

Abstract
Gene therapy provides powerful new approaches to cure a large variety of diseases, which are being explored in ongoing worldwide clinical trials. To overcome the limitations of viral gene delivery systems, synthetic nonviral vectors such as cationic liposomes (CLs) are desirable. However, improvements of their efficiency at reduced toxicity and a better understanding of their mechanism of action are required. We present the efficient synthesis of a series of degradable multivalent cationic lipids (CMVLn, n=2 to 5) containing a disulfide bond spacer between headgroup and lipophilic tails. This spacer is designed to be cleaved in the reducing milieu of the cytoplasm and thus decrease lipid toxicity. Small angle X-ray scattering demonstrates that the initially formed lamellar phase of CMVLn–DNA complexes completely disappears when reducing agents such as DTT or the biologically relevant reducing peptide glutathione are added to mimic the intracellular milieu. The CMVLs (n=3 to 5) exhibit reduced cytotoxicity and transfect mammalian cells with efficiencies comparable to those of highly efficient non-degradable analogues and benchmark commercial reagents such as Lipofectamine 2000. Thus, our results demonstrate that degradable disulfide spacers may be used to reduce the cytotoxicity of synthetic nonviral gene delivery carriers without compromising their transfection efficiency.
1. Introduction
Gene therapy has the potential to address a wide variety of diseases at their root cause. These include cancer, hereditary disorders, and many others [1-4]. However, despite massive efforts to the contrary, including numerous clinical trials worldwide [5, 6], widespread therapeutic success has remained elusive. A key challenge for gene therapy is to develop efficient and safe gene delivery methods. Viral vectors, such as engineered adenoviruses and retroviruses, are very efficient (particularly in vivo) and have resulted in gene therapy's first successes [7], but they suffer from a number of drawbacks. A primary concern is vector safety: in clinical trials, viral vectors have led to serious adverse effects, owing to immunogenicity and non-specific integration of their genetic cargo into the host genome [8-11]. Other limitations are the small size of viral genomes and the comparatively difficult vector production. Synthetic nonviral vectors, on the other hand, lack immunogenic protein components, do not impose a size limit on the nucleic acid to be transferred, and are more easily prepared and modified. Their main limitation is their lower transfection efficiency (TEa, a measure of the amount of successfully transferred and transcribed DNA) and the onset of toxicity with increasing cationic lipid/nucleic acid mole ratios, which needs to be improved in order to compete with viral vectors [12-15].
Cationic liposome (CL)–nucleic acid (NA) complexes are one of the main classes of nonviral vectors [13, 16-20]. They form spontaneously when cationic liposomes (typically containing a neutral lipid (NL) as well as a cationic lipid) are combined with NAs. A large number of cationic lipids have been synthesized [21-23] to enhance the efficiency of CL–DNA complexes and uncover structure–TE relationships, albeit with limited success. Thus, further development of cationic lipid-based vectors is required to match the TE of viral vectors. Among the chemical approaches to improve the TE of CL–DNA complexes, the synthesis of cationic lipids with multivalent head groups has been particularly promising [24-30].
The development of cationic lipids with low cytotoxicity is an important objective, especially for applications such as gene silencing which require higher cationic lipid/nucleic acid ratios [27]. The toxicity of CL–NA complexes is known to be due to the cationic lipid component of the complex [27, 31]. Thus, we hypothesized that triggered disintegration of an appropriately designed, degradable multivalent cationic lipid component of the CL–DNA complex upon entry into the cytoplasm should reduce cytotoxicity. At the same time, such lipids should retain high TE levels comparable to those of recently synthesized non-degradable multivalent lipids [24-30] because the key steps of lipid-mediated DNA delivery prior to cytoplasmic release—DNA compaction, complex uptake and endosomal escape [32, 33]—will not be compromised by the lipid degradation process.
In order to test this hypothesis and potentially decrease CL-induced cytotoxicity of lipid vectors, we prepared a series of cationic lipids designed to quickly degrade in the cytoplasm. As we describe, degradable multivalent lipids exhibit reduced toxicity with increasing cationic lipid/nucleic acid mole ratio while maintaining the high TE levels of previously synthesized multivalent lipids [24-30], consistent with our hypothesis.
Cells maintain a high redox potential gradient between the intracellular (reducing) environment and extracellular (oxidizing) space. The main intracellular reducing agent is the short peptide glutathione (GSH), which is continually recycled [34]. The intracellular concentration of GSH varies for different tissues but can reach up to 5 mM (e.g. in the liver) [35, 36]. The redox potential gradient can be exploited to trigger, e.g., the cleavage of disulfide bonds in molecules once they reach the cytoplasm. This mechanism is used by biological toxins [37] as well as in drug delivery and diagnostic imaging [38-42]. A few studies have also applied the concept to gene delivery vectors [43-46], including CL–DNA complexes [47-50].
We report the convenient and efficient synthesis of a series of new degradable multivalent cationic lipids, termed CMVLn (n = 2 to 5). These CMVLs contain a reductively cleavable disulfide bond in the spacer between the hydrophobic moiety and the cationic head group. Reducing conditions result in separation of the hydrophobic tail from the headgroup. Except for the spacer, the CMVLs are identical to a previously synthesized series of multivalent lipids [32]. Of these, MVL5 in particular is a highly efficient vector for both DNA and siRNA [26, 27, 32]. The headgroup charge of the CMVLs was measured using an ethidium bromide (EtBr) displacement assay. CL–DNA complexes prepared from mixtures of the CMVLs with neutral 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC) were studied using small-angle X-ray scattering (SAXS) under reducing and non-reducing conditions, as well as measurements of cytotoxicity and TE. The CMVLs were compared with commercially available 2,3-dioleoyloxypropyltrimethylammonium chloride (DOTAP) and Lipofectamine 2000 as well as with non-degradable MVL5. All CMVL/DOPC–DNA complexes form lamellar phases which can be completely disassembled by addition of reducing agents such as GSH and DTT. Importantly, the complexes of CMVLn (n = 3, 4, 5) exhibit high TE at reduced cytotoxity when compared to non-degradable analogs and commercial lipid reagents.
2. Materials and Methods
2.1. Materials and reagents
MVL5 was prepared as described [26]. Opti-MEM cell culture medium and Lipofectamine 2000 were purchased from Invitrogen. The lipids DOTAP and DOPC were purchased from Avanti Polar Lipids. The chemical structures of MVL5, DOTAP and DOPC are shown in the Supporting Information, Figure S1. For X-ray samples and ethidium bromide displacement assay, highly polymerized calf thymus DNA (Sigma-Aldrich) was used. Luciferase plasmid DNA (pGL3 Control Vector, Promega) for microscopy, dynamic light scattering, cytotoxicity, and transfection assays was propagated in E. coli and isolated using a Qiagen Giga Kit. Other chemicals were purchased from σ–Aldrich unless otherwise stated and were used as received.
2.2. General methods
NMR spectroscopy was carried out on Bruker Avance instruments (200 MHz and 500 MHz). Detection of the spots in thin-layer chromatography was achieved by UV absorption and ninhydrin reagent (300 mg in 95 mL of 2-propanol and 5 mL of acetic acid) or phosphomolybdic acid in ethanol (10 wt%). Silica gel (Merck) with a mesh size of 200–425 was used for flash chromatography [51].
2.3. Lipid suspensions
Lipid stock solutions in chloroform (for DOTAP and DOPC) or chloroform/methanol (9:1, v/v; for MVL5 and all CMVLs) were combined at the appropriate ratios in glass vials. The obtained lipid solutions were dried, first by a stream of nitrogen and subsequently in a vacuum for 12 h, to form a thin lipid film. Sterile high resistivity (18.2 MΩ) water was added and the mixture was incubated at 37 °C for at least 12 h to give suspensions of a final concentration of 50 mM for X-ray samples. For transfection, aqueous lipid suspensions were prepared at 0.5 mM. Lipid mixtures containing higher mole fractions of DOPC formed opaque suspensions which were tip-sonicated to clarity (Vibra-cell, Sonics Materials). The aqueous lipid suspensions were stored at 4 °C until use.
2.4. Ethidium bromide displacement assay
Wells in a 96-well plate were preloaded with a mixture 2.4 μg of DNA and 0.28 μg of EtBr in 100 μL of water. To the wells, varied amounts of a 0.2 mM aqueous lipid solution (at lipid mole fraction of DOPC (ΦDOPC) = 0) were added, preparing at least duplicate samples for each data point. Fluorescence was measured on a 1420 Multilabel counter Victor 3V (Perkin Elmer) plate reader. The results were normalized to the fluorescence reading in the absence of lipid. To calculate the lipid headgroup charge, Z, the following equation was used: at the isoelectric point (determined from the data as described [32]), , where N+ and N– are the number of cationic charges on the lipid and of negative charges on the DNA, respectively; mCL and mDNA are the mass of cationic lipid and of DNA respectively; and MCL and MDNA-bp are the molecular weight of cationic lipid and DNA basepair, respectively.
2.5. Optical microscopy
For imaging of complexes with optical microscopy, 0.2 weight-% (of total lipid) DHPE-Texas Red (Avanti Polar Lipids) was added to the lipid mixture (before evaporation of the organic solvent). This fluorescent dye has an excitation/emission peak at 585/615 nm. Cy5 (Mirus Bio LLC), with an excitation/emission peak at 605/670 nm, was conjugated to DNA using the manufacturer's protocol. Complexes were then prepared as for transfection and imaged in Opti-MEM, 20 minutes after complex formation. Fluorescence intensity profiles of particles were obtained with the ImageJ Color Profiler software tool.
2.6. Dynamic light scattering (DLS)
Complexes for DLS were prepared as for transfection but at a final concentration of 2 μg/mL of DNA (twice the concentration used for transfection, to achieve a sufficient particle concentration for the DLS measurement). The complexes were analyzed by DLS in Opti-MEM, 20 minutes after complex formation using a Malvern Nano ZS (173 degree scattering angle). The plot shows the z-average hydrodynamic diameter as obtained from the instrument. Data points shown are the average of duplicate measurements of the same sample, with error bars showing the standard deviation.
