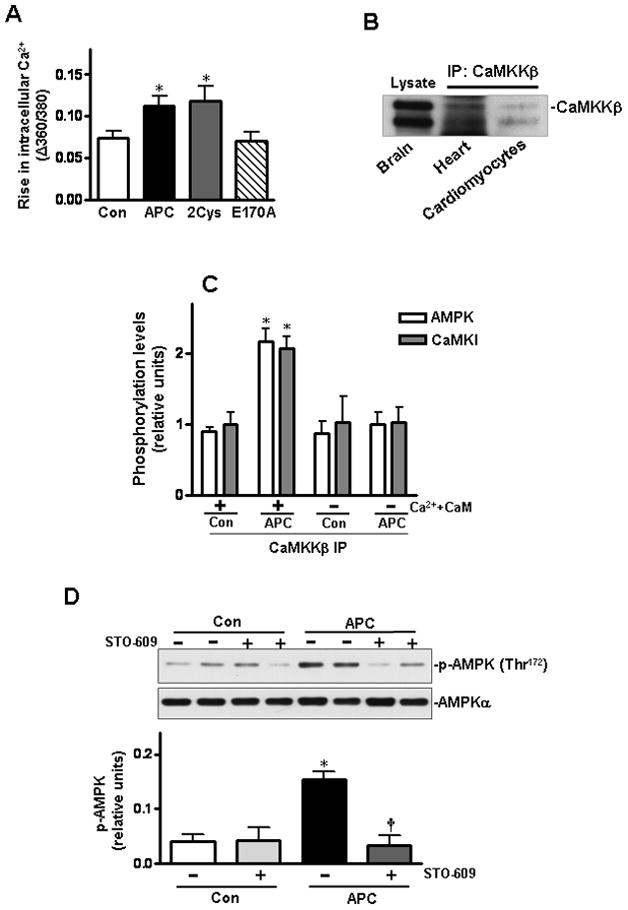
Activation of AMPK by APC is mediated via the Ca2+-CaMKKβ pathway. (A) Isolated cardiomyocytes were treated with APC derivatives or saline. The rise in intracellular Ca2+ was recorded in response to electrical stimulus. Values are means ± S.E., n = 60–90 cells per group, *p< 0.05 vs. control. (B) Immunoblotting of CaMKKβ in heart and cardiomyocytes after immunoprecipitation. (C) Densitometric scanning of immunoprecipitated CaMKKβ from saline or APC-treated heart tissues following incubation with immunoprecipitated AMPK from basal saline-treated hearts with or without Ca2+ and calmodulin. (D) The phosphorylation of AMPK in mouse hearts by APC with or without pre-treatment with CaMKKβ inhibitor, STO-609. Values are means ± S.E., n=4. *p<0.05 vs. control, †p<0.05 vs. APC alone.
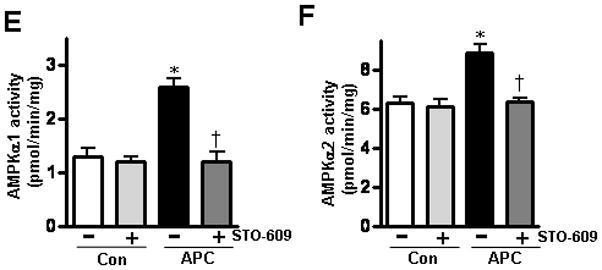
APC improves the contractile properties of cardiomyocytes after exposure to hypoxia
We next evaluated the effect of APC on the contractile properties of isolated cardiomyocytes under normal and hypoxic conditions by using an extracellular Ca2+ concentration of 1.0 mmol/L and a stimulus frequency of 0.5 Hz [26]. As shown in Figure 5, APC did not significantly affect cardiomyocyte contractility under normal conditions, though an enhanced trend on maximal velocity of shortening/re-lengthening (±dL/dt) was observed with both APC and APC-2Cys treatment groups (Figures 5C and 5D). However, cardiomyocytes displayed markedly impaired peak shortening and reduced maximal velocity of shortening/re-lengthening after exposure to hypoxia (Figures 5B-D). Interestingly, both APC and 2Cys significantly improved the contractility of cardiomyocytes exposed to hypoxia (Figure 5B-D). To determine whether cardiac AMPK signaling triggered by APC is responsible for ameliorating cardiomyocyte contractile dysfunction exposed to hypoxic condition, isolated cardiomyocytes were pre-treated with AMPK inhibitor, Compound C [36], prior to exposure to hypoxia. Compound C abolished protective effects of APC and APC-2Cys on cardiomyocytes’ mechanical properties in response to hypoxia (Figure 5B-D). These results suggest that the AMPK signaling function of APC protects cardiomyocytes from hypoxic injury.
