

Abstract
Cancer is a disease initiated and driven by the accumulation and interplay of genetic and epigenetic mutations of genes involved in the regulation of cell growth and signaling. Dysregulation of these genes and pathways in a cell leads to a growth advantage and clonal expansion. The epigenetic alterations involved in the initiation and progression of cancer are DNA methylation and histone modifications which interact to remodel chromatin, as well as RNA interference. These alterations can be used as candidate targets in molecular tests for risk, early detection, prognosis, prediction of response to therapy, and monitoring, as well as new therapeutic targets in cancer. In this review, we discuss the rationale, studies to date, and issues in the translational application of epigenetics using epithelial ovarian cancer as a specific example of all types of cancer.
Keywords: methylation, histone modifications, microRNAs, translational research, ovarian cancer
Introduction
The three main epigenetic mechanisms are DNA methylation and histone modifications as well as RNA interference. DNA methylation involves the chemical modification of cytosoine by the addition of a methyl group to the 5′ carbon of the cytosine base in CG dinucleotides. Compared to normal cells, cancer cells show global hypomethylation mainly of repetitive elements in the DNA sequence but also localized hypermethylation. In particular, CpG islands in the promoter regions of certain genes can be hypermethylated with associated loss of expression [1].
Histone modifications act together with DNA methylation at promoters to organize chromatin and thereby regulate gene expression. Histone complexes consist of two subunits each of the H2A, H2B, H3 and H4 histones forming an octamer that wraps 146 bps of DNA- referred to as a nucleosome. The H1 linker histone binds to the outside of the nucleosome and seals two turns of DNA. The nucleosome allows for large quantities of DNA to be packaged into a small area. Histone tails remain external to the core and are the substrate of modification. As a result, how the histone tail modifications interact with DNA can influence chromatin condensation, stability and structure. Modifications to the histone core components as well as the histone linker have been demonstrated as crucial in the regulation of different cellular processes [2–4]. Alterations in the status of histone modifications are evident in tumor cells [1].
MicroRNAs (miRNA) are short non-coding RNA sequences that regulate gene expression through targeted degradation of mRNA transcripts [5]. The alterations in gene expression that result from miRNA-initiated destruction have significant impact on cell function and survival. Atypical expression of miRNAs has been demonstrated in cancer cells and is known to be involved in tumorigenesis. To date, almost a thousand miRNAs have been identified in human cells. MiRNAs are the result of several processing events. Initially, primary transcripts, known as pri-miRNAs, are transcribed and cleaved by the enzyme Drosha into a 60–70 nucleotide sequence that forms a stem loop structure. These pri-miRNAs are then cleaved to short sequences (about 21–25 nucleotides in length) called pre-miRNAs by the enzyme Dicer. These sequences are then associated with RISC (RNA induced silencing complex) to initiate mRNA degradation. The expression levels of certain miRNAs have been shown be altered in tumor cells compared to the normal progenitor cells [6–8].
Epigenetic alterations present in neoplastic cells are potential molecular tools to detect and manage cancer. Epithelial ovarian cancer is a disease that is neither common nor rare, has no specific symptoms and is mostly detected at an advanced stage, and sooner or later resistant to chemotherapy [9]. Here, we use epithelial ovarian cancer as an example to discuss the epigenetic alterations and their translational application to risk, detection, prognosis, prediction of response, and therapy for cancer.
Risk of Developing Cancer
The identification of individuals at a higher risk of developing a particular cancer might allow lifestyle intervention, chemoprevention, and screening for early detection. The etiology of ovarian cancer is not well understood but known modulators of risk include a woman’s reproductive history and increasing age. Pregnancy at a younger age as well as number of pregnancies, oral contraceptive use, tubal ligation and hysterectomy have all been associated with a lower risk of ovarian cancer [10]. To begin to examine if epigenetics underlies this protective effect, it is necessary to have a definitive identification of the normal progenitor cells from which epithelial ovarian cancer develops. A consensus on the origin of ovarian tumors has not yet been formed. Normal human ovarian surface epithelial (OSE) cells have been most widely used in laboratory studies. More recently, inclusion cysts or cells from the fimbria of the fallopian tube that implant in the ovary have been proposed as progenitors. A mullerian origin for ovarian tumor cells has also been argued for [10–13]}.
A related barrier to the study of the role of epigenetic alterations in risk is the inability to access the ovaries, fallopian tubes or peritoneum in a non-invasive manner in order to obtain specimens from women at progressive age cohorts whom subsequently develop ovarian cancer and women whom never develop ovarian cancer. Few prophylactic oophorectomies from high-risk women and even fewer from average-risk women with benign disease show clear evidence of early ovarian cancer. Some other anatomical sites are more amenable to sampling for risk e.g. oral cancer by simple visual examination and bladder cancer from urothelial cells shed in a simple urine specimen. To overcome this issue, animal studies in which environmental factors or carcinogens are administered over time followed by ovariectomy and analysis of the epigenome may be considered.
These two issues of definitive identification of the normal progenitor cells and non-invasive access to those cells at a time point before diagnosis of cancer apply to some other cancer types and need to be overcome before exploratory studies of global methylation, histone modifications and miRNA expression profiles can be performed. Peripheral blood lymphocytes have been suggested as surrogate cells for the study of environmentally-induced alterations in cancer. The biological rationale for lymphocytes over normal progenitor cells of epithelial cancer is not compelling and is likely made partly to circumvent the issue of invasive access and also because lymphocytes have been banked in large population studies and are therefore available to researchers. Since it is known that epigenetic alterations can occur in premalignant lesions or the earliest identified stages of neoplasia of many epithelial cancers [14,15], these alterations are candidate targets for chemoprevention. There is some pre-clinical evidence for prevention by epigenetic drugs in animal models of neoplasia [16–19].
Early Detection
Diagnosis of Ovarian Cancer
Detection of cancer at an early stage generally correlates with good prognosis and higher survival rates. Ovarian cancer is diagnosed by a physical exam, computerized tomography (CT) scan, ultrasound, and the CA-125 serum marker. All these methods have been limited in success for early detection. Regular pelvic exams miss early stage tumors and the specificity of ultrasonography for ovarian cancer is limited by detection of cysts and solid benign lesions. Serum levels of CA-125 are elevated in only approximately 80% of ovarian cancer patients and lack specificity. CA-125 is elevated in other types of cancer, can be present in women with benign ovarian conditions and in up to 0.5–1% of the normal population. Therefore this blood test cannot be used alone to diagnose ovarian cancer. Exploratory surgery (laparoscopy) is often necessary to diagnose epithelial ovarian cancer, since ovarian masses may also be caused by benign cysts, other primary ovarian cancers such as germ cell or stromal cell, or by metastasis to the ovaries [20].