2.7. Small-angle X-ray scattering
High-resolution SAXS experiments were performed at the Stanford Synchrotron Radiation Lightsource. X-ray samples were prepared from 50 mM liposome solutions and 4 mg/mL DNA solutions, using 100 μg of DNA. CL–DNA complexes were formed by combining the CL and DNA solutions in microcentrifuge tubes, diluting 1-fold with Opti-MEM and incubating at 4 °C for at least 3 days after extensive centrifugation. For the samples containing reducing agents, GSH and DTT stock solutions were prepared at 300 mM and added to each sample after complex formation (after centrifugation) at a molar ratio of ≥ 10 over the CMVLs. Samples were then transferred into 1.5 mm quartz capillaries (Hilgenberg) and flame-sealed. Typically, the CL–DNA complexes formed a white opaque precipitate. Samples containing reducing agents occasionally exhibited phase separation of the initial precipitate. SAXS measurements were performed at multiple positions throughout the samples and representative scans are shown.
To assess the stability of CMVL-containing CL–DNA complexes (cf. Supplementary Material), each X-ray sample was measured at least twice (unless the sample capillary was inadvertently broken during handling). The second measurements were performed (on the same sealed sample, but at a different location within the capillary) no earlier than one month later. Repeat experiments (of independently prepared samples), performed for a number of lipid compositions of CMVL5, resulted in scans that matched the original ones.
2.8. Cytotoxicity
Cytotoxicity was measured using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega). Mouse fibroblast L-cells were seeded in 96-well plates at approximately 15 000 cells per well. After 18 h of incubation, the cells were treated with CL–DNA complexes in Opti-MEM using a total of 0.08 μg DNA per well. After 6 h incubation, the CL–DNA complex solution was replaced by a mixture of 100 μl of Opti-MEM and 20 μl of the Cell Proliferation Assay, followed by 2 h of incubation. Cytotoxicity was then determined by colorimetric measurement on a 1420 Multilabel counter Victor 3V (Perkin Elmer) plate reader as per the assay manufacturer's instructions. The experiment was performed simultaneously for all lipids and the results were normalized to control wells, which differed from the experimental wells only in that they were treated with Opti-MEM instead of complexes. All data points were measured as quadruplicates.
2.9. Cell transfection
Mouse fibroblast L-cells were cultured and transfected as previously reported [52]. Briefly, cells in 24-well plates were incubated for 6 h with CL–DNA complexes containing a constant 0.4 μg of plasmid DNA, washed with phosphate-buffered saline, and the complex-containing medium replaced by serum-containing culture medium. For samples containing Lipofectamine 2000, 1 μg reagent per sample was used. Cells were then incubated for 18 h before luciferase expression was measured using the luciferase assay (Promega) as per manufacturer's instructions. Light output readings were measured as relative light units (RLU) on a 1420 Multilabel counter Victor 3V (Perkin Elmer) plate reader. These readings were normalized to the weight of total cellular protein determined using Bio-Rad Protein Assay Dye Reagent. Data points shown in the same plot were measured on the same day, and all data points were measured in duplicate.
2.7. Synthesis
General procedure for the deprotection of Boc-protected lipids
The protected lipid (0.05–0.25 g) was cooled on an ice bath under a nitrogen atmosphere and dissolved with stirring in cooled trifluoroacetic acid (10 mL/g lipid; from Fisher Scientific) that had been saturated with nitrogen gas. The reaction was stirred in a water bath at room temperature for 30 min and all volatiles evaporated in vacuo overnight.
Oleyl alcohol (1)
To a solution of 12.5 g (44.3 mmol) oleic acid (Alfa Aesar) in 150 mL of ether was added with stirring a mixture of 2.15 g (56.7 mmol) lithium aluminum hydride and 30 mL of THF in portions. When refluxing had subsided, the mixture was heated to reflux for 45 minutes. Excess lithium aluminum hydride was hydrolyzed by the addition of wet ether, then water, and finally 2 M hydrochloric acid. The resulting mixture was acidified with conc. hydrochloric acid and the phases separated. The aqueous phase was extracted twice with CH2Cl2, and the combined organic phases were washed twice with water, dried (Na2SO4), and evaporated in vacuo to yield 11.7 g (43.4 mmol, 98%) of oleyl alcohol as a colorless liquid: Rf = 0.23 (cyclohexane/ethylacetate 4:1), identical to a commercial sample.
3,4-Di(oleyloxy)benzoic acid (DOB; 2)
Under cooling with an ice/water bath, 4.97 g (27.3 mmol) 3,4-dihydroxy-benzoic acid ethyl ester (TCI America) was added to a solution of 21.49 g (81.94 mmol) of triphenylphosphine in 246 mL of THF. After stirring for 30 minutes, a total of 37.3 mL of a 40 wt % solution of DEAD (81.9 mmol) and a solution of 22.0 g (81.9 mmol) of 1 in 41 mL of toluene were added dropwise (using two separate addition funnels), maintaining an internal temperature of less than 5 °C. After stirring for 18 h at room temperature, the reaction mixture was heated to 50 °C, and stirring was continued for another 3 hours. The solvents were evaporated and the residue suspended in 44 mL of ether by stirring for 10 min. A total of 22 mL of hexane was added and the mixture was incubated for 15 min with sonication (bath sonicator), filtered, and the residue washed with 110 mL ether/hexane (1:1, v/v). The combined filtrates were evaporated in vacuo, a solution of 7.69 g (137 mmol) of KOH in 210 mL ethanol was added and the mixture was heated to reflux for 4 hours under nitrogen. The hot reaction mixture was added to 260 mL of water and filtered. The residue was washed three times with water and recrystallized from 75 mL of ethanol to yield 9.8 g (14.34 mmol, 52.5%) of DOB [53] as a colorless solid: Rf = 0.26 (cyclohexane/ethylacetate 4:1), NMR spectra identical to a genuine sample [53].
[2-(2-Amino-ethyldisulfanyl)-ethyl]-carbamic acid tert-butyl ester (3) [54]
To a solution of 8.0 g (36 mmol) cystamine bis hydrochloride in 200 mL methanol were added 14.9 mL (107 mmol) of triethylamine and 7.7 g (36 mmol) of di-tert-butyldicarbonate (Fluka). The mixture was stirred for 30 min at room temperature and the solvent evaporated in vacuo. To the residue, 80 mL of 1 M aq NaH2PO4 (pH = 4.2) was added and the mixture was extracted twice with ether to remove di-Boc-cystamine. The aqueous solution was basified (pH = 9) with 1 M NaOH and extracted twice with EtOAc. The combined EtOAc phases were washed twice with water, dried (Na2SO4) and evaporated in vacuo to yield 4.10 g (16.2 mmol, 45% ) of 3 as a white solid: Rf = 0.47 (CHCl3/MeOH 9:1, 1% concentrated NH4OH); NMR spectra matching literature data [54].
[2-(2-{[3,4-Di(oleyloxy)benzoyl]amino}-ethyldisulfanyl)ethyl]-carbamic acid tert-butyl ester (4)
Under a nitrogen atmosphere, 91 mL (5.4 mmol) of N,N-diisopropylethylamine (DIEA) were added with stirring to a mixture of 0.971 g (3.03 mmol) of 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU) and 1.771 g (2.703 mmol) of 2 in CH2Cl2. After 20 minutes, 0.761 g (3.03 mmol) of 3 in CH2Cl2 was added and stirring was continued for 18 h. The reaction mixture was extracted twice with 1 M aq NaH2PO4, twice with water, twice with 1 M aq NaHCO3, and twice more with water, dried (Na2SO4), and evaporated in vacuo to yield 2.02 g (2.27 mmol, 84%) of 4 as an off-white solid: Rf = 0.26 (CHCl3/MeOH 100:1); 1H NMR (CDCl3, 200 MHz): δ = 0.87 (“t”, J = 6.4 Hz, 6 H, CH2CH3), 1.1-2.2 (several m, lg. peaks @ 1.27, 1.31, 1.44, 1.82 (bm), 2.00, 2.02, 65 H, C-CH2-C, C(CH3)3), 2.80 (t, J = 6.5 Hz, 2 H, S-CH2), 2.90 (t, J = 6.0 Hz, 2 H, S-CH2), 3.45, 3.74 (2 “q”, J ≈ 6.1 Hz, 4 H, N-CH2), 4.03 (“t”, J = 5.6 Hz, 4 H, O-CH2), 5.08 (b, 1 H, NH), 5.3-5.6 (m, 4H, =CH), 6.87 (d, 3J = 8.4 Hz, 1 H, Har m-C(O)), 6.97 (b, 1 H, NH), 7.35-7.5 (m, 2 H, Har); 13C NMR (125 MHz, CDCl3): δ = 14.2 (CH2CH3), 22.8, 26.1, 27.3, 29.4, 29.6, 29.9, 32.0 (C-CH2-C), 28.5 ((CH3)3C), 38.4 (b), 39.1, 39.6 (S-CH2, CH2-N), 69.1, 69.3 (CH2-O), 79.4 ((CH3)3C), 112.4, 113.0, 119.8 (CarH), 126.9 (Car-C(O)), 129.9, 130.0 (=CH), 149.1, 152.1 (Car-O), 154.2 (C(O)O), 167.5 (Car-C(O)).
N-[2-(2-Amino-ethyldisulfanyl)-ethyl]-3,4-di(oleyloxy)benzamide trifluoroacetate (5)
As described in the general procedure, 1.9 g (2.1 mmol) of 4 were deprotected to yield 1.76 g (1.95 mmol, 91%) of 5 as an off-white solid: Rf = 0.3 (CHCl3/MeOH 9:1); 1H NMR (200 MHz, CDCl3): δ = 0.91 (“t”, J = 6.4 Hz, 6 H, CH3), 1.0-2.2 (m, lg. peaks at 1.25, 1.80 (b), 2.04, 2.06, 56 H, C-CH2-C), 2.7-3.1 (b, 4 H, S-CH2), 3.2-3.5, 3.5-3.8 (2 b, 4 H, N-CH2), 3.8-4.2 (m, 4 H, O-CH2), 5.0-5.25 (m, 5 H, CH-N), 5.25-5.55 (m, 4 H, =CH), 6.83 (d, 3J = 8.1 Hz, 1 H, Har m-C(O)), 7.2-7.5 (m, 2 H, Har), 8.2 (b, 4 H, NH); 13C NMR (50 MHz, CDCl3): δ = 14.2 (CH2CH3), 22.8, 26.2, 27.4, 29.5, 29.7, 29.9, 32.0 (C-CH2-C), 37.3 (2C), 39.2, 39.6 (S-CH2, CH2-N), 69.3, 69.6 (CH2-O), 112.4, 112.8, 120.7 (CarH), 126.2 (Car-C(O)), 129.9, 130.0 (=CH), 149.1, 152.5 (Car-O), 168.2 (C(O)N).