Figure 5.
Effect of APC on the contractile properties of cardiomyocytes after exposure to hypoxia. (A) Resting cell length; (B) peak shortening (PS, normalized to cell length); (C) maximal velocity of shortening (+dL/dt) and (D) re-lengthening (-dL/dt) were calculated as described under Methods. Values are means ± S.E., n = 50–100 cells per group, *p<0.01 vs. Con (normoxia), †p<0.05 vs. Con (hypoxia), #p<0.05 vs. APC (hypoxia) or APC-2Cys (hypoxia).
APC ameliorates post-ischemic cardiac dysfunction and injury
To further investigate whether APC can improve the recovery of post-ischemic heart, we performed ex vivo ischemic experiments using the langendorff ischemia/reperfusion system. Isolated mouse hearts underwent 20 min global ischemia followed by reperfusion with the perfusate containing either saline or APC derivatives. There were no significant differences in the basal parameters among the four groups with respect to heart rate and left ventricular pressure (Figure 6A). However, during the reperfusion, the heart rate-left ventricle pressure product was significantly better with both APC and APC-2Cys, but not with E170A treatment compared to the control group (Figure 6A). The TTC staining after 2h perfusion indicated that APC-mediated AMPK signaling contributes to the recovery from myocardial injury as evidenced by the reduced myocardial infarct size with both APC and APC-2Cys treated but not with the APC-E170A treated groups (Figure 6B). The phosphorylation of AMPK and acetyl-CoA carboxylase and specific activities of both AMPKα1 and AMPKα2 were enhanced in APC and APC-2Cys but not in APC-E170A treated ex vivo ischemic heart tissues (Figures 6C and D), suggesting that the APC-AMPK signaling pathway contributes to protection against ischemic heart injury.
Figure 6.
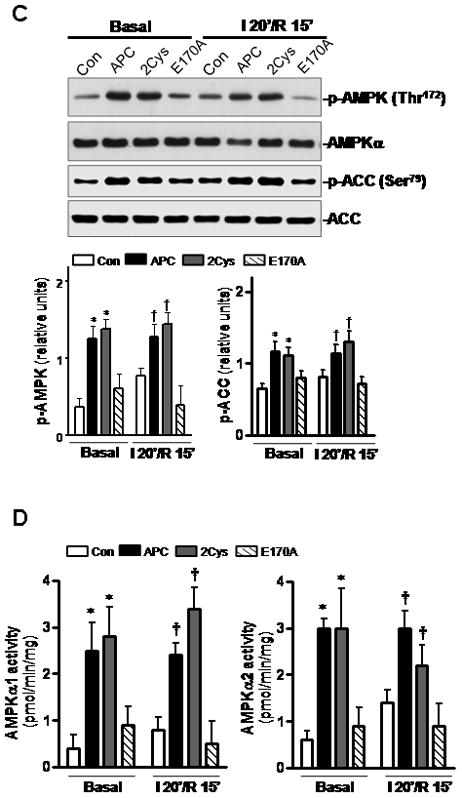
APC ameliorates post-ischemic cardiac dysfunction and injury. (A) The heart rate and left ventricular pressure during baseline perfusion and post-ischemic reperfusion with or without administration of APC derivatives were assessed as described under Methods. Values are means ± S.E., n=4–6 hearts for each group. *p<0.05 APC vs. Con; †p<0.05 APC-2Cys vs. Con during reperfusion. (B) The ratio of infarct size of isolated mouse hearts after 2h reperfusion with APC derivatives or vehicle (Con). Values are means ± S.E., n=4 per group. *p<0.05 vs. Con. (C) Phosphorylation of AMPK and ACC and (D) activation of AMPKα1 (left panel) and AMPKα2 (right panel) during ex vivo ischemia reperfusion with or without APC. Values are means ± S.E., n=4 per group, *p< 0.05 vs. basal control, †p<0.05 vs. I/R control.