Nucleic Acid Based Detection of Cancer in Body Fluids
Alterations at the DNA or RNA level are promising markers for molecular diagnosis and prognosis because they can precede obvious cancer, are highly specific, can be detected by PCR-based techniques at extremely sensitive levels, and can provide diagnostic and prognostic information simultaneously. Nucleic acid-based markers have been found in body fluids that surround or drain from the organ of interest [21,22]. Women at high risk, i.e. familial ovarian, BRCA1, BRCA2 and hereditary non-polyposis colorectal cancer (HNPCC) carriers, may be willing to undergo a semi-invasive peritoneal needle or catheter wash for early diagnosis. However, serum is a readily available, non-invasive fluid that contains circulating tumor cells and/or free tumor DNA and could potentially be used for early detection in both high risk and sporadic ovarian cancer [23] (Figure 1A).
Figure 1.
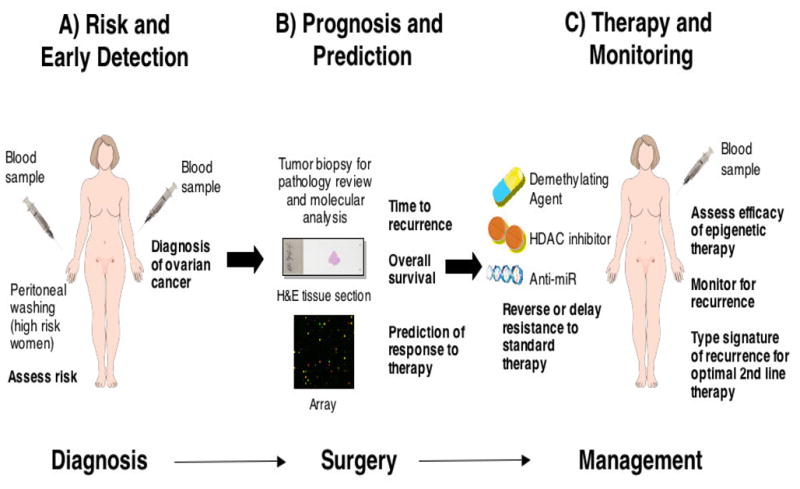
Translational Applications of Epigenetic Alterations in Cancer
Aberrant Promoter Hypermethylation in Blood
Aberrant DNA hypermethylation of CpG islands in the promoter region of genes is well established as a common mechanism for the silencing of tumor suppressor genes in cancer cells and serves as an alternative mechanism to point mutation or deletion for functional inactivation [24]. The classical tumor suppressor genes BRCA1, p16INK4a and mutL homolog1 (MLH1) as well as the putative tumor suppressor genes RAS association domain family protein 1A (RASSF1A) and opioid binding protein/cell adhesion molecule like (OPCML) among others have been identified as hypermethylated with associated loss of expression in ovarian cancer [25–32]. The availability of a common and early alteration, such as promoter hypermethylation, to target the presence of cancer combined with the availability of technology, e.g. methylation specific PCR (MSP), which is capable of the detection of few methylated alleles from neoplastic cells in a background of many unmethylated alleles from normal or non-neoplastic cells, led to studies of the feasibility of methylation-based detection of cancer in body fluids or biopsies [33].
An initial study obtained matched tumor, pre-operative serum or plasma, and peritoneal fluid (washes or ascites) DNAs from 50 patients with epithelial ovarian tumors. Microdissected tissue DNA and body fluid DNA was analyzed by conventional gel-based MSP for hypermethylation status of the normally unmethylated BRCA1 and RASSF1A, adenomatous polyposis coli (APC), p14ARF, p16INK4a or death associated protein-kinase (DAP-Kinase) tumor suppressor genes. Hypermethylation of one or more of the gene panel was found in all 50 tumor DNAs (100% diagnostic coverage) and in all histological cell types, grades and stages of epithelial ovarian tumor examined. An identical pattern of gene hypermethylation was found in the matched pre-operative serum DNA from 41 of 50 patients (82% sensitivity) including all 8 cases of stage I disease. Hypermethylation was detected in 27 of 29 peritoneal fluid DNAs from stage Ic-IV patients, including 3 cases with negative or atypical cytology. In contrast, no hypermethylation was observed, albeit in a relatively small number of controls that included non-neoplastic tissue, peritoneal fluid or serum from age-matched women with no evidence of ovarian cancer [23].
Histone Modifications and miRNA Expression for Early Detection
The western, immunohistochemistry (IHC), and chromatin immunoprecipitation (ChIP) assays currently used to examine histone modifications are generally not amenable to the detection of alterations from few tumor cells in a mixture of predominantly normal cells. These techniques may be more readily applied to tumor cell-rich tissue specimens obtained at time of surgical resection and therefore, at present, histone modifications seem more relevant as markers for prognosis and prediction.
The demonstration that miRNA expression profiles are different in many types of cancer compared to the normal progenitor cells suggests that a neoplastic cell-specific profile could be used for detection and prognosis [6–8]. Furthermore, there is evidence that miRNAs are more stable and/or more resistant to RNase activity than are mRNAs in tissue and body fluid specimens thereby facilitating analysis [6,34–36].
A study of ovarian patients prior to chemotherapy found 8 miRNAs with dysregulated expression in patient sera compared to normal controls [37]. The dysregulated miRNAs included miR-29a, miR-93 and miR-155 previously reported to be dysregulated in primary ovarian tumors [7,37–39]. Another study examining the miRNA profiles in 5mls of blood of 24 patients with recurrence of ovarian cancer (mostly of serous histology) found 147 miRNAs dysregulated in the ovarian cancer compared to normal control blood specimens [40]. The list included miR-16, miR-29a, miR-106b and miR-155 also previously shown to be dysregulated in ovarian cancer [7,37–39].
The precursor genomic sequence of some microRNAs are located in CpG islands that can be methylated [41–43]. Such methylation, if aberrant in neoplastic cells, is another potential target for molecular detection.