{(S)-1-[2-(2-{(3,4-Di(oleyloxy)benzoyl)amino}-ethyldisulfanyl)-ethylcarbamoyl]-4-tert-butoxycarbonylamino-butyl}-carbamic acid tert-butyl ester (BocCMVL2)
To a mixture of 0.54 g (1.61 mmol) of Nα,Nδ-Bis(Boc)-ornithine [28] and 0.57 g (1.8 mmol) of TBTU in CH2Cl2 were added 422 μL (2.42 mmol) of DIEA with stirring. After 10 minutes, the resulting solution was added to a mixture of 1.605 g (1.78 mmol) of 5 and 422 μL (2.42 mmol) of DIEA. After stirring for 18 h, the reaction mixture was extracted three times with 1 M aq NaH2PO4, twice with water, twice with 5% NaHCO3 and twice more with water. The organic phase was dried (Na2SO4) and evaporated in vacuo and the residue purified by flash chromatography on 150 g of silica gel using CHCl3/MeOH (100:1, v/v) as the eluent to yield 1.502 g (1.361 mmol, 84%) of BocCMVL2 as a colorless solid: Rf = 0.28 (CHCl3/MeOH 40:1); 1H NMR (200 MHz, CDCl3): δ = 0.81 (“t”, J = 6.3 Hz, 6 H, CH2CH3), 0.94-2.1 (several m, lg. peaks @ 1.20, 1.25, 1.36, 1.75 (b), 1.93, 1.96, 78 H, C-CH2-C, C(CH3)3), 2.7-3.4, 3.4-3.7 (several m, 6+4 H, S-CH2, N-CH2), 3.8-4.1 (m, 4 H, O-CH2), 4.1-4.3, 4.5-4.8 (2 bm, 2 H, NH), 4.8-5.1 (m, 1 H, CH), 5.1-5.4 (m, 4 H, =CH-), 6.79 (d, 3J = 8.4 Hz, 1 H, Har m-C(O)), 6.89, 7.11 (2 bt, 2 H, NH), 7.3-7.5 (m, 2 H, Har); 13C NMR (50 MHz, CDCl3): δ = 14.0 (CH2CH3), 22.5, 25.9, 26.2, 27.1, 29.0, 29.1, 29.2, 29.26, 29.29, 29.4, 29.6, 31.8, 33.6 (C-CH2-C), 28.2, 28.3 ((CH3)3C), 37.6, 37.9, 38.2, 39.1 (S-CH2, CH2-N), 53.2 (CH), 69.0, 69.3 (CH2-O), 79.1, 79.7 (C(CH3)3), 112.2, 112.8, 119.8 (CarH), 126.4 (Car-C(O)), 129.6, 129.8 (=CH), 148.8, 152.0 (Car-O), 155.7, 156.3 (C(O)O), 167.4, 172.6 (C(O)N).
N-(2-((2-((2S)-2,5-Diaminopentanamido)ethyl)disulfanyl)ethyl)-{3,4-di(oleyloxy) benzamide} di-trifluoroacetate (CMVL2)
As described in the general procedure, 50 mg (45 μmol) of BocCMVL2 were deprotected using 800 μL of TFA to yield 50 mg (45 μmol, 98%) of CMVL2 as a colorless solid: Rf = 0.33 (CHCl3/MeOH/NH4OH 12:4:1). 1H NMR (500 MHz, CDCl3/MeOH-d4 9:1): δ = 0.72 (“t”, J = 5.8 Hz, 6 H, CH3), 0.9-2.0 (several m, lg. peaks @ 1.12, 1.17, 1.32 (b), 1.66, 1.87, 60 H, C-CH2-C), 2.65-2.85 (2 m, 6 H, S-CH2, CH2-N), 3.35-3.6, (2 m, 4H, CH2-N), 3.7-3.85 (m, 1 H, CH), 3.85-3.95 (m, 4 H, O-CH2), 5.1-5.3 (m, 4 H, =CH), 6.74 (d, 1 H, 3J = 8.3 Hz, Har m-C(O)), 7.15-7.3 (m, 2 H, Har); 13C NMR (125 MHz, CDCl3/MeOH-d4 9:1): δ = 13.8 (CH3), 22.5, 25.89, 25.93, 27.0, 28.0, 29.02, 29.12, 29.2, 29.26, 29.29, 29.7, 31.8 (C-CH2-C), 37.1, 37.2, 38.3, 38.5, 39.1 (S-CH2, N-CH2), 52.3 (N-CH), 69.0, 69.5 (CH2-O), 112.3, 112.9, 120.4 (Car-H), 116.3 (q, J = 299 Hz , F3C), 126.2 (Car-CO), 129.7, 129.8 (=CH-CH2), 148.6, 152.2 (Car-O), 161.6 (q, J = 35.5 Hz , F3C-C), 168.5, 168.6(C(O)-N).
{(S)-1-[2-(2-{(3,4-Di(oleyloxy)benzoyl)amino}-ethyldisulfanyl)-ethylcarbamoyl]-4-tert-butoxycarbonylamino-butyl}-(3-tert-butoxycarbonylamino-propyl)-carbamic acid tert-butyl ester (BocCMVL3)
To a mixture of 0.38 g (0.78 mmol) of (S)-2-(tert-butoxycarbonyl(3-(tert-butoxycarbonylamino)propyl)amino)-5-(tertbutoxycarbonylamino)pentanoic acid and 0.27 g (0.85 mmol) of TBTU in CH2Cl2 were added 203 μL (1.16 mmol) of DIEA with stirring. After 10 minutes, the resulting solution was added to a mixture of 0.77 g (0.85 mmol) of 5 and 203 μL (1.16 mmol) of DIEA in CH2Cl2 and stirring was continued for 18 h. The reaction mixture was extracted twice with 1 M aq NaH2PO4, once with water, twice with 5% NaHCO3, and once more with water. The organic phase was dried (Na2SO4), evaporated in vacuo and the residue purified by flash chromatography on 85 g of silica gel using CHCl3/MeOH (100:1) as the eluent to yield to yield 0.747 g (0.580 mmol, 72%) of BocCMVL3 as a colorless solid: Rf = 0.41 (CHCl3/MeOH 20:1); 1H NMR (500 MHz, CDCl3/MeOH-d4 9:1) δ =0.72 (“t”, J = 6.0 Hz, 6 H, CH2CH3), 0.9-2.0 (several m, 89 H, lg. peaks @ 1.12, 1.17, 1.27, 1.31, 1.53 (b), 1.68 (b), 1.87, C-CH2-C, C(CH3)3), 2.6-2.7 (2 H), 2.7-2.85 (2 H), 2.85-3.0 (4 H), 3.0-3.15 (2 H) (4 bm, S-CH2, N-CH2), 3.35-3.45 (bm, 2 H), 3.5-3.6 (bm, 2 H) (CH2-N), 3.8-3.95 (m, 4 H, O-CH2), 4.23 (b, 1 H, CH), 5.3-5.7 (m, 4 H, CH), 5.4 (b, 1 H, NH), 6.73 (“d”, 3J = 8.2 Hz, 1 H, Har m-C(O)), 7.2-7.3 (m, 2 H, CarH), 7.53 (b, 1 H, NH); 13C NMR (125 MHz, CDCl3/MeOH-d4 9:1):δ = 13.9 (CH2CH3), 22.5, 25.90, 25.94, 27.1, 29.1, 29.19, 29.23, 29.30, 29.34, 29.4, 29.5, 29.6, 31.8 (C-CH2-C), 28.09, 28.12 ((CH3)3C), 37.2, 37.6, 38.0 (b), 38.1 (b), 39.0, 39.7 (CH2-N, S-CH2), 58.5 (CH-N), 69.1, 69.4 (CH2-O), 79.1, 81.1 ((CH3)3C), 112.5, 113.0, 120.3 (CarH), 126.4 (Car-CO), 129.8, 129.9 (=CH2), 148.7, 152.0 (Car-O), 156.60, 156.64 (C(O)O), 168.0, 171.9 (C(O)-N).
N-(2-((2-(5-Amino-(2S)-2-(3-aminopropylamino)pentanamido)ethyl)disulfanyl) ethyl)- {3,4-di(oleyloxy)benzamide} tri-trifluoroacetate (CMVL3)
As described in the general procedure, 225 mg (175 μmol) of BocCMVL3 were deprotected to yield220 mg (165 μmol, 94%) of CMVL3 as a colorless solid: Rf = 0.46 (CHCl3/MeOH/NH4OH 6:3:1); 1H NMR (500 MHz, CDCl3/MeOH-d4 9:1): δ = 0.77 (“t”, 6 H, J = 6.8 Hz, CH2CH3), 1.0-2.1 (several m, 62 H, lg. peaks at 1.16, 1.21, 1.35 (m), 1.67 (m), 1.86 (m), C-CH2-C), 2.76 (t, J = 6.3 Hz), 2.81 (t, J = 6.8 Hz), 2.85 (t, J = 7.0 Hz), 2.87-3.1 (2m), (12 H, S-CH2, N-CH2), 3.6-3.75 (2 m, 4 H, CH2-N), 3.8-4.0 (m, 4 H, O-CH2), 4.85-5.15 (m, 1 H, CH), 5.15-5.3 (m, 4H, =CH), 6.78 (d, 3J = 8.5 Hz, 1 H, Har m-C(O)), 7.2-7.3 (m, 2 H, Har ); 13C NMR (125 MHz, CDCl3/MeOH-d4 9:1): δ =13.9 (CH3), 22.4, 22.6, 24.9, 26.0, 27.0, 27.1, 27.2, 29.1, 29.2, 29.25, 29.29, 29.4, 29.5, 30.7, 31.7, 31.8, 31.9, 33.6 (C-CH2-C), 36.5, 37.23, 37.27, 38.4, 38.6, 39.0, 43.4 (S-CH2, CH2-N), 59.6 (CH-N), 69.1, 69.5 (CH2-O), 112.4, 113.0, 120.4 (CarH), 126.3 (Car-CO), 129.8, 129.9 (=CH), 148.7, 152.3 (Car-O), 167.3, 168.3 (C(O)N).