APC reduces cardiac inflammatory mediators in response to ischemia/reperfusion in vivo
APC is known to exert its cytoprotective and anti-inflammatory effects via the down-regulation of the NF-κB pathway and inhibition of the proinflammatory cytokines [6,7]. Both APC and APC-2Cys down-regulated the NF-κB pathway in heart tissues during ischemia/reperfusion (Figure 7A). Moreover, we also detected a reduction of the c-Jun N-terminal kinase (JNK) phosphorylation as well as its downstream target c-Jun phosphorylation with both APC derivatives during ischemia/reperfusion (Figure 7B). JNK is a stress-activated protein kinase whose phosphorylation is known to be stimulated by inflammatory stress [37]. The activation and subsequent translocation of JNK into the nucleus phosphorylates c-Jun, a transcription factor that is known to regulate the expression of proinflammatory cytokines [38]. The attenuation of JNK phosphorylation contributed to reduced production of inflammatory cytokines including TNFα and IL-6 in heart tissues after long time reperfusion (Figures 7C and D).
Figure 7.
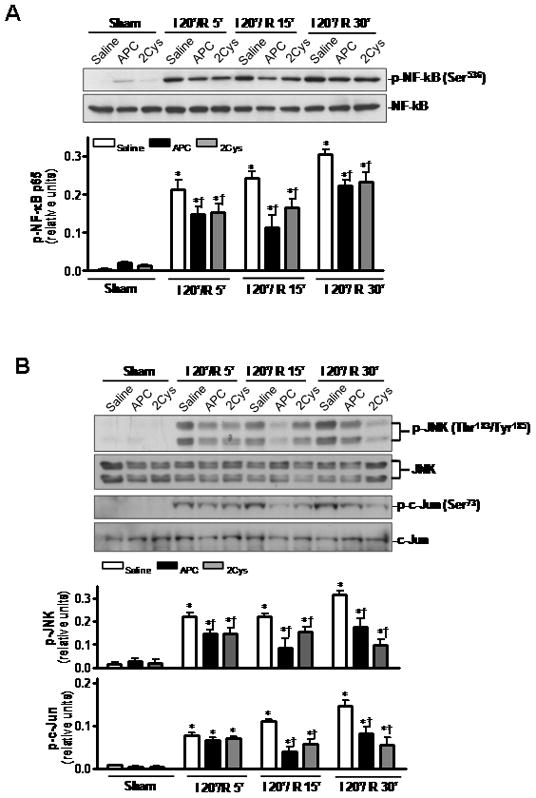
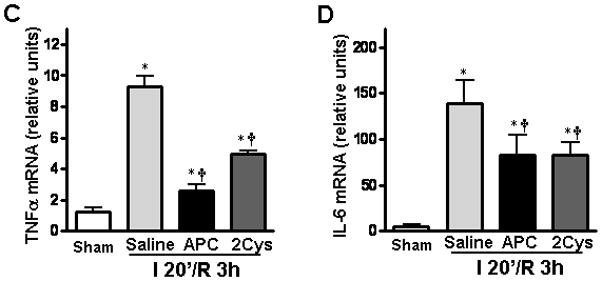
APC reduces cardiac inflammatory responses after ischemia/ reperfusion. (A) Phosphorylation of NF-κB is decreased by both APC and 2Cys during ischemia/reperfusion. (B) Both APC and APC-2Cys attenuate JNK and c-Jun phosphorylation during in vivo ischemia/reperfusion. (C) Both APC derivatives also reduce the expression of TNFα and IL-6 mRNA in hearts. Values are means ± S.E., n=3. *p<0.01 vs. sham, respectively, †p<0.05 vs. I/R saline, respectively.
Discussion
The ability of APC to limit the myocardial ischemia/reperfusion injury was reported previously [19–21], however, the mechanism through which APC exerts a cardioprotective effect in response to ischemic stress has not been fully understood. In this study, we demonstrated that APC confers protection against acute myocardial injury by activating AMPK by a mechanism that is largely independent of its anticoagulant function. This was evidenced by the observation that administration of both APC and the anticoagulant-defective APC-2Cys, but not the signaling-defective APC-E170A, reduced myocardial infarction and restored cardiac function in the ischemic mouse heart by activating AMPK in both in vivo and ex vivo model systems. Our results indicated that cardiomyocytes express EPCR and that both APC and APC-2Cys directly trigger AMPK phosphorylation in cardiomyocytes by enhancing the Ca2+/CaMKKβ activity by EPCR and PAR-1-dependent mechanisms without affecting the AMP/ATP ratio (Figure 8).