Issues of Epigenetic Alterations in Early Detection
There are several barriers to the validation of gene methylation for early detection of ovarian cancer. To examine the specificity of an alteration for cancer, it is useful to determine if a gene is methylated only in neoplastic cells. It is therefore important to examine normal progenitor cell DNA to see if a gene is imprinted, has tissue specific methylation or shows age-related methylation. As well as the question of origin of ovarian tumors, there is the further problem of obtaining sufficient cells from a surgical specimen for molecular analysis. A brushing of OSE cells from normal ovary or microdissection of fimbria, do not contain sufficient numbers of cells to permit more than minimal molecular analysis. SV40-immortalized human OSE cell lines do provide sufficient amounts of DNA to examine gene methylation however while these cell lines may be non-tumorigenic they cannot be considered biologically normal. Similarly, the amount of DNA in 1ml of serum is limits analysis. For retrospective validation, curators of large serum specimen banks are reluctant to release 1ml of serum for study of a nucleic acid marker when proteomic studies typically require far less specimen. Prospective collection of serum would allow sufficient amounts for molecular studies, for example several mls may be discarded at time of a CA-125 test, but this requires time to accrue large numbers of samples and so has proved a delay to validation.
By definition, a circulating body fluid can be accessed by many different anatomical sites. The use of serum means differential diagnosis of the organ site from which the positive methylation result originated is needed. To date, few genes have demonstrated specificity of aberrant hypermethylation to a single cell type or anatomical site. One example is BRCA1 hypermethylation which is restricted to ovarian and breast tumors. Another is GSTP1 methylated in breast tumors but rarely, if ever, in ovarian cancer [28]. It is possible that genes that show hypermethylation specific to epithelial ovarian cancer remain to be discovered. Several such genes included in a panel with BRCA1 and GSTP1 might allow development of an algorithm sufficiently accurate for differential diagnosis of ovarian cancer. In the near term an epigenetic marker would likely be used alongside CA-125 to improve the accuracy of CA-125 screening. Arguments against screening the general population for the >90% of ovarian cancer that is sporadic include that there are no or minimal risk factors, a false positive can mean an unnecessary laparotomy, and that the disease is relatively uncommon [20]. It may be that current cost benefit analysis would only allow screening in the general population if ovarian cancer was ‘bundled” with screening for other more common cancers. This would place further importance on differential diagnosis.
Alterations in miRNA expression would appear to be a more abundant target for early detection strategies in body fluids as each tumor cell contains hundreds or thousands of molecules of a particular miRNA but only one to several (in aneuploid cells) methylated alleles of a gene. That initial miRNA studies used several mls of serum/plasma would question such abundance and suggest similar issues to limiting amounts of DNA [34,44–46]. The ratio of few tumor cells to many normal cells in blood is another issue depending on the relative difference in miRNA expression between normal and tumor cells..
It has been stated that miRNAs are stable i.e. resistant to degradation through secondary structure and/or being contained in exosomes [47]. While miRNA is more stable than mRNA, in a clinical specimen miRNA should survive no better or worse than short pieces of degraded DNA currently used in forensics and archeology. Similarly, exosomes proposed to protect miRNA also contain DNA [48]. Overall it seems that miRNA will have the same practical issues as DNA methylation as a target for early detection.
Many other methylated tumor suppressor and cancer genes as well as individual miRNAs important in ovarian tumorigenesis likely remain to be identified. Most useful for early detection are genes with aberrant methylation or microRNAs with aberrant expression that is early and frequent in tumorigenesis, neoplastic cell-specific, organ type-specific, biologically relevant, capable of providing simultaneous diagnostic and prognostic information, and amenable to technology that can detect the gene methylation or miRNA expression at a sensitive level in a non-invasive specimen with sufficient lead time for effective treatment, at an acceptable financial cost.
Prognosis
A prognostic marker can be defined as a characteristic associated with prognosis or outcome, usually in terms of relative hazard of failure either time to recurrence/progression or death, whereas a predictive marker is defined as a characteristic that predicts the differential efficacy (benefit) of a particular therapy based on marker status, that is, only patients with the marker will respond to the specific treatment or will respond to a greater degree than individuals without the marker [49]. Currently, the amount of ovarian tumor remaining after cytoreduction in combination with patient response to platinum drugs are the best factors to gauge ovarian cancer patient prognosis [50,51]. That almost all epithelial ovarian cancer patients receive chemotherapy means that it can be difficult to distinguish a marker for prognosis from a marker for prediction of chemoresponse. An example of where the epigenetic basis for the biology of tumor aggressiveness can be separated from response to therapy is bladder cancer. A subset of superficial bladder tumors are cured after initial resection, others recur, while a minority both recur and progress. Some of these superficial bladder tumors are not treated with chemotherapy and the role of epigenetic alterations in the behavior of such tumors could be studied to identify markers of prognosis.
Aberrant DNA Methylation and Prognosis
The methylation status of individual genes has been investigated as a marker for prognosis of ovarian cancer. One study looked at 235 ovarian cancer patients and found that the association between hypermethylation of the IGFBP-3 gene and poor prognosis was stronger in women with early stage disease [52]. In a subsequent study, promoter methylation of IGFBP-3 in combination with CDKN2A, BRCA1 or MLH1 was reported to increase the risk of disease progression [53]. In another study, progression free survival of ovarian cancer patients was associated with hypomethylation of satellite DNA repeat sequences located on chromosome1 [54] and hypermethylation of 18S and 28S rDNA was also indicative of progression free survival [55]. Determining a methylation signature that predicts the amount of time until relapse and/or overall survival would greatly impact individualized care regimens. CpG island microarray analysis has identified additional candidate methylation markers of prognosis in ovarian cancer towards the development of such signatures [56–58].
Histone Modifications and Prognosis
Histone core subunits share a common structure including an extended tail that is the site of post-translation modifications. The most common modifications to histone tails include acetylation, methylation, phosphorylation and ubiquitylation. These modifications have the ability to enhance or block transcription factor binding and transcription initiation. As a result, dysregulated modifications in the histone code can lead to aberrant gene expression in tumor cells and likely contribute to the disease [1].
H3 histone can be methylated up to three times at K4, K9, K27, K36 and K79. Trimethylation of H3K4 is linked to gene expression while H3K27 trimethylation is associated with gene silencing. Histones are methylated at specific lysines and arginines by Histone Methyl Transferases (HMTs). Two common HMTs are mixed lineage leukemia (MLL) and Su(var)3–9, Enhancer of zeste, and trithorax (SET1) [59]. Histones are demethylated by Histone Demethylases (HDMs) including lysine specific demethylase 1 (LSD1) and jumonji AT-rich interactive domain 1 (JARIDI1) [60].