{(S)-1-[2-(2-{(3,4-Di(oleyloxy)benzoyl)amino}-ethyldisulfanyl)-ethylcarbamoyl]-4-[tert-butoxycarbonyl-(3-tert-butoxycarbonylamino-propyl)-amino]-butyl}-(3-tert-butoxycarbonylamino-propyl)-carbamic acid tert-butyl ester(BocCMVL4)
To a mixture of 0.46 g (0.71 mmol) of Nα,Nδ-Bis(Boc)-Nα,Nδ-bis(3-[Boc-amino]propyl)ornithine [52] and 0.25 g (0.78 mmol) TBTU in CH2Cl2 were added 184 μL (1.06 mmol) of DIEA with stirring. After 10 minutes, the resulting solution was added to a mixture of 0.70 g (0.78 mmol) of 5 and 184 μL (1.06 mmol) of DIEA in CH2Cl2 and stirring was continued for 18 h. The reaction mixture was extracted twice with 1 M aq NaH2PO4, once with water, twice with 5% NaHCO3, and once more with water. The organic phase was dried (Na2SO4), evaporated in vacuo and the residue purified by flash chromatography on 85 g of silica gel using CHCl3/MeOH (100:1) as the eluent to yield 0.720 g (0.507 mmol, 70%) of BocCMVL4 as a colorless solid: Rf = 0.26 (CHCl3/MeOH 40:1); 1H NMR (500 MHz, MeOH-d4): δ = 0.89 (“t”, 6 H, J = 6.4 Hz, CH2-CH3), 1.1-2.2 (several m, lg. peaks @ 1.28, 1.34, 1.40, 1.43, 1.45, 1.47, 1.69 (b), 1.79 (b), 2.03 (b), 100 H, C-CH2-C, C(CH3)3), 2.86 (t, J = 6.0 Hz, 2H), 2.95 (t, J = 6.4 Hz, 2H), 3.03 (t, J = 6.0 Hz, 4 H), 3.1-3.4 (m, 6 H + MeOH-d3), 3.52 (t, J = 6.0 Hz, 2H), 3.67 (t, J = 6.6 Hz, 2 H) (S-CH2, CH2-N), 4.0-4.1 (m, 4 H, O-CH2), 4.52 (b, 1 H, CH), 5.3-5.45 (m, 4 H, =CH), 6.94 (d, 3J = 8.8 Hz, 1 H, Har m-C(O)), 7.45 (b, 2 H, Har); 13C NMR (125 MHz, MeOH-d4): δ = 14.8 (CH2CH3), 23.9, 27.5, 28.4, 30.5, 30.6, 30.66, 30.73, 30.8, 30.9, 31.1, 33.3 (C-CH2-C), 29.0, 29.08, 29.12 ((CH3)3CO), 38.6, 38.8, 39.0 (b), 39.3 (b), 39.7 (b), 40.6 (S-CH2, CH2-N), 70.3, 70.7 (C-CH2-O), 80.1 (b), 81.2, 82.0 (b) ((CH3)3C), 113.9, 114.5, 122.2 (CarH), 127.9 (Car-C(O)), 131.0, 131.1 (=CH), 150.3, 153.8 (Car-O), 157.5, 158.5 (2 b, C(O)O), 169.8, 173.7 (b) (C(O)N).
N-(2-(((2S)-2-(2,5-Bis(3-aminopropylamino)pentanamido)ethyl)disulfanyl) ethyl)- {3,4-di(oleyloxy)benzamide} tetra-trifluoroacetate (CMVL4)
As described in the general procedure, 232 mg (164 μmol) of BocCMVL4 were deprotected to yield 238 mg 161 μmol, 98%) of CMVL4 as a colorless solid: Rf = 0.41 (CHCl3/MeOH/25% NH4OH 6:2:1). 1H NMR (500 MHz, MeOH-d4): δ = 0.81 (“t”, 6 H, J = 7 Hz, CH2CH3), 1.1-2.1 (several m, 64 H, lg. peaks at 1.21, 1.27, 1.37, 1.44 (m), 1.59 (b), 1.73 (m), 1.95, 2.00 (m) C-CH2-C), 2.78-2.92 (m, 4 H, S-CH2), 2.92-3.15 (m, 10 H, N-CH2), 3.42-3.68 (m, 4 H, HN-CH2), 3.87 (t, 1 H, J = 6.3, +H2N-CH), 3.9-4.02 (m, 4 H, O-CH2), 5.2-5.35 (m, 4 H, =CH), 6.90 (d, 3J = 8.1 Hz, 1 H, Har m-C(O)), 7.4-7.5 (m, 2 H, Har), 8.0 (b, NH); 13C NMR (125 MHz, MeOH-d4): δ = 14.6 (CH3), 22.9, 23.9, 25.48, 25.53, 26.3, 27.5, 28.3, 28.7, 30.7, 30.78, 30.82, 30.9, 30.94, 30.97, 31.0, 31.04, 33.2, 34.9 (C-CH2-C), 38.0, 38.52, 38.55, 39.69, 40.54, 45.1, 46.0, 48.3 (CH2-N, S-CH2), 61.5 (CH), 70.3, 70.8 (CH2-O), 113.9, 114.4, 122.3 (CarH), 118.0 (q, J = 290 Hz, CF), 127.8 (Car-C(O)), 130.9, 131.0 (=CH), 150.3, 154.0 (Car-O), 162.6 (q, J = 34.2 Hz, F3C-C(O)), 168.6, 170.1 (C(O)).
{4-[Bis-(3-tert-butoxycarbonylamino-propyl)amino](S)-1-[2-(2-{(3,4-di(oleyloxy)benzoyl)amino}-ethyldisulfanyl)-ethylcarbamoyl]-butyl}-(3-tert-butoxycarbonylamino-propyl)-carbamic acid tert-butyl ester (BocCMVL5)
To a mixture of 575 mg (817 μmol) of Nα-Boc-Nα,Nδ,Nδ-tris(3-[Boc-amino]propyl)ornithine [26] and 289 mg (899 μmol) of TBTU in CH2Cl2 were added 116 μL (899 μmol) of DIEA with stirring. After 10 minutes, the resulting solution was added to a mixture of 812 mg (899 μmol) of 5 and 116 μL (899 μmol) of DIEA in CH2Cl2 and stirring was continued for 18 h. The reaction mixture was extracted twice with 1 M aq NaH2PO4, once with water, twice with 5% NaHCO3, and once more with water. The organic phase was dried (Na2SO4), evaporated in vacuo and the residue purified by flash chromatography on 120 g of silica gel using CHCl3/MeOH (100:1) as the eluent to yield 643 mg (436 μmol, 52%) of BocCMVL5 as a colorless solid: Rf = 0.32 (CHCl3/MeOH 9:1); 1H NMR (200 MHz, CDCl3) δ = 0.75 (“t”, J = 6.4 Hz, 6 H, CH2-CH3), 0.9-2.3 (several m, lg. peaks @ 1.26, 1.31, 1.43, 1.47; 102 H, C-CH2-C, C(CH3)3), 2.46 (b, 6 H), 2.82 (t, 2 H), 2.93 (t, 2 H), 3.0-3.35 (m, 8 H) 3.45-3.85 (2 “q”, 4 H) (S-CH2, N-CH2), 3.9-4.15 (m, 4 H, O-CH2), 4.37 (b, 1 H), 4.95 (b, 1 H), 5.1-5.5 (m, 5 H) (NH, N-CH, =CH), 6.84 (d, 3J = 8.4 Hz, 1 H, Har m-C(O)), 7.00 (b, 1 H, NH), 7.34 (dd, 3J = 8.4 Hz, 4J = 1.9 Hz, 1 H, Har p-OR), 7.44 (d, 4J = 1.9 Hz, 1 H, Har o-C(O), o-OR); 13C NMR (125 MHz, CDCl3): δ = 14.6 (CH2CH3), 22.8, 26.2, 27.4, 29.3, 29.5, 29.6, 29.7, 30.0, 32.1 (C-CH2-C), 28.7 ((CH3)3C), 37.7, 38.1, 38.4, 39.1, 51.8, 53.4 (S-CH2, CH2-N), 53.4 (CH-N), 69.3, 69.5 (C-CH2-O), 79.2 (b), 81.2 ((CH3)3C), 112.5, 113.2, 119.9 (CarH), 126.9 (Car-C(O)), 130.0, 130.1 (=CH), 149.1, 152.2 (Car-O), 156.28, 156.31 (C(O)O), 167.5, 171.9 (C(O)N).
N-(2-((2-((2S)-2-(3-Aminopropylamino)-5-(bis(3-aminopropyl)amino) pentanamido)ethyl)disulfanyl)ethyl)- {3,4-di(oleyloxy)benzamide} penta-trifluoroacetate (CMVL5)
As described in the general procedure, 63 mg (43 μmol) of BocCMVL5 were deprotected using 2 mL of TFA. The obtained residue was dissolved in 100 μL methanol, 20 mL of diethylether/pentane (1:1, v/v) were added and the mixture was incubated for 16 h at 4 °C. The resulting precipitate was isolated by decanting, washed twice with 5 mL diethylether/pentane (1:1, v/v) and dried to yield 67 mg (41 μmol, 95 %) of CMVL5 as a colorless solid: Rf = 0.32 (CHCl3/MeOH/25% NH4OH 40:24:10); 1H NMR (500 MHz, MeOH-d4): δ = 0.82 (b, 6 H, CH2CH3), 1.01-2.49 (several m, 66 H, lg. peaks at 1.22, 1.28, 1.45, 1.60, 1.74, 1.86, 1.97, 2.09 C-CH2-C), 2.75-3.35 (22 H, S-CH2, N-CH2), 3.42-3.72 (m, 4 H, O-CH2), 3.8-4.1 (b, 5 H, NH, CH), 4.92-5.2 (b, 4 H, =CH), 6.9 (d, 3J = 5 Hz, 1 H, Har m-C(O)), 7.2-7.6 (m, 2 H, Har); 13C NMR (125 MHz, MeOH-d4 9): δ = 14.6 (CH3), 20.7, 23.3, 23.9, 25.5, 26.2, 26.3, 27.5, 28.3, 28.7, 30.47, 30.52, 3.57, 30.6, 30.7, 30.8, 31.0, 31.05 (C-CH2-C), 33.2, 33.8, 34.9, 38.0, 38.5, 39.4, 40.5, 45.2, 51.4, 53.7 (S-CH2, CH2-N), 61.5 (CH), 70.3, 70.8 (CH2-O), 113.9, 114.4, 122.3 (CarH), 127.8 (Car-C(O)), 131.0, 131.04 (=CH), 150.3, 154.0 (Car-O-CH2), 163.0 (q, J = 34.8 Hz, F3C-C(O)), 168.7, 170.1 (C(O)N).