Figure 8.
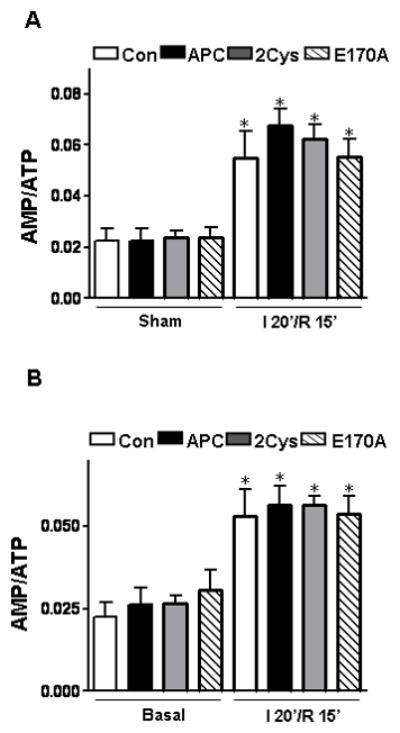
The effect of APC on the AMP/ATP ratio. (A) Myocardial AMP/ATP ratios during sham or ischemia/reperfusion in vivo. (B) Myocardial AMP/ATP ratios at baseline perfusion and following ischemia/reperfusion in the ex vivo hearts. The amount of AMP and ATP was measured in neutralized perchloric acid extracts of frozen tissues by HPLC, n=4 per group. *p<0.05 vs. sham or basal, respectively.
The discovery of APC activation of the cardiac AMPK opens up a whole new avenue for understanding the mechanism by which APC may exert its cytoprotective function in ischemic stress. AMPK is an energy sensor protein which is activated in response to ATP depletion [29]. The downstream effects of AMPK include mediating the translocation of the glucose transporter, GLUT4, onto the cell membrane to enhance glucose uptake [29,38]. Additionally, AMPK can facilitate glycolysis and fatty acid oxidation to generate more ATP, while inhibiting fatty acid biosynthesis [29,38]. Noting that the lethality of severe sepsis syndrome is the result of multiple organ failure due to ischemic stress, our findings suggest that APC may exert a cytoprotective activity through the regulation of metabolic pathways.
In addition to the up-regulation of the AMPK activity, our results further demonstrated that APC attenuates ischemia/reperfusion injury by down-regulating the NF-κB and JNK signaling pathways, thereby inhibiting the expression of inflammatory mediators including TNFα and IL-6 in the ischemic heart tissues. APC is also known to decrease the expression level of inflammatory cytokines such as TNFα, IL-6, and IL-8 in sepsis [4] and renal ischemic conditions [39]. Previous results have established that APC decreases inflammatory cytokines by inhibiting the NF-κB pathway 6,7]. Based on our data in this study, we now demonstrate that inhibition of JNK phosphorylation by APC also contributes to its protective effect in cardiac ischemia/reperfusion injury. JNK is an inflammation-triggered MAP kinase that can be activated via various extracellular stimuli such as ischemic stress and LPS [40]. Upon activation, JNK is translocated into the nucleus where it phosphorylates c-Jun, a transcription factor that leads to production of inflammatory mediators [37]. A comparable activity for both APC and APC-2Cys suggests that the signaling activity of APC is sufficient for its cardioprotective activity in this acute ischemic injury model.
As indicated above, the FDA has approved APC for treating adult patients with severe sepsis, which reduces the overall mortality by 19% [14]. However, because of its potent anticoagulant activity, APC-therapy is also associated with an increased risk (~ 2%) of bleeding [6,14]. This problem may be greater than it appears since bleeding also limits the threshold of APC dose in treatments, thus potentially masking the beneficial effect of APC in a higher number of patients. Recent in vivo studies using mouse mutants of APC which were defective in either signaling or the anticoagulant activity provided strong support for the hypothesis that the signaling activity of APC is sufficient for its cytoprotective and antiinflammatory activity in mouse models of severe sepsis [17,41] and ischemic stroke [42]. The observation of this study that human APC-2Cys also protects the heart from ischemic/reperfusion injury and dysfunction in an acute mouse model reinforces the idea that it may be feasible to develop non-anticoagulant APC therapeutics for treating patients with cardiac/brain infarction, severe sepsis and other inflammatory disorders without increasing the risk of bleeding.