The availability of tumor tissue at time of surgery means that histone modifications can be examined and any association with prognosis studied. A particular pattern of histone marks is associated with hypermethylated CpG islands in promoters of genes in tumor cells. For example, the repression of tight junction proteins, claudin-3 and claudin-4, is reversed in ovarian cancer cell lines through loss of H3K27me3 and H4K20me3 [61]. While this was demonstrated in ovarian cancer cell lines, claudin-3 and claudin-4 are consistently over-expressed in primary ovarian tumors and claudin-3 over-expression is associated with poor prognosis, supporting potential clinical relevance [62,63]. Additionally, the methylatransferase responsible for H3K27 methylation, Enhancer of Zeste Homologue 2 (EZH2), is over-expressed in cells resistant to cisplatin. Inhibition of EZH2 activity can resensitize ovarian cancer cells to cisplatin treatment suggesting that histone methylation at H3K27 plays a critical role in gene silencing [64].
MiRNA Expression and Prognosis
The expression levels of miRNAs as well as that of the enzyme machinery for processing of miRNAs have been examined for association with prognosis of ovarian cancer. A study analyzing miRNA expression profiles of 34 ovarian cancer tissues and 10 ovarian cancer cell lines found that miR-221 is the most consistently over-expressed miRNA in ovarian cancer [65]. In another study, when ovarian carcinomas were compared to non-immortalized primary ovarian surface epithelial cultures miR-221 was under-expressed in ovarian carcinomas [66]. These conflicting results are likely due to differences between the normal comparison samples but supports the hypothesis that miR-221 is likely dysregulated in ovarian cancer. MiR-221 along with miR-222 regulate cell cycle progression and directly target CDK1B (p27) and CDK1C (p57) [67,68]. The levels of both CDK1B and CDK1C are under-expressed in ovarian cancer suggesting that miR-221 and miR-222 may be playing a role in their mRNA transcript degradation. Mir-221 and miR-222 are both transcribed on the X chromosome so their expression levels are typically associated. Interestingly, ovarian cancer patients with a worse overall survival had a lower ratio of miR-221 to miR-222 [69].
While dysregulation of miRNAs is clearly involved in tumorogenesis [70], it is currently unclear how miRNAs are dysregulated however incorrect processing of miRNA transcripts likely has a role. In the nucleus, miRNA transcripts are processed into ~70 nt stem-loop pre-miRNA by the enzyme Drosha. Following this step the pre-miRNA is transported into the cytoplasm where it is processed by the enzyme Dicer into a 17–25 nt mature miRNA [71]. In a study of 111 epithelial ovarian cancers, Drosha expression was decreased 51% and Dicer expression decreased by 60% compared to benign ovarian epithelial tissue. In addition, decreased Dicer expression correlated with advanced tumor stage and decreased Drosha expression was associated with suboptimal surgical cytoreduction [72]. The findings in this study suggest that a decrease in miRNA processing may affect the outcome of patients with ovarian cancer. In support of these findings, a separate study found that decreased Dicer expression in ovarian tumors was correlated with lower patient survival time. MiRNA analysis of the Dicer positive tumors versus Dicer negative patients indicated that miRNA expression was lower in Dicer negative samples [73]. While it is possible that regulation of miRNA processing differs between tissue types, other studies have conversely reported high Dicer expression to be associated with poor prognosis in patients with prostate and esophageal cancer [74,75].
Prediction of Response to Therapy
Chemotherapy for Ovarian Cancer
Understanding and overcoming resistance to chemotherapy is central to improving survival of cancer patients. The basis of chemoresistance is multifactorial and is often thought of as intrinsic or acquired. While mutation of individual genes such as RAS or p53 can be relatively common in a tumor type, overall there is considerable heterogeneity between the genetic, epigenetic and pre- and post-translational alterations in the tumor cells of one patient to another patient [76]. This is particularly true in high grade serous carcinomas, the most common and most lethal subtype of ovarian cancer, and a tumor marked with significant genomic instability [77]. A very large collection of conventional chemotherapy agents and a growing list of molecular targeted agents occasionally provide significant tumor cytoreduction and clinical benefit yet the selection of ‘best agents’ for a given individual’s tumor is typically accomplished by trial and error. Women who are diagnosed with ovarian cancer are given a platinum-based drug therapy and those whose cancer progresses while on therapy or who recur within 6 months are deemed platinum resistant. Only at this time are the patients given alternative agents. Thus, it is critical to identify biomarkers that can predict patient response to platinum drugs so that optimal treatment can be administered immediately.
The power of predictive biomarkers is now confirmed in multiple epithelial and mesenchymal tumors with examples including the presence of Her-2-neu amplification, c-Kit mutation, EGFR mutations, or BCR-Abl rearrangement all of which have transformed the treatment of certain subtypes of breast cancer, sarcoma, lung cancer and leukemia, respectively [78–80]. Such potent molecular based predictive biomarkers do not exist in epithelial ovarian cancer with the possible exception of germline mutations in BRCA genes which greatly increase the risk of subsequent serous carcinomas of the ovary and predict for response to platinum [81,82] and Poly(ADP-ribose) polymerase (PARP) inhibitors [83]. Figure 1B demonstrates potential approaches for women diagnosed with ovarian cancer when predictive markers are available. Following diagnosis, a biopsy of the tumor is reviewed by a pathologist and tumor cell DNA, RNA and/or protein are analysed by global technologies to identify the patient’s time to recurrence, overall survival as well as prediction of response to therapy based on the molecular signature of the tumor.
In a recent paper, Boettcher and colleagues used microarray technology to compare methylation profiles from doxorubicin sensitive and resistant breast and ovarian cancer cell lines. Specifically breast cancer cell line MCF-7_wt (doxorubicin sensitive) was compared to MCF-7_ADR (a selected sub-line of MCF-7_wt that is doxorubicin resistant). In addition, the methylation profiles of ovarian cancer cell lines OVCAR-4 (inherently resistant), OVCAR-5 (sensitive) and NCI/ADR-RES (doxorubicin selected resistance) were also compared. Hypermethylation of BRCA1, CDH1, DNAFC15 and SULF2 and hypomethylation of ABC1, APC and HIC1 were observed in doxorubicin resistant cell lines. Quantitative RT-PCR was used to detect changes in gene expression which in most cases was associated with methylation status of the corresponding gene [84]. This study suggests that there are key hypomethylated or hypermethylated genes involved in drug resistance which are potential indicators of chemoresistance that could be used to determine appropriate therapy.