3. Results and discussion
3.1. Lipid design and synthesis
The CMVLn (n = 2 to 5) lipids presented in this study were designed to be stable in the extracellular environment but readily degrade in the reducing environment of the cytoplasm. We anticipated that this would reduce lipid toxicity and enhance nucleic acid release from their DNA complexes. The former is of practical importance (in particular for siRNA delivery [2] while the latter should shed light on the delivery pathway of multivalent lipids, particularly the limitations observed at high membrane charge density (σM) [32]. As evident from their chemical structures shown in Table 1, the CMVLs are closely related to the MVL series of lipids previously synthesized in our group [26, 32, 55]: MVLs and CMVLs differ only in the spacer connecting lipid tails and headgroup, which is slightly longer and incorporates a reductively degradable disulfide bond in the CMVLs. The synthesis of the CMVLs made use of the headgroup building blocks developed for the synthesis of the MVLs [26, 28] as well as the same hydrophobic moiety (3,4-di(oleoyloxy)benzoic acid). However, a new synthesis of DOB was devised to circumvent the use of expensive oleyl bromide as the starting material. Thus, oleyl alcohol was synthesized by reduction of oleic acid and used to alkylate ethyl(3,4-(dihydroxy))benzoate in a Mitsunobu reaction [56, 57] to yield DOB (2) after saponification (cf. Figure 1). The synthesis proceeded by coupling DOB with mono-Boc-protected cystamine 3 [54] and Boc-deprotection to yield 5, the common precursor for the CMVLs. As exemplified for CMVL4 in Figure 1, precursor 5 was coupled with the Boc-protected headgroup building blocks, and the resulting Boc-protected CMVLs were purified extensively and finally deprotected with TFA.
Table 1.
Chemical Structures, Abbreviated Names, and Headgroup Charges of the Synthesized Degradable Cationic Lipids.
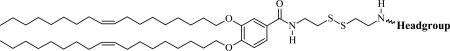
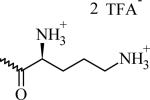

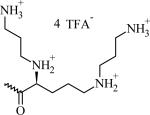
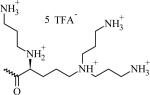
At full protonation.
Figure 1.

Synthesis of reductively degradable CMVL4, starting from oleic acid. Reaction conditions: (a) LiAlH4; (b) 1. PPh3, DEAD 2. KOH; (c) Boc2O, Et3N; (d) TBTU, DIEA; (e) TFA; (f) See ref [28]. DEAD: diethylazodicarboxylate; DIEA: N,N-diisopropylethylamine; TBTU: O-benzotriazol-1-yl-N,N,N′,N′-tetramethyluronium tetrafluoroborate; TFA: trifluoroacetic acid.
CL–DNA complexes were prepared from mixtures of the CMVLs and DOPC at varied mole fractions of CMVL (ΦCMVL = 1 – ΦDOPC) and using varying amounts of DNA. The complexes were characterized using small-angle X-ray scattering, optical microscopy, and dynamic light scattering, and their cytotoxicity and transfection efficiency were assessed and compared to both MVL5 (a highly efficient, non-degradable analog of CMVL5 [26, 27, 32] and commercially available transfection agents (DOTAP and Lipofectamine 2000). The chemical structures of MVL5, DOTAP and DOPC are shown in the Supporting Information, Figure S1.
3.2. Determination of the headgroup charge
An EtBr displacement assay [58, 59] was used to estimate the head group charge of the cationic lipids as reported previously [32]. EtBr fluoresces when intercalated into DNA but self-quenches in solution. Therefore, the fluorescence of a mixture of DNA and EtBr decreases when cationic lipids complex the DNA and displace the intercalated EtBr. When EtBr displacement is complete, the fluorescence reaches a baseline. The isoelectric point, where the positive charges on the lipids match the negative charges on the DNA, was determined as the onset of the baseline value as described [32]. Figure 2 displays the results of the EtBr displacement assay for the CMVLs, plotting relative fluorescence against the weight ratio of CMVL to DNA. Dashed vertical lines mark the isoelectric point for each lipid. The resulting headgroup charges are Z = 1.4 for CMVL2, Z = 1.9 for CMVL3, Z = 2.4 for CMVL4, and Z = 3.1 for CMVL5, with an estimated accuracy of ± 0.1.
Figure 2.

Data from the EtBr displacement assay used to measure the charge of CMVLs in CL–DNA complexes, obtained at ΦCMVL = 1. The dashed lines indicate the location of the isoelectric point, which was determined as described previously [32]. The resulting headgroup charges are Z = 1.4 for CMVL2, Z = 1.9 for CMVL3, Z = 2.4 for CMVL4, and Z = 3.1 for CMVL5, with an estimated accuracy of ± 0.1. Error bars show the standard deviation of duplicate measurements.
3.3. Characterization of CL–DNA complexes by dynamic light scattering and optical microscopy
To unambiguously demonstrate that the lipids and DNA form complexes and to further characterize these complexes, we performed optical microscopy and dynamic light scattering (DLS) experiments. Figure 3 shows optical micrographs for CMVL5/DOPC–DNA complexes with ΦDOPC = 0.6 and L/D = 10 (the optimal composition for transfection, see below). The differential interference contrast image (showing all observable particles) and the two fluorescence mode images (showing lipid and DNA, respectively) fully overlap. This is demonstrated more quantitatively by the intensity profiles obtained from the fluorescence mode images. Thus, optical microscopy proves that all observed particles contain both lipid and DNA.
Figure 3.

Optical micrographs of CMVL5/DOPC–DNA complexes at ΦCMVL5 = 0.6 and L/D = 10. (a) Differential interference contrast image. (b,c) Fluorescence mode images showing lipid and DNA label, respectively. Scale bar, 10 μm (valid for all three micrographs). (d, e) Intensity profiles along the dotted lines in the fluorescence mode images of parts b and c. Colocalization of the lipid and DNA fluorescence in all observed particles unambiguously demonstrates complex formation.
Figure 4 shows the results of DLS measurements of DNA complexes of the CMVLs at the L/D optimal for transfection (see below), corresponding to L/D = 10.0 for CMVL5, L/D = 10.7 for CMVL4, L/D = 20.2 for CMVL3, and L/D = 13.7 for CMVL2. For all complexes, the size increases with the mole fraction of CMVL. The measured hydrodynamic diameter is between 100 and 200 nm for most of the samples, and only high mole fractions of CMVL2 yield a diameter larger than 300 nm. The size of complexes at a given mole fraction of CMVL shrinks with headgroup valency for CMVL5 to CMVL3, but CMVL2 deviates from this trend, forming the largest complexes by far, in particular at high mole fractions of CMVL2.
Figure 4.

Hydrodynamic diameter (z-average) of CMVL/DOPC–DNA complexes at ΦCMVL = 0.6 in Opti-MEM 20 minutes after complex formation. The samples were prepared at values of L/D optimal for transfection (L/D = 10.0 for CMVL5, L/D = 10.7 for CMVL4, L/D = 20.2 for CMVL3, and L/D = 13.7 for CMVL2, see below). Data points shown are the average of duplicate measurements of the same sample, with error bars showing the standard deviation.
3.4. Small-angle X-ray scattering
We used SAXS to probe the structures of CMVL/DOPC–DNA complexes in the absence and presence of reducing agents. No signs of lipid phase separation could be detected; hence the complexes contained both CMVL and DOPC at the nominal ratios. Figure 5 shows SAXS scans obtained for CMVL2 and CMVL5 at ΦCMVL = 0.6 with and without reducing agents (DTT and GSH). CMVL/DOPC–DNA complexes prepared in non-reducing environments are highly stable as judged by SAXS scans. As shown in the Supplementary Material, the lamellar structure shows no signs of disintegration after 6 months and (for CMVL5, the only lipid investigated over such a long time period) predominates even after 41 months. The scattering patterns of these complexes exhibit a series of sharp peaks that are labeled q001, q002, q003, q004. These are indicative of the lamellar structure, , with a repeat distance d = 2π/q001. The peaks at qDNA (marked by arrows) reflect the one-dimensional ordering of the DNA molecules within the lamellar complex and correspond to an interhelical spacing dDNA = 2π/qDNA. The experimental values obtained in non-reducing media are d = 71.7 Å and dDNA = 30.3 Å for CMVL2, and d = 74.8 Å and dDNA = 27.3 Å for CMVL5. Complexes containing MVL5 (the non-degradable analog of CMVL5) also are lamellar and their structure is unaffected by the presence of a strong reducing agent such as DTT. This is demonstrated in Figure 5b which shows the SAXS pattern obtained for MVL5/DOPC–DNA complexes at ΦMVL5 = 0.6 after addition of DTT. The characteristic pattern of complexes is preserved, with d = 71.3 Å and dDNA = 27.3 Å. In contrast, addition of reducing agents drastically changes the structure of CMVL/DOPC–DNA complexes. As evident from the scans shown at the bottom of Figure 5, the characteristic peaks of the lamellar structure disappear after treatment with DTT or GSH. In the case of CMVL2, only a single broad feature remains: a broad peak at q ≈ 0.143 Å-1 for DTT-treated complexes, and a less symmetric, broad feature at about the same position for GSH-treated complexes. In the case of CMVL5, similar broad features are seen, albeit at a lower q ≈ 0.135 Å-1. In addition, a sharp peak at q = 0.231 Å-1 appears for complexes treated with DTT, while a broader peak at higher q is observed after treatment with GSH.