Supplementary Material
Acknowledgments
We would like to thank Audrey Rezaie for proofreading the manuscript. This work was supported by grants awarded by the National Heart, Lung, and Blood Institute of the National Institutes of Health HL 101917 and HL 68571 to ARR; and by grants awarded by American Heart Association SDG0835169N to JL and Pre-doctoral Fellowship 10PRE4190043 to JW.
References
- 1.Esmon CT. Molecular events that control the protein C anticoagulant pathway. Thromb Haemost. 1993;70(1):29–35. [PubMed] [Google Scholar]
- 2.Walker FJ, Fay PJ. Regulation of blood coagulation by the protein C system. FASEB J. 1992;6(8):2561–2567. doi: 10.1096/fasebj.6.8.1317308. [DOI] [PubMed] [Google Scholar]
- 3.Taylor FB, Jr, Chang A, Esmon CT, D’Angelo A, Vigano-D’Angelo S, Blick KE. Protein C prevents the coagulopathic and lethal effects of Escherichia coli infusion in the baboon. J Clin Invest. 1987;79(3):918–925. doi: 10.1172/JCI112902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor FB, Jr, Stearns-Kurosawa DJ, Kurosawa S, Ferrell G, Chang AC, Laszik Z, Kosanke S, Peer G, Esmon CT. The endothelial cell protein C receptor aids in host defense against Escherichia coli sepsis. Blood. 2000;95(5):1680–1686. [PubMed] [Google Scholar]
- 5.Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296(5574):1880–1882. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- 6.Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109(8):3161–3172. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 7.Joyce DE, Grinnell BW. Recombinant human activated protein C attenuates the inflammatory response in endothelium and monocytes by modulating nuclear factor-kappaB. Crit Care Med. 2002;30(5 Suppl):S288–293. doi: 10.1097/00003246-200205001-00019. [DOI] [PubMed] [Google Scholar]
- 8.Finigan JH, Dudek SM, Singleton PA, Chiang ET, Jacobson JR, Camp SM, Ye SQ, Garcia JG. Activated protein C mediates novel lung endothelial barrier enhancement: role of sphingosine 1-phosphate receptor transactivation. J Biol Chem. 2005;280(17):17286–17293. doi: 10.1074/jbc.M412427200. [DOI] [PubMed] [Google Scholar]
- 9.Nick JA, Coldren CD, Geraci MW, Poch KR, Fouty BW, O’Brien J, Gruber M, Zarini S, Murphy RC, Kuhn K, Richter D, Kast KR, Abraham E. Recombinant human activated protein C reduces human endotoxin-induced pulmonary inflammation via inhibition of neutrophil chemotaxis. Blood. 2004;104(13):3878–3885. doi: 10.1182/blood-2004-06-2140. [DOI] [PubMed] [Google Scholar]
- 10.Elphick GF, Sarangi PP, Hyun YM, Hollenbaugh JA, Ayala A, Biffl WL, Chung HL, Rezaie AR, McGrath JL, Topham DJ, Reichner JS, Kim M. Recombinant human activated protein C inhibits integrin-mediated neutrophil migration. Blood. 2009;113(17):4078–4085. doi: 10.1182/blood-2008-09-180968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feistritzer C, Riewald M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood. 2005;105(8):3178–3184. doi: 10.1182/blood-2004-10-3985. [DOI] [PubMed] [Google Scholar]
- 12.Cheng T, Liu D, Griffin JH, Fernandez JA, Castellino F, Rosen ED, Fukudome K, Zlokovic BV. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med. 2003;9(3):338–342. doi: 10.1038/nm826. [DOI] [PubMed] [Google Scholar]
- 13.Griffin JH, Fernandez JA, Liu D, Cheng T, Guo H, Zlokovic BV. Activated protein C and ischemic stroke. Crit Care Med. 2004;32(5 Suppl):S247–253. doi: 10.1097/01.ccm.0000126127.87484.2b. [DOI] [PubMed] [Google Scholar]