The BRCA/HR Pathway and Chemoresponse
The biological rationale for a better response to platinum or PARP inhibitors in women with germline BRCA mutations or “BRCAness” is that the BRCA1 and BRCA2 genes are important for double-strand break (DSB) repair by homologous recombination (HR). Cells defective in BRCA1 or BRCA2 are more sensitive to cisplatin and carboplatin, which cause covalent cross-links between bases on the two opposite DNA strands termed interstrand DNA cross-links. BRCA1 or BRCA2 negative cells show even greater hypersensitivity to inhibition of the PARP enzyme that is involved in base excision repair, a key pathway in the repair of DNA single strand breaks. During S phase, it is believed that partially processed cross-links cause stalling of the DNA replication machinery and fork collapse. Base excision with incomplete repair can be converted into DSB with progression of the DNA replication machinery. These DSB would normally be repaired by HR in cells where BRCA1 and BRCA2 are functional [85]. The impairment of base excision repair is not lethal in cells with alternative mechanisms of DNA repair but proves insurmountable in cells deficient in HR, such as is seen in tumors with loss of BRCA1 or BRCA2 function, providing a therapeutic opportunity
Epigenetic markers can also serve as predictive biomarkers, with an example being the O6-methyl-guanine-DNA-methyltransferase (MGMT) gene, a DNA repair protein that removes mutagenic and cytotoxic adducts from O6-guanine in DNA. The silencing of the MGMT gene by the epigenetic mutation of hypermethylation in gliomas is an independent predictor of a favorable response to carmustine [29] or Temozolomide [86]. The epigenome of the cancer cell likely underlies at least a component of intrinsic and acquired chemoresistance since some of the aberrant hypermethylation in the ovarian cancer genome inactivates tumor suppressors which result in the loss of function of key genes (such as BRCA1) or the impairment of key pathways (such as BRCA/HR) [29,87].
BRCA1 and Other HR Gene Methylation in Cancer
The unexpected absence of inactivating point mutations of BRCA1 in sporadic breast and ovarian cancers and the findings of methylation as an alternative mechanism of inactivation of the VHL, p16 and MLH1 classical tumor suppressor genes led to studies of the methylation status of the BRCA1 promoter region in sporadic breast and ovarian tumors reviewed in [88]. Several studies reported that BRCA1 was hypermethylated in 10–15% of sporadic ovarian tumors and that hypermethylation was strongly associated with loss of expression at the RNA or protein level. It was also reported that patients with hypermethylation of BRCA1 in ovarian tumors had a better survival [88]. The subset of sporadic breast tumors with BRCA1 methylation show a similar phenotype to familial BRCA1 patient breast tumors termed “BRCAness” [88] Sporadic ovarian tumors with functional inactivation of BRCA1 by hypermethylation will also have the BRCA deficiency phenocopy [88]. The loss of BRCA1 expression caused by promoter hypermethylation will disrupt BRCA-associated DNA repair and may sensitize tumors to BRCA-directed therapies. The model for sensitivity to PARP inhibition is dependent upon homologous recombination deficiency rather than inherited BRCA mutation. Therefore PARP inhibitors can be applicable to sporadic ovarian tumors with functional impairment of the HR pathway other than loss of BRCA1 or BRCA2 [85]. The methylation, and thereby functional, status of other genes implicated in the wider BRCA/HR pathway may also be relevant to suitability of PARP inhibitor therapy.
The BRCA2 gene appears to be unmethylated in ovarian and other cancers [89–91]. A Fanconi’s Anemia gene, FANC-F, was reported to be frequently methylated in ovarian cancer and to be associated with cisplatin resistance [92] but subsequent studies have reported a lower frequency [28,93] and no association with response to cisplatin [94]. The partner and localizer of BRCA2 (PALB2) gene [95] occasionally shows aberrant promoter hypermethylation associated with loss of expression in sporadic ovarian tumors mainly non-serous histologies. Ongoing genome-wide studies e.g. The Cancer Genome Atlas (TCGA), should reveal the methylation status of other BRCA/HR pathway genes in sporadic ovarian cancer.
MiRNA and Chemoresponse
MiRNAs have also been investigated as potential biomarkers of chemoresistance [96]. To identify miRNAs that are dysregulated in patients with resistance to platinum therapy, Eitan et al (2009) preformed microarray analysis of microRNA expression in patients with stage III ovarian tumors sensitive to platinum-based therapies compared to platinum-resistant patients. This study highlighted several miRNAs that were differentially expressed at a significant level including miR-27a, miR-378 and miR-23a [97]. In another study, decreased expression of let-7i was associated with shorter progression-free survival identifying let-7i as a candidate marker to detect patients who will recur sooner [98].
Epigenetics and Radioresponse
Tumor cells can also be resistant to radiation therapy. For head and neck cancer, radiation therapy is the standard adjuvant treatment. Aberrant methylation of negative regulators of the Ras/PI3K/AKT pathway has been investigated for association with outcome after radiation therapy for oral cancer [99].
Cancer stem cells have the ability to progress through the cell cycle to produce new tumor cells. It has been proposed that cancer stem cells are responsible for cancer recurrence [100]. Cancer stem cells, which by definition are epigenetic, have recently been shown to be more resistant to chemotherapy than other cells in the tumor and potentially more resistant to radiation therapy as well [101–105].
The association of miRNA expression levels and response to radiation therapy is an emerging area of research.
Recurrent Ovarian Tumors May Have Acquired Additional Epigenetic Alterations
The genotype of the cancer cell is dynamic. There is clonal selection for, and outgrowth of, a tumor cell with an acquired mutation that confers a growth advantage over other tumor cells. While a biopsy of a patient’s ovarian tumor obtained at initial diagnosis is available to type for methylation status, it is possible that at the time of recurrence after (first line) therapy, the epigenome of the dominant tumor cell clone has changed. A non-invasive analysis of gene methylation in the recurrent tumor might allow better stratification of therapy options at time of recurrence (Figure 1C). In this regard, further development and testing of a blood-based assay for gene hypermethylation that we previously reported [106] would be of interest.
Acquired resistance to platinum or PARP-inhibitors could arise from a defect in methylation maintenance leading to global demethylation or, more likely, clonal outgrowth of cells in a tumor that no longer have hypermethylation of BRCA1 for example. A solitary study of BRCA1 in the literature found that the methylation status (2 positive, 4 negative) was retained in 6 recurrent tumors after interim chemotherapy [25].
Epigenetic Therapy
Demethylating Agents
That epigenetic alterations are more plastic than genetic alterations, the availability of epigenetic modulating drugs and that these drugs have previously been used for cytotoxic therapy in the clinic together have led to consideration and testing of epigenetic therapy for cancer. The demethylating drugs azacitidine and decitabine have been used as cytotoxic agents but demonstrated limited efficacy in clinical trials though with a likely sub-optimal regimen. More recently, a strategy of a low dose of azacytidine, better tolerated by patients, to increase sensitivity or resensitize (delay or reverse resistance) to standard therapy has been advocated [107–109]. The sensitivity would presumably derive from reactivation of genes epigenetically silenced in the tumor cells (but not surrounding normal tissue). Few genes have been implicated in the chemoresponse of ovarian cancer, although genes functioning in DNA damage response pathways are likely candidates given the mode of action of platinum therapy. Pre-clinical studies have examined the mismatch repair gene MLH1 and the RASSF1A gene implicated in chemosensitivity by a putative role in the control of microtubule stability [110,111].