Figure 5.

SAXS data of CL–DNA complexes before and after addition of reducing agents. (a) Data for CMVL2/DOPC–DNA complexes at ΦCMVL2 = 0.6 as prepared (blue). (b) Data for CMVL5/DOPC–DNA complexes at ΦCMVL5 = 0.6 as prepared (blue) and after addition of DTT (red) or GSH (green). Also shown is data for MVL5/DOPC–DNA complexes at ΦCMVL5 = 0.6 after addition of DTT (brown).
As confirmed by TLC analysis (see Supplementary Material), reducing agents such as GSH and DTT are able to reduce (break) the disulfide spacer between the positively charged headgroup and the hydrophobic tail of the CMVLs. The SAXS data shows that the CMVLs are also reduced when assembled into CL–DNA complexes, as indicated by the disappearance of the lamellar structure (which remains intact under the same conditions for non-degradable MVL5). The products of the reductive cleavage of CMVL/DOPC–DNA complexes are DNA, multivalent ions (the former lipid headgroup), the lipid tail and DOPC. Theoretical as well as experimental studies have shown that multivalent ions are able to condense DNA into bundled aggregates if their valency, Z, is at least 3 [60-64]. Taken together with the EtBr assay data, this means that only the cleaved headgroup of CMVL5 is able to condense the DNA. This permits assignment of the additional peak observed after reductive treatment. The position of this peak is consistent with the formation of hexagonal DNA bundles, as seen with, e.g., spermine, spermidine, and small poly(propyleneimine) dendrimers [65]. For the DTT-treated CMVL5/DOPC–DNA complexes, the observed peak corresponds to an interhelical distance of dDNA = 4π/(3)1/2q = 31.2 Å in the bundles. A detailed, quantitative SAXS study of all CMVLs and their DNA complexes is in progress, and will be reported in due course.
3.5. Cytotoxicity
Cytotoxicity was measured by assessing cell viability with a tetrazolium salt-based assay [66]. Figure 6a shows the resulting data for DNA complexes (at ΦDOPC = 0) of CMVLn (n = 2 to 5), MVL5, DOTAP, and Lipofectamine 2000 as a function of lipid/DNA weight ratio (to accommodate Lipofectamine 2000). Up to a cationic lipid/DNA weight ratio (L/D) of 18, none of the reagents exhibits appreciable cytotoxicity, with CMVL4 and CMVL5 affecting cell viability the least. Beyond this value, however, toxicity increases strongly for DOTAP and Lipofectamine 2000, and somewhat less strongly for CMVL2 and MVL5. In contrast, cell viability stays near 90% up to L/D = 28 for CMVL3 and L/D = 36 for CMVL4 and CMVL5. Thus, with the exception of CMVL2, the CMVLs showed a remarkable decrease in cytotoxicity over important commercially available reagents as well as MVL5, the CMVL5 analog without a reductively cleavable disulfide bond.
Figure 6.

(a) Cytotoxicity (in mouse L-cells) of CMVL–DNA complexes and other vectors at ΦDOPC = 0. Cell viability is plotted as a function of lipid/DNA weight ratio (L/D). (b) Cytotoxicity of CMVL–DNA complexes and other vectors for optimal or near-optimal (for CMVL2 and CMVL3) formulation (ΦDOPC = 0.4 for MVL5 and the CMVLs; ΦDOPC = 0 for DOTAP and Lipofectamine 2000). Cell viability is plotted as a function of total lipid/DNA weight ratio (TL/D; for DOTAP and Lipofectamine 2000, L/D = TL/D). Note the change in scale for the x-axis. For both plots, error bars show the standard deviation of quadruplicate measurements.
Comparing optimal or near-optimal (for CMVL2 and CMVL3) formulations, Figure 6b shows cytotoxicity data taken at ΦDOPC = 0.4 for the CMVLs and MVL5 in comparison with the data for DOTAP and Lipofectamine 2000 from Figure 6a. The data is plotted as a function of total lipid/DNA ratio (TL/D; for DOTAP and Lipofectamine 2000, L/D = TL/D). The addition of neutral lipid extends the onset of toxicity as a function of TL/D. This effect is strongest for CMVL2, which behaves very similarly to the other CMVLs at this ΦDOPC. For MVL5, toxicity increases sharply for TL/D > 33; for the CMVLs, toxicity increases less sharply and at much higher TL/D. At very high TL/D values, the toxicity of the CMVLn lipids decreases with increasing n.
The comparison of MVL5 with CMVL5 is the most relevant in terms of deducing structure–toxicity relationships, because the only difference between those lipids is in the spacer, which in the case of CMVL5 provides an established means of cleavage in the cytoplasm. In addition, the fact that both CMVL3 and CMVL4 show a toxicity profile similar to that of CMVL5 for both ΦDOPC investigated suggests that for these three lipids, the headgroup size and charge play only a minor role in determining the toxicity. On the other hand, CMVL2–DNA complexes (at ΦDOPC = 0) are unexpectedly toxic. The source of this high toxicity is not clear, and a subject of ongoing investigations. We have found that similar divalent lipids (MVL2 and a lipid with a slightly different, longer spacer) are also unexpectedly toxic (MVL2 in siRNA delivery) or unexpectedly inefficient DNA vectors. Thus, we believe that the deviating behavior of CMVL2 is not related to an effect of the spacer and rather due to effects specific to divalent (or possibly ornithine-based) lipids. Of note, CMVL2 forms comparatively large DNA complexes (cf. DLS results in Figure 4), in particular at ΦDOPC = 0 where it is most toxic. Summarizing the cytotoxicity results, the design of the CMVLs, with the disulfide-spacer that connects headgroup and lipid tails acting as a built-in weak link that can be reductively cleaved in the cytoplasm, appears to be successful in reducing lipid and CL–DNA complex cytotoxicity. Of note, MVL5 has previously been found to be a much better vector for gene silencing using siRNA than DOTAP, in part due to the lower toxicity of MVL5 at increased lipid/DNA charge ratios. The cleavable disulfide linker of CMVL5 reduced this toxicity even further. Studies of the CMVLs as siRNA vectors are currently ongoing.
3.6. Cell transfection
Transfection efficiency of CL–DNA complexes at varied L/D and ΦDOPC was measured using a luciferase assay. Figure 7 shows the resulting data for complexes of CMVLn (n = 2 to 5) and MVL5 at ΦDOPC = 0.4 as a function of L/D. For the multivalent lipids, ΦDOPC = 0.4 was chosen because it yielded optimal or near-optimal TE for the MVLn lipid series (n = 2, 3, 5) [32]. Lipofectamine 2000 was used as per manufacturer's instructions and its TE as well as that of optimized DOTAP/DOPC complexes is plotted for comparison. As observed with other lipids [26, 28, 52, 67], the TE of complexes of the CMVLs and MVL5 initially increases with L/D, then reaches a plateau, and (in the case of CMVL2 and CMVL5) finally drops again for even higher L/D. The plateau of optimal TE spans L/D ≈ 9–20 for CMVL2, L/D ≈ 18–24 for CMVL3, L/D ≈ 10–21 for CMVL4, L/D ≈ 7–13 for CMVL5, and L/D = 7–15 for MVL5. The lipids with higher valency (CMVL4, CMVL5, MVL5) reach a maximum TE that is very high and comparable to that of Lipofectamine 2000. The optimal TE for CMVL2 is only slightly lower than that for CMVL3, which in turn is about an order of magnitude lower than that of the high-valency CMVLs.
Figure 7.

Transfection efficiency of CMVL-based CL–DNA complexes compared with non-degradable lipid MVL5 and commercial vectors. The amount of DNA was constant for all data points. (top) TE as a function of L/D at a constant ΦDOPC = 0.4. TE for DOTAP and Lipofectamine 2000 is for optimized formulations. (bottom) TE at optimal L/D as a function of mole fraction of cationic lipid. Specifically, data was taken at L/D = 10.0 for CMVL5, L/D = 10.7 for CMVL4, L/D = 13.7 for CMVL2, and L/D = 7.4 for MVL5 (each approximately corresponding to a lipid/DNA charge ratio of 6) as well as L/D = 20.2 for CMVL3 and L/D = 5.9 for DOTAP. For both plots, error bars show the standard deviation of duplicate measurements.
To investigate the effect of membrane charge density, we varied ΦDOPC at an optimal L/D. We chose L/D = 10.0 for CMVL5, L/D = 10.7 for CMVL4, L/D = 13.7 for CMVL2, and L/D = 7.4 for MVL5 (each approximately corresponding to a lipid/DNA charge ratio of 6) as well as L/D = 20.2 for CMVL3 and L/D = 5.9 for DOTAP. The results for TE at the optimal L/D as a function of mole fraction of cationic lipid are shown in Figure 7b. As observed previously for DOTAP and the MVL series [32], TE initially increases with increasing mole fraction of cationic lipid to a maximum. For DOTAP, this maximum is at ΦDOTAP = 0.8 to 1. For the multivalent lipids, the maximum TE is reached at a lower Φ(C)MVL of 0.6 for CMVL4 and CMVL5 and of 0.8 for CMVL2 and CMVL3; TE then drops again. At the optimal composition, TE for complexes prepared from CMVL2 and CMVL3 is slightly closer to the TE achievable with CMVL4 and CMVL5 and exceeds the TE for optimized DOTAP complexes. Thus, as observed for the MVL series [32], the TE of CMVL/DOPC–DNA complexes exhibits three regimes as a function of increasing ΦCMVL: initial increase, optimum TE, and a decrease at very high σM. Regarding the transfection mechanism, this suggests that lipid degradability (and, judging from the SAXS and cytotoxicity results, most likely intracellular degradation) does not affect the transfection mechanism. It appears that release of DNA from the lipids is not the factor limiting TE, since the drop in TE is not only observed for CMVL5 (for which condensation by the cleaved headgroup would still limit DNA release) but also for the other, less highly charged CMVLs.
In the light of their reduced cytotoxicity, the finding that the CMVLs with higher valency transfect cells as efficiently as non-degradable MVL5 and benchmark commercial lipids represents a major success. It suggests that the toxicity of a given lipid can be effectively reduced without negative impact on its efficiency by inserting a disulfide spacer between the lipid tails and headgroup.