- 14.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher CJ., Jr Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344(10):699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 15.Guo H, Liu D, Gelbard H, Cheng T, Insalaco R, Fernandez JA, Griffin JH, Zlokovic BV. Activated protein C prevents neuronal apoptosis via protease activated receptors 1 and 3. Neuron. 2004;41(4):563–572. doi: 10.1016/s0896-6273(04)00019-4. [DOI] [PubMed] [Google Scholar]
- 16.Iwaki T, Cruz DT, Martin JA, Castellino FJ. A cardioprotective role for the endothelial protein C receptor in lipopolysaccharide-induced endotoxemia in the mouse. Blood. 2005;105(6):2364–2371. doi: 10.1182/blood-2004-06-2456. [DOI] [PubMed] [Google Scholar]
- 17.Kerschen EJ, Fernandez JA, Cooley BC, Yang XV, Sood R, Mosnier LO, Castellino FJ, Mackman N, Griffin JH, Weiler H. Endotoxemia and sepsis mortality reduction by non-anticoagulant activated protein C. J Exp Med. 2007;204(10):2439–2448. doi: 10.1084/jem.20070404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niessen F, Furlan-Freguia C, Fernandez JA, Mosnier LO, Castellino FJ, Weiler H, Rosen H, Griffin JH, Ruf W. Endogenous EPCR/aPC-PAR1 signaling prevents inflammation-induced vascular leakage and lethality. Blood. 2009;113(12):2859–2866. doi: 10.1182/blood-2008-12-192385. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Pirat B, Muderrisoglu H, Unal MT, Ozdemir H, Yildirir A, Yucel M, Turkoglu S. Recombinant human-activated protein C inhibits cardiomyocyte apoptosis in a rat model of myocardial ischemia-reperfusion. Coron Artery Dis. 2007;18(1):61–66. doi: 10.1097/MCA.0b013e328010a44a. [DOI] [PubMed] [Google Scholar]
- 20.Loubele ST, Spek CA, Leenders P, van Oerle R, Aberson HL, Hamulyak K, Ferrell G, Esmon CT, Spronk HM, ten Cate H. Activated protein C protects against myocardial ischemia/reperfusion injury via inhibition of apoptosis and inflammation. Arterioscler Thromb Vasc Biol. 2009;29(7):1087–1092. doi: 10.1161/ATVBAHA.109.188656. [DOI] [PubMed] [Google Scholar]
- 21.Medina P, Navarro S, Corral J, Zorio E, Roldan V, Estelles A, Santamaria A, Marin F, Rueda J, Bertina RM, Espana F. Endothelial protein C receptor polymorphisms and risk of myocardial infarction. Haematologica. 2008;93(9):1358–1363. doi: 10.3324/haematol.13066. [DOI] [PubMed] [Google Scholar]
- 22.Li J, Miller EJ, Ninomiya-Tsuji J, Russell RR, 3rd, Young LH. AMP-activated protein kinase activates p38 mitogen-activated protein kinase by increasing recruitment of p38 MAPK to TAB1 in the ischemic heart. Circ Res. 2005;97(9):872–879. doi: 10.1161/01.RES.0000187458.77026.10. [DOI] [PubMed] [Google Scholar]
- 23.Miller EJ, Li J, Leng L, McDonald C, Atsumi T, Bucala R, Young LH. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature. 2008;451(7178):578–582. doi: 10.1038/nature06504. [DOI] [PubMed] [Google Scholar]
- 24.Jensen TE, Rose AJ, Jorgensen SB, Brandt N, Schjerling P, Wojtaszewski JF, Richter EA. Possible CaMKK-dependent regulation of AMPK phosphorylation and glucose uptake at the onset of mild tetanic skeletal muscle contraction. Am J Physiol Endocrinol Metab. 2007;292(5):E1308–1317. doi: 10.1152/ajpendo.00456.2006. [DOI] [PubMed] [Google Scholar]
- 25.Zhao P, Wang J, Ma H, Xiao Y, He L, Tong C, Wang Z, Zheng Q, Dolence EK, Nair S, Ren J, Li J. A newly synthetic chromium complex-Chromium (d-phenylalanine)(3) activates AMP-activated protein kinase and stimulates glucose transport. Biochem Pharmacol. 2009;77(6):1002–1010. doi: 10.1016/j.bcp.2008.11.018. [DOI] [PubMed] [Google Scholar]
- 26.Dong F, Zhang X, Yang X, Esberg LB, Yang H, Zhang Z, Culver B, Ren J. Impaired cardiac contractile function in ventricular myocytes from leptin-deficient ob/ob obese mice. J Endocrinol. 2006;188(1):25–36. doi: 10.1677/joe.1.06241. [DOI] [PubMed] [Google Scholar]