Clinical trials testing efficacy of demethylating agents exist for several types of leukemia and exhibited promising results. These studies are reviewed in [107]. In a phase 1b–2a clinical study, ovarian cancer patients with resistance to carboplatinum therapy received azacitidine and a partial reversal of platinum resistance was reported [112]. A phase 1 trial to assess the ability of decitabine to reverse platinum sensitivity through global demethylation found that after 8 days of treatment the HOX1A and BRCA1 genes were unmethylated in plasma [113]. While there is no direct evidence, it is conceivable that reactivation of tumor suppressor genes with functions outside of DNA repair might also lead to growth inhibition of tumor cells. As the ovarian cancer cell methylome is further elucidated it should become clearer which key genes in ovarian tumorigenesis are under epigenetic control and their biological function will provide a better rationale for the potential of epigenetic therapy.
It has been argued that through the induction of genome-wide hypomethylation, demethylating drug treatment could potentially have detrimental effects for a cancer patient. For example, reactivation of viral elements integrated into human DNA has been postulated although this may not be possible since the DNA sequence will likely have accumulated mutation over millennia. The biological effects of a 2-fold increase in gene dosage of imprinted genes might not be a major issue as many imprinted genes code for proteins with function in fetal development [114]. Global hypomethylation has also been associated with chromosomal instability (CIN) although it may already exist in the advanced stage ovarian tumor that would potentially receive demethylating treatment. Another issue is that, as discussed above, aberrant methylation of some genes may confer an increased response to chemotherapy for example, ovarian tumors with BRCA1, GSTP1 or MGMT methylation [28]. Since the primary ovarian tumor biopsy would likely be available from surgery, ovarian cancer patients could be typed for methylation of BRCA1, GSTP1 or MGMT or other “beneficial” methylation and stratified for suitability to demethylating drug therapy. Perhaps of more immediate relevance are the more practical issues of delivery and half-life of epigenetic drugs as well as evidence of rapid remethylation after the end of treatment [24,107].
Histone Deacetylating Agents
Acetylation and deacetylation of histones occurs by histone acetylatransferases (HATs) and deacetylases (HDACs). Acetylation of the lysine residue causes DNA to become more relaxed and transcription factor binding competent. Conversely, deacetylation compacts chromatin structure and results in gene silencing [115,116]. Acetylation of H2A induces a conformational change of the nucleosome and acts as a gene regulation switch [117]. Laboratory-based studies of HDAC inhibitors alone and in combination with demethylating or other agents have demonstrated anti-tumor effects and reactivation of cancer genes. Although, some genes can be reactivated in cultured cells by treatment with an HDAC inhibitor alone e.g. p21, typically, histone deacetylation is involved with DNA methylation for transcriptional regulation at gene promoters. Clinical trials support a method of treating patients with combinations of demethylation agents or DNMT inhibitors followed by or in combination with HDAC inhibitors [118]. Experience with HDAC inhibitors and ovarian cancer in the clinic is reviewed by [119].
MiRNA as Targets of Therapy
The deregulation of miRNA expression in tumor cells makes miRNAs potential therapeutic targets [7,8,34,120,121]. Over-expression of miRNAs that act as oncogenes can be targeted for down-regulation by anti-miRNA oligonucleotides, miRNA masking, miRNA sponges or small molecule inhibitors. Anti-miRNA oligonucleotides are complementary sequences to miRNA and block the miRNA’s ability to interact with its target mRNA transcript [122,123]. The downside of this approach is that since an individual miRNA is involved in post-transcriptional control of many different genes and pathways, there will likely be significant off-target effects. However, miRNA masking blocks the miRNA-mRNA interaction through complementation but with more specificity since the complement is to the mRNA of interest [124]. This approach depends on the ability to develop a sequence within the miRNA binding site that is unique. A miRNA sponge contains several binding sites and is capable of blocking many miRNAs at a time [125]. Small molecule inhibitors of miRNAs are also being investigated, such as azobenzene which was identified as a mir-21 blocking agent [126]. MiRNA technology is being tested for efficacy in treating leukemia, prostate cancer, and skin cancer and information on these clinical trials is reviewed in [127]. Restoring the activity of tumor suppressor miRNAs can lead to inhibition of tumor growth and apoptosis. Here miRNA mimics could be administered to restore miRNA activity. Several strategies for delivery of RNAi therapeutics are under development [72,128].