4. Conclusions
We have presented the efficient synthesis of a series of new degradable multivalent cationic lipids, termed CMVLn (n = 2 to 5), which contain a disulfide-bond spacer between headgroup and the hydrophobic moiety. This spacer is cleaved by reducing agents such as the biologically relevant reducing peptide glutathione or DTT. SAXS demonstrates that that the initially formed lamellar phase () of CMVLn–DNA complexes completely disappears upon reductive cleavage of the CMVLs. Importantly, while the CMVLs (n = 3 to 5) transfect mammalian cells with efficiencies comparable to highly efficient non-degradable analogues as well as benchmark commercial reagents such as Lipofectamine 2000, they do so at significantly reduced cytotoxicity. DNA condensation by the cleaved head group—observed for CMVL5, but not CMVL2—does not reduce the transfection efficiency. This demonstrates that triggered disintegration of degradable disulfide spacers may be used to reduce the cytotoxicity of synthetic nonviral vectors without compromising their transfection efficiency. The fact that transfection efficiency remains high also shows that the key steps of lipid-mediated DNA delivery prior to cytoplasmic release—DNA compaction, complex uptake and endosomal escape [32, 33]—are not compromised by the lipid degradation process.
Supplementary Material
Acknowledgement
This work was supported by NIH GM-59288. Cecília Leal was funded by the Swedish Research Council (VR) and in part by DOE BES DE-FG-02-06ER46314. Some aspects of the synthesis involving design of environmentally responsive lipid materials were also supported by BES DE-FG-02-06ER46314. NMR characterization was performed using the Central Facilities of the Materials Research Laboratory at UCSB which are supported by the MRSEC Program of the NSF under Award No. DMR05-20415; a member of the NSF-funded Materials Research Facilities Network (www.mrfn.org). SAXS experiments were carried out at the Stanford Synchrotron Radiation Lightsource which is supported by the Department of Energy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Degradable Multivalent Lipids for Gene Delivery
Abbreviations: Boc, tertButoxycarbonyl; CL, cationic liposome; DEAD, diethylazodicarboxylate; DIEA, N,N-diisopropylethylamine; DLS, dynamic light scattering; DOB, 3,4-Di(oleyloxy)benzoic acid; DOPC, 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine; DOTAP, 2,3-dioleoyloxypropyltrimethylammonium chloride; DTT, dithiothreitol; EtBr, ethidium bromide; GSH, glutathione; L/D, cationic lipid/DNA weight ratio; NA, nucleic acid; NL, neutral lipid; RLU, relative light units; SAXS, small-angle X-ray scattering; TBTU, 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate; TE, transfection efficiency; TFA, trifluoroacetic acid; TLC, thin-layer chromatography; TL/D, total lipid/DNA weight ratio; Φi, mole fraction of lipid i in the lipid mixture; σM, membrane charge density.
Supplementary Material Available: Chemical structures of DOTAP, DOPC, and MVL5; details and results of TLC experiments showing the degradation of the CMVLs by reducing agents; assessment of CMVL/DOPC–DNA complex stability over time (in absence of reducing agents) by SAXS.
REFERENCES
- 1.Li S-D, Huang L. Gene therapy progress and prospects: non-viral gene therapy by systemic delivery. Gene Ther. 2006;13:1313–1319. doi: 10.1038/sj.gt.3302838. [DOI] [PubMed] [Google Scholar]
- 2.Miller A. Human gene therapy comes of age. Nature. 1992;357:455–460. doi: 10.1038/357455a0. [DOI] [PubMed] [Google Scholar]
- 3.Crystal R. Gene therapy strategies for pulmonary disease. Am. J. Med. 1992;92:44S–52S. doi: 10.1016/0002-9343(92)90607-d. [DOI] [PubMed] [Google Scholar]
- 4.Anderson W. Human gene therapy. Science. 1992;256:808–813. doi: 10.1126/science.1589762. [DOI] [PubMed] [Google Scholar]
- 5.Edelstein M, Abedi M, Wixon J, Edelstein R. Gene therapy clinical trials worldwide 1989–2004—an overview. J. Gene. Med. 2004;6:597–602. doi: 10.1002/jgm.619. [DOI] [PubMed] [Google Scholar]
- 6.Edelstein M, Abedi M, Wixon J. Gene therapy clinical trials worldwide to 2007—an update. J. Gene. Med. 2007;9:833–842. doi: 10.1002/jgm.1100. [DOI] [PubMed] [Google Scholar]
- 7.Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, Gross F, Yvon E, Nusbaum P, Selz F, Hue C, Certain S, Casanova J, Bousso P, Deist F, Fischer A. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288:669–672. doi: 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- 8.Marshall E. Gene therapy on trial. Science. 2000;288:951–957. [PubMed] [Google Scholar]
- 9.Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, Radford I, Villeval J, Fraser C, Cavazzana-Calvo M, Fischer A. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003;348:255–256. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 10.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack M, Wulffraat N, Leboulch P, Lim A, Osborne C, Pawliuk R, Morillon E, Sorensen R, Forster A, Fraser P, Cohen J, de Saint Basile G, Alexander I, Wintergerst U, Frebourg T, Aurias A, Stoppa-Lyonnet D, Romana S, Radford-Weiss I, Gross F, Valensi F, Delabesse E, Macintyre E, Sigaux F, Soulier J, Leiva L, Wissler M, Prinz C, Rabbitts T, Le Deist F, Fischer A, Cavazzana-Calvo M. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 11.Raper S, Chirmule N, Lee F, Wivel N, Bagg A, Gao G, Wilson J, Batshaw M. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 2003;80:148–158. doi: 10.1016/j.ymgme.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 12.Friedmann T. Overcoming the obstacles to gene therapy. Sci. Am. 1997;276:96–101. [PubMed] [Google Scholar]
- 13.Ferber D. Gene therapy. Safer and virus-free? Science. 2001;294:1638–1642. doi: 10.1126/science.294.5547.1638. [DOI] [PubMed] [Google Scholar]
- 14.Miller AD. The Problem with Cationic Liposome/Micelle-Based Non-Viral Vecto Systems for Gene Therapy. Curr. Med. Chem. 2003;10:1195–1211. doi: 10.2174/0929867033457485. [DOI] [PubMed] [Google Scholar]
- 15.Chesnoy S, Huang L. Structure and function of lipid-DNA complexes for gene delivery. Annu. Rev. Biophys. Biomol. Struct. 2000;29:27–47. doi: 10.1146/annurev.biophys.29.1.27. [DOI] [PubMed] [Google Scholar]
- 16.Huang L, Huang M-C, Wagner E, editors. Non-Viral Vectors for Gene Therapy, Advances in Genetics Volume 53. Elsevier; San Diego: 2005. [Google Scholar]
- 17.Niidome T, Huang L. Gene therapy progress and prospects: nonviral vectors. Gene. Ther. 2002;9:1647–1652. doi: 10.1038/sj.gt.3301923. [DOI] [PubMed] [Google Scholar]
- 18.Felgner P, Rhodes G. Gene therapeutics. Nature. 1991;349:351–352. doi: 10.1038/349351a0. [DOI] [PubMed] [Google Scholar]
- 19.Wheeler C, Felgner P, Tsai Y, Marshall J, Sukhu L, Doh S, Hartikka J, Nietupski J, Manthorpe M, Nichols M, Plewe M, Liang X, Norman J, Smith A, Cheng S. A novel cationic lipid greatly enhances plasmid DNA delivery and expression in mouse lung. Proc. Natl. Acad. Sci. U.S.A. 1996;93:11454–11459. doi: 10.1073/pnas.93.21.11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li W, Szoka FC. Lipid-based Nanoparticles for Nucleic Acid Delivery. Pharm. Res. 2007;24:438–449. doi: 10.1007/s11095-006-9180-5. [DOI] [PubMed] [Google Scholar]
- 21.Behr J. Gene transfer with synthetic cationic amphiphiles: prospects for gene therapy. Bioconjugate Chem. 1994;5:382–389. doi: 10.1021/bc00029a002. [DOI] [PubMed] [Google Scholar]
- 22.Zabner J. Cationic lipids used in gene transfer. Adv. Drug Deliv. Rev. 1997;27:17–28. doi: 10.1016/s0169-409x(97)00019-7. [DOI] [PubMed] [Google Scholar]
- 23.Miller AD. Cationic Liposomes for Gene Therapy. Angew. Chem. Int. Ed. 1998;37:1768–1785. [Google Scholar]
- 24.Behr J, Demeneix B, Loeffler J, Perez-Mutul J. Efficient gene transfer into mammalian primary endocrine cells with lipopolyamine-coated DNA. Proc. Natl. Acad. Sci. U.S.A. 1989;86:6982–6986. doi: 10.1073/pnas.86.18.6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pitard B, Aguerre O, Airiau M, Lachagès A, Boukhnikachvili T, Byk G, Dubertret C, Herviou C, Scherman D, Mayaux J, Crouzet J. Virus-sized self-assembling lamellar complexes between plasmid DNA and cationic micelles promote gene transfer. Proc. Natl. Acad. Sci. U.S.A. 1997;94:14412–14417. doi: 10.1073/pnas.94.26.14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ewert K, Ahmad A, Evans HM, Schmidt H-W, Safinya CR. Efficient synthesis and cell-transfection properties of a new multivalent cationic lipid for nonviral gene delivery. J. Med. Chem. 2002;45:5023–5029. doi: 10.1021/jm020233w. [DOI] [PubMed] [Google Scholar]
- 27.Bouxsein NF, McAllister C, Ewert KK, Samuel CE, Safinya CR. Structure and gene silencing activities of monovalent and pentavalent cationic lipid vectors complexed with siRNA. Biochemistry. 2007;46:4785–4792. doi: 10.1021/bi062138l. [DOI] [PubMed] [Google Scholar]
- 28.Ewert KK, Evans HM, Zidovska A, Bouxsein NF, Ahmad A, Safinya CR. A columnar phase of dendritic lipid-based cationic liposome-DNA complexes for gene delivery: hexagonally ordered cylindrical micelles embedded in a DNA honeycomb lattice. J. Am. Chem. Soc. 2006;128:3998–4006. doi: 10.1021/ja055907h. [DOI] [PubMed] [Google Scholar]