- 27.Bae JS, Yang L, Manithody C, Rezaie AR. Engineering a disulfide bond to stabilize the calcium-binding loop of activated protein C eliminates its anticoagulant but not its protective signaling properties. J Biol Chem. 2007;282(12):9251–9259. doi: 10.1074/jbc.M610547200. [DOI] [PubMed] [Google Scholar]
- 28.Yang L, Bae JS, Manithody C, Rezaie AR. Identification of a specific exosite on activated protein C for interaction with protease-activated receptor 1. J Biol Chem. 2007;282(35):25493–25500. doi: 10.1074/jbc.M702131200. [DOI] [PubMed] [Google Scholar]
- 29.Young LH, Li J, Baron SJ, Russell RR. AMP-activated protein kinase: a key stress signaling pathway in the heart. Trends Cardiovasc Med. 2005;15(3):110–118. doi: 10.1016/j.tcm.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 30.Domotor E, Benzakour O, Griffin JH, Yule D, Fukudome K, Zlokovic BV. Activated protein C alters cytosolic calcium flux in human brain endothelium via binding to endothelial protein C receptor and activation of protease activated receptor-1. Blood. 2003;101(12):4797–4801. doi: 10.1182/blood-2002-12-3680. [DOI] [PubMed] [Google Scholar]
- 31.Bretschneider E, Uzonyi B, Weber AA, Fischer JW, Pape R, Lotzer K, Schror K. Human vascular smooth muscle cells express functionally active endothelial cell protein C receptor. Circ Res. 2007;100(2):255–262. doi: 10.1161/01.RES.0000255685.06922.c7. [DOI] [PubMed] [Google Scholar]
- 32.Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280(32):29060–29066. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- 33.Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2(1):21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 34.Ren J, Privratsky JR, Yang X, Dong F, Carlson EC. Metallothionein alleviates glutathione depletion-induced oxidative cardiomyopathy in murine hearts. Crit Care Med. 2008;36:2106–2116. doi: 10.1097/CCM.0b013e31817bf925. [DOI] [PubMed] [Google Scholar]
- 35.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2(1):9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 36.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, Shepherd EG, Nelin LD. MAPK phosphatases--regulating the immune response. Nat Rev Immunol. 2007;7(3):202–212. doi: 10.1038/nri2035. [DOI] [PubMed] [Google Scholar]
- 38.Young LH. AMP-activated protein kinase conducts the ischemic stress response orchestra. Circulation. 2008;117(6):832–840. doi: 10.1161/CIRCULATIONAHA.107.713115. [DOI] [PubMed] [Google Scholar]
- 39.Mizutani A, Okajima K, Uchiba M, Noguchi T. Activated protein C reduces ischemia/reperfusion-induced renal injury in rats by inhibiting leukocyte activation. Blood. 2000;95(12):3781–3787. [PubMed] [Google Scholar]
- 40.Swantek JL, Cobb MH, Geppert TD. Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for lipopolysaccharide stimulation of tumor necrosis factor alpha (TNF-alpha) translation: glucocorticoids inhibit TNF-alpha translation by blocking JNK/SAPK. Mol Cell Biol. 1997;17(11):6274–6282. doi: 10.1128/mcb.17.11.6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mosnier LO, Zampolli A, Kerschen EJ, Schuepbach RA, Banerjee Y, Fernandez JA, Yang XV, Riewald M, Weiler H, Ruggeri ZM, Griffin JH. Hyperantithrombotic, noncytoprotective Glu149Ala-activated protein C mutant. Blood. 2009;113(23):5970–5978. doi: 10.1182/blood-2008-10-183327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo H, Singh I, Wang Y, Deane R, Barrett T, Fernandez JA, Chow N, Griffin JH, Zlokovic BV. Neuroprotective activities of activated protein C mutant with reduced anticoagulant activity. Eur J Neurosci. 2009;29(6):1119–1130. doi: 10.1111/j.1460-9568.2009.06664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.