Conclusions
The plasticity, the identification of epigenetic alterations in the earliest known stages of cancer, and available sensitive technology provide a compelling rationale for the utility of epigenetic alterations for risk over lifetime and for early detection. The availability of tumor tissue at time of biopsy or surgery allows for wider study of epigenetic alterations and is not subject to the sampling issues inherent to risk and early detection studies. Therefore, prognostic indexes and prediction of response to therapy may have the most immediate traction in translational epigenetic research. Epigenetic alterations identified at time of biopsy could be used for monitoring of tumor burden, screening for recurrence, and assessment of efficacy of therapy. Plasticity again, and the availability of epigenetic modulating drugs have led to early studies of epigenetic therapy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–59. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 2.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 3.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 4.Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10:457–69. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–31. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 6.Li J, et al. Comparison of miRNA expression patterns using total RNA extracted from matched samples of formalin-fixed paraffin-embedded (FFPE) cells and snap frozen cells. BMC Biotechnol. 2007;7:36. doi: 10.1186/1472-6750-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu J, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 8.Calin GA, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353:1793–801. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- 9.Bast RC, Jr, et al. Prevention and early detection of ovarian cancer: mission impossible? Recent Results Cancer Res. 2007;174:91–100. doi: 10.1007/978-3-540-37696-5_9. [DOI] [PubMed] [Google Scholar]
- 10.Schorge JO, Modesitt SC, Coleman RL, Cohn DE, Kauff ND, Duska LR, Herzog TJ. SGO White Paper on ovarian cancer: etiology, screening and surveillance. Gynecol Oncol. 2010;119:7–17. doi: 10.1016/j.ygyno.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 11.Dubeau L. The cell of origin of ovarian epithelial tumours. Lancet Oncol. 2008;9:1191–7. doi: 10.1016/S1470-2045(08)70308-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurman RJ, McConnell TG. Characterization and comparison of precursors of ovarian and endometrial carcinoma: parts I and II. Int J Surg Pathol. 2010;18:181S–189S. doi: 10.1177/1066896910370881. [DOI] [PubMed] [Google Scholar]
- 13.Kurman RJ, Shih Ie M. The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am J Surg Pathol. 2010;34:433–43. doi: 10.1097/PAS.0b013e3181cf3d79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Issa JP. Cancer prevention: epigenetics steps up to the plate. Cancer Prev Res (Phila) 2008;1:219–22. doi: 10.1158/1940-6207.CAPR-08-0029. [DOI] [PubMed] [Google Scholar]
- 15.Kopelovich L, Crowell JA, Fay JR. The epigenome as a target for cancer chemoprevention. J Natl Cancer Inst. 2003;95:1747–57. doi: 10.1093/jnci/dig109. [DOI] [PubMed] [Google Scholar]
- 16.Laird PW, Jackson-Grusby L, Fazeli A, Dickinson SL, Jung WE, Li E, Weinberg RA, Jaenisch R. Suppression of intestinal neoplasia by DNA hypomethylation. Cell. 1995;81:197–205. doi: 10.1016/0092-8674(95)90329-1. [DOI] [PubMed] [Google Scholar]
- 17.Eads CA, Nickel AE, Laird PW. Complete genetic suppression of polyp formation and reduction of CpG-island hypermethylation in Apc(Min/+) Dnmt1-hypomorphic Mice. Cancer Res. 2002;62:1296–9. [PubMed] [Google Scholar]
- 18.Belinsky SA, Klinge DM, Stidley CA, Issa JP, Herman JG, March TH, Baylin SB. Inhibition of DNA methylation and histone deacetylation prevents murine lung cancer. Cancer Res. 2003;63:7089–93. [PubMed] [Google Scholar]
- 19.Yoo EJ, Park SY, Cho NY, Kim N, Lee HS, Kang GH. Helicobacter pylori-infection-associated CpG island hypermethylation in the stomach and its possible association with polycomb repressive marks. Virchows Arch. 2008;452:515–24. doi: 10.1007/s00428-008-0596-7. [DOI] [PubMed] [Google Scholar]
- 20.Clarke-Pearson DL. Clinical practice. Screening for ovarian cancer. N Engl J Med. 2009;361:170–7. doi: 10.1056/NEJMcp0901926. [DOI] [PubMed] [Google Scholar]
- 21.Cairns P, Sidransky D. Molecular methods for the diagnosis of cancer. Biochim Biophys Acta. 1999;1423:C11–8. doi: 10.1016/s0304-419x(99)00006-2. [DOI] [PubMed] [Google Scholar]
- 22.Sidransky D. Emerging molecular markers of cancer. Nat Rev Cancer. 2002;2:210–9. doi: 10.1038/nrc755. [DOI] [PubMed] [Google Scholar]
- 23.Ibanez L, Valls C, Cabre S, De Zegher F. Flutamide-metformin plus ethinylestradiol-drospirenone for lipolysis and antiatherogenesis in young women with ovarian hyperandrogenism: the key role of early, low-dose flutamide. J Clin Endocrinol Metab. 2004;89:4716–20. doi: 10.1210/jc.2004-0047. [DOI] [PubMed] [Google Scholar]
- 24.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baldwin RL, Nemeth E, Tran H, Shvartsman H, Cass I, Narod S, Karlan BY. BRCA1 promoter region hypermethylation in ovarian carcinoma: a population-based study. Cancer Res. 2000;60:5329–33. [PubMed] [Google Scholar]
- 26.Catteau A, Harris WH, Xu CF, Solomon E. Methylation of the BRCA1 promoter region in sporadic breast and ovarian cancer: correlation with disease characteristics. Oncogene. 1999;18:1957–65. doi: 10.1038/sj.onc.1202509. [DOI] [PubMed] [Google Scholar]
- 27.Sellar GC, et al. OPCML at 11q25 is epigenetically inactivated and has tumor-suppressor function in epithelial ovarian cancer. Nat Genet. 2003;34:337–43. doi: 10.1038/ng1183. [DOI] [PubMed] [Google Scholar]
- 28.Teodoridis JM, et al. CpG island methylation of DNA damage response genes in advanced ovarian cancer. Cancer Res. 2005;65:8961–7. doi: 10.1158/0008-5472.CAN-05-1187. [DOI] [PubMed] [Google Scholar]
- 29.Esteller M, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000;92:564–9. doi: 10.1093/jnci/92.7.564. [DOI] [PubMed] [Google Scholar]
- 30.McCluskey LL, Chen C, Delgadillo E, Felix JC, Muderspach LI, Dubeau L. Differences in p16 gene methylation and expression in benign and malignant ovarian tumors. Gynecol Oncol. 1999;72:87–92. doi: 10.1006/gyno.1998.5235. [DOI] [PubMed] [Google Scholar]
- 31.Rathi A, et al. Methylation profiles of sporadic ovarian tumors and nonmalignant ovaries from high-risk women. Clin Cancer Res. 2002;8:3324–31. [PubMed] [Google Scholar]