- 29.Ewert KK, Samuel CE, Safinya CR. Lipid-DNA Interactions, Structure-Function Studies of Nanomaterials for Gene Delivery. In: Dias R, Lindman B, editors. Interaction of DNA with Surfactant and Polymers. Blackwell; Boston: 2008. pp. 377–404. [Google Scholar]
- 30.Ewert KK, Zidovska A, Ahmad A, Bouxsein NF, Evans HM, McAllister CS, Samuel CE, Safinya CR. Cationic Lipid–Nucleic Acid Complexes for Gene Delivery and Silencing: Pathways and Mechanisms for Plasmid DNA and siRNA. In: Bielke W, Erbacher C, editors. Topics in Current Chemistry – Nucleic Acid Transfection. Springer; New York: 2010. pp. 191–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lv H, Zhang S, Wang B, Cui S, Yan J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release. 2006;114:100–109. doi: 10.1016/j.jconrel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 32.Ahmad A, Evans HM, Ewert K, George CX, Samuel CE, Safinya CR. New multivalent cationic lipids reveal bell curve for transfection efficiency versus membrane charge density: lipid-DNA complexes for gene delivery. J. Gene Med. 2005;7:739–748. doi: 10.1002/jgm.717. [DOI] [PubMed] [Google Scholar]
- 33.Lin AJ, Slack NL, Ahmad A, George CX, Samuel CE, Safinya CR. Three-dimensional imaging of lipid gene-carriers: membrane charge density controls universal transfection behavior in lamellar cationic liposome-DNA complexes. Biophys. J. 2003;84:3307–3316. doi: 10.1016/S0006-3495(03)70055-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meister A, Anderson M. Glutathione. Annu Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 35.Voet DJ, Voet JG. Biochemistry. second ed. Wiley; New York: 1996. [Google Scholar]
- 36.Han D, Hanawa N, Saberi B, Kaplowitz N. Mechanisms of liver injury. III. Role of glutathione redox status in liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2006;291:G1–G7. doi: 10.1152/ajpgi.00001.2006. [DOI] [PubMed] [Google Scholar]
- 37.Falnes P, Sandvig K. Penetration of protein toxins into cells. Curr. Opin. Cell Biol. 2000;12:407–413. doi: 10.1016/s0955-0674(00)00109-5. [DOI] [PubMed] [Google Scholar]
- 38.Saito G, Swanson J, Lee K. Drug delivery strategy utilizing conjugation via reversible disulfide linkages: role and site of cellular reducing activities. Adv. Drug Deliv. Rev. 2003;55:199–215. doi: 10.1016/s0169-409x(02)00179-5. [DOI] [PubMed] [Google Scholar]
- 39.Lu Z, Wang X, Parker D, Goodrich K, Buswell H. Poly(l-glutamic acid) Gd(III)-DOTA conjugate with a degradable spacer for magnetic resonance imaging. Bioconjugate Chem. 2003;14:715–719. doi: 10.1021/bc0340464. [DOI] [PubMed] [Google Scholar]
- 40.Ke T, Feng Y, Guo J, Parker D, Lu Z. Biodegradable cystamine spacer facilitates the clearance of Gd(III) chelates in poly(glutamic acid) Gd-DOTA conjugates for contrast-enhanced MR imaging. Magn. Reson. Imaging. 2006;24:931–940. doi: 10.1016/j.mri.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 41.Meng F, Hennink W, Zhong Z. Reduction-sensitive polymers and bioconjugates for biomedical applications. Biomaterials. 2009;30:2180–2198. doi: 10.1016/j.biomaterials.2009.01.026. [DOI] [PubMed] [Google Scholar]
- 42.Boutorine A, Kostina E. Reversible covalent attachment of cholesterol to oligodeoxyribonucleotides for studies of the mechanisms of their penetration into eucaryotic cells. Biochimie. 1993;75:35–41. doi: 10.1016/0300-9084(93)90022-k. [DOI] [PubMed] [Google Scholar]
- 43.Chittimalla C, Zammut-Italiano L, Zuber G, Behr J. Monomolecular DNA nanoparticles for intravenous delivery of genes. J. Am. Chem. Soc. 2005;127:11436–11441. doi: 10.1021/ja0522332. [DOI] [PubMed] [Google Scholar]
- 44.Dauty E, Remy J, Zuber G, Behr J. Intracellular delivery of nanometric DNA particles via the folate receptor. Bioconjugate Chem. 2002;13:831–839. doi: 10.1021/bc0255182. [DOI] [PubMed] [Google Scholar]
- 45.Soundara Manickam D, Oupický D. Polyplex gene delivery modulated by redox potential gradients. J. Drug Target. 2006;14:519–526. doi: 10.1080/10611860600834409. [DOI] [PubMed] [Google Scholar]
- 46.Park Y, Kwok K, Boukarim C, Rice K. Synthesis of sulfhydryl cross-linking poly(ethylene glycol)-peptides and glycopeptides as carriers for gene delivery. Bioconjugate Chem. 2002;13:232–239. doi: 10.1021/bc010070a. [DOI] [PubMed] [Google Scholar]
- 47.Tang F, Hughes J. Introduction of a disulfide bond into a cationic lipid enhances transgene expression of plasmid DNA. Biochem. Biophys. Res. Commun. 1998;242:141–145. doi: 10.1006/bbrc.1997.7923. [DOI] [PubMed] [Google Scholar]
- 48.Tang F, Hughes J. Use of dithiodiglycolic acid as a tether for cationic lipids decreases the cytotoxicity and increases transgene expression of plasmid DNA in vitro. Bioconjugate Chem. 1999;10:791–796. doi: 10.1021/bc990016i. [DOI] [PubMed] [Google Scholar]
- 49.Byk G, Wetzer B, Frederic M, Dubertret C, Pitard B, Jaslin G, Scherman D. Reduction-sensitive lipopolyamines as a novel nonviral gene delivery system for modulated release of DNA with improved transgene expression. J. Med. Chem. 2000;43:4377–4387. doi: 10.1021/jm000284y. [DOI] [PubMed] [Google Scholar]
- 50.Wetzer B, Byk G, Frederic M, Airiau M, Blanche F, Pitard B, Scherman D. Reducible cationic lipids for gene transfer. Biochem. J. 2001;356:747–756. doi: 10.1042/0264-6021:3560747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Still WC, Kahn M, Mitra A. Rapid chromatographic technique for preparative separations with moderate resolution. J. Org. Chem. 1978;43:2923–2925. [Google Scholar]
- 52.Ewert KK, Evans HM, Bouxsein NF, Safinya CR. Dendritic cationic lipids with highly charged headgroups for efficient gene delivery. Bioconjugate Chem. 2006;17:877–888. doi: 10.1021/bc050310c. [DOI] [PubMed] [Google Scholar]
- 53.Schulze U, Schmidt H-W, Safinya CR. Synthesis of novel cationic poly(ethylene glycol) containing lipids. Bioconjugate Chem. 1999;10:548–552. doi: 10.1021/bc9801068. [DOI] [PubMed] [Google Scholar]
- 54.Jacobson K, Fischer B, Ji X. “Cleavable trifunctional” approach to receptor affinity labeling: chemical regeneration of binding to A1-adenosine receptors. Bioconjugate Chem. 1995;6:255–263. doi: 10.1021/bc00033a004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ewert K, Slack NL, Ahmad A, Evans HM, Lin AJ, Samuel CE, Safinya CR. Cationic lipid-DNA complexes for gene therapy: understanding the relationship between complex structure and gene delivery pathways at the molecular level. Curr. Med. Chem. 2004;11:133–149. doi: 10.2174/0929867043456160. [DOI] [PubMed] [Google Scholar]
- 56.Mitsunobu O. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural-products. Synthesis-Stuttgart. 1981:1–28. [Google Scholar]
- 57.Hughes DL. The Mitsunobu Reactions. In: Beak P, editor. Organic Reactions. Wiley; New York: 1992. pp. 335–656. [Google Scholar]
- 58.Boger D, Fink B, Brunette S, Tse W, Hedrick M. A simple, high-resolution method for establishing DNA binding affinity and sequence selectivity. J. Am. Chem. Soc. 2001;123:5878–5891. doi: 10.1021/ja010041a. [DOI] [PubMed] [Google Scholar]
- 59.Ham Y, Tse W, Boger D. High-resolution assessment of protein DNA binding affinity and selectivity utilizing a fluorescent intercalator displacement (FID) assay. Bioorg. Med. Chem. Lett. 2003;13:3805–3807. doi: 10.1016/j.bmcl.2003.06.002. [DOI] [PubMed] [Google Scholar]
- 60.Bloomfield V. Condensation of DNA by multivalent cations: considerations on mechanism. Biopolymers. 1991;31:1471–1481. doi: 10.1002/bip.360311305. [DOI] [PubMed] [Google Scholar]
- 61.Wilson R, Bloomfield V. Counterion-induced condensation of deoxyribonucleic acid – a light-scattering study. Biochemistry. 1979;18:2192–2196. doi: 10.1021/bi00578a009. [DOI] [PubMed] [Google Scholar]
- 62.Bloomfield V. DNA condensation. Curr. Opin. Struct. Biol. 1996;6:334–341. doi: 10.1016/s0959-440x(96)80052-2. [DOI] [PubMed] [Google Scholar]
- 63.Bloomfield V. DNA condensation by multivalent cations. Biopolymers. 1997;44:269–282. doi: 10.1002/(SICI)1097-0282(1997)44:3<269::AID-BIP6>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 64.Pelta J, Livolant F, Sikorav J. DNA aggregation induced by polyamines and cobalthexamine. J. Biol. Chem. 1996;271:5656–5662. doi: 10.1074/jbc.271.10.5656. [DOI] [PubMed] [Google Scholar]
- 65.Evans HM. Ph.D. Thesis. University of California; Santa Barbara, CA: 2005. Structure-Function Investigations of DNA Condensing Agents with Application to Gene Delivery. [Google Scholar]
- 66.Cory A, Owen T, Barltrop J, Cory J. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991;3:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- 67.Lin AJ, Slack NL, Ahmad A, Koltover I, George CX, Samuel CE, Safinya CR. Structure and structure-function studies of lipid/plasmid DNA complexes. J. Drug Target. 2000;8:13–27. doi: 10.3109/10611860009009206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.