- 32.Strathdee G, Appleton K, Illand M, Millan DW, Sargent J, Paul J, Brown R. Primary ovarian carcinomas display multiple methylator phenotypes involving known tumor suppressor genes. Am J Pathol. 2001;158:1121–7. doi: 10.1016/S0002-9440(10)64059-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cairns P. Gene methylation and early detection of genitourinary cancer: the road ahead. Nat Rev Cancer. 2007;7:531–43. doi: 10.1038/nrc2170. [DOI] [PubMed] [Google Scholar]
- 34.Mitchell PS, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105:10513–8. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nelson PT, Baldwin DA, Kloosterman WP, Kauppinen S, Plasterk RH, Mourelatos Z. RAKE and LNA-ISH reveal microRNA expression and localization in archival human brain. RNA. 2006;12:187–91. doi: 10.1261/rna.2258506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xi Y, Nakajima G, Gavin E, Morris CG, Kudo K, Hayashi K, Ju J. Systematic analysis of microRNA expression of RNA extracted from fresh frozen and formalin-fixed paraffin-embedded samples. RNA. 2007;13:1668–74. doi: 10.1261/rna.642907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Resnick KE, Alder H, Hagan JP, Richardson DL, Croce CM, Cohn DE. The detection of differentially expressed microRNAs from the serum of ovarian cancer patients using a novel real-time PCR platform. Gynecol Oncol. 2009;112:55–9. doi: 10.1016/j.ygyno.2008.08.036. [DOI] [PubMed] [Google Scholar]
- 38.Jiang Q, et al. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009;37:D98–104. doi: 10.1093/nar/gkn714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nam EJ, Yoon H, Kim SW, Kim H, Kim YT, Kim JH, Kim JW, Kim S. MicroRNA expression profiles in serous ovarian carcinoma. Clin Cancer Res. 2008;14:2690–5. doi: 10.1158/1078-0432.CCR-07-1731. [DOI] [PubMed] [Google Scholar]
- 40.Hausler SF, et al. Whole blood-derived miRNA profiles as potential new tools for ovarian cancer screening. Br J Cancer. 2010;103:693–700. doi: 10.1038/sj.bjc.6605833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saito Y, Jones PA. Epigenetic activation of tumor suppressor microRNAs in human cancer cells. Cell Cycle. 2006;5:2220–2. doi: 10.4161/cc.5.19.3340. [DOI] [PubMed] [Google Scholar]
- 42.Brueckner B, Stresemann C, Kuner R, Mund C, Musch T, Meister M, Sultmann H, Lyko F. The human let-7a-3 locus contains an epigenetically regulated microRNA gene with oncogenic function. Cancer Res. 2007;67:1419–23. doi: 10.1158/0008-5472.CAN-06-4074. [DOI] [PubMed] [Google Scholar]
- 43.Lujambio A, et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007;67:1424–9. doi: 10.1158/0008-5472.CAN-06-4218. [DOI] [PubMed] [Google Scholar]
- 44.Heneghan HM, Miller N, Kerin MJ. Circulating miRNA signatures: promising prognostic tools for cancer. J Clin Oncol. 2010;28:e573–4. doi: 10.1200/JCO.2010.29.8901. author reply e575–6. [DOI] [PubMed] [Google Scholar]
- 45.Lawrie CH, et al. Detection of elevated levels of tumour-associated microRNAs in serum of patients with diffuse large B-cell lymphoma. Br J Haematol. 2008;141:672–5. doi: 10.1111/j.1365-2141.2008.07077.x. [DOI] [PubMed] [Google Scholar]
- 46.Brase JC, Wuttig D, Kuner R, Sultmann H. Serum microRNAs as non-invasive biomarkers for cancer. Mol Cancer. 2010;9:306. doi: 10.1186/1476-4598-9-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Skog J, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10:1470–6. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Balaj L, Lessard R, Dai L, Cho YJ, Pomeroy SL, Breakefield XO, Skog J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat Commun. 2011;2:180. doi: 10.1038/ncomms1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sargent DJ, Conley BA, Allegra C, Collette L. Clinical trial designs for predictive marker validation in cancer treatment trials. J Clin Oncol. 2005;23:2020–7. doi: 10.1200/JCO.2005.01.112. [DOI] [PubMed] [Google Scholar]
- 50.Bristow RE, Tomacruz RS, Armstrong DK, Trimble EL, Montz FJ. Survival effect of maximal cytoreductive surgery for advanced ovarian carcinoma during the platinum era: a meta-analysis. J Clin Oncol. 2002;20:1248–59. doi: 10.1200/JCO.2002.20.5.1248. [DOI] [PubMed] [Google Scholar]
- 51.Winter WE, 3rd, et al. Prognostic factors for stage III epithelial ovarian cancer: a Gynecologic Oncology Group Study. J Clin Oncol. 2007;25:3621–7. doi: 10.1200/JCO.2006.10.2517. [DOI] [PubMed] [Google Scholar]
- 52.Wiley A, Katsaros D, Fracchioli S, Yu H. Methylation of the insulin-like growth factor binding protein-3 gene and prognosis of epithelial ovarian cancer. Int J Gynecol Cancer. 2006;16:210–8. doi: 10.1111/j.1525-1438.2006.00299.x. [DOI] [PubMed] [Google Scholar]
- 53.Wiley A, et al. Aberrant promoter methylation of multiple genes in malignant ovarian tumors and in ovarian tumors with low malignant potential. Cancer. 2006;107:299–308. doi: 10.1002/cncr.21992. [DOI] [PubMed] [Google Scholar]
- 54.Widschwendter M, et al. DNA hypomethylation and ovarian cancer biology. Cancer Res. 2004;64:4472–80. doi: 10.1158/0008-5472.CAN-04-0238. [DOI] [PubMed] [Google Scholar]
- 55.Chan MW, et al. Hypermethylation of 18S and 28S ribosomal DNAs predicts progression-free survival in patients with ovarian cancer. Clin Cancer Res. 2005;11:7376–83. doi: 10.1158/1078-0432.CCR-05-1100. [DOI] [PubMed] [Google Scholar]
- 56.Huang TH, Perry MR, Laux DE. Methylation profiling of CpG islands in human breast cancer cells. Hum Mol Genet. 1999;8:459–70. doi: 10.1093/hmg/8.3.459. [DOI] [PubMed] [Google Scholar]
- 57.Wei SH, et al. Prognostic DNA methylation biomarkers in ovarian cancer. Clin Cancer Res. 2006;12:2788–94. doi: 10.1158/1078-0432.CCR-05-1551. [DOI] [PubMed] [Google Scholar]
- 58.Wei SH, et al. Methylation microarray analysis of late-stage ovarian carcinomas distinguishes progression-free survival in patients and identifies candidate epigenetic markers. Clin Cancer Res. 2002;8:2246–52. [PubMed] [Google Scholar]
- 59.Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 60.Klose RJ, Zhang Y. Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol. 2007;8:307–18. doi: 10.1038/nrm2143. [DOI] [PubMed] [Google Scholar]
- 61.Kwon MJ, et al. Derepression of CLDN3 and CLDN4 during ovarian tumorigenesis is associated with loss of repressive histone modifications. Carcinogenesis. 2010;31:974–83. doi: 10.1093/carcin/bgp336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Choi YL, et al. Expression profile of tight junction protein claudin 3 and claudin 4 in ovarian serous adenocarcinoma with prognostic correlation. Histol Histopathol. 2007;22:1185–95. doi: 10.14670/HH-22.1185. [DOI] [PubMed] [Google Scholar]
- 63.Morin PJ. Claudin proteins in human cancer: promising new targets for diagnosis and therapy. Cancer Res. 2005;65:9603–6. doi: 10.1158/0008-5472.CAN-05-2782. [DOI] [PubMed] [Google Scholar]
- 64.Hu S, Yu L, Li Z, Shen Y, Wang J, Cai J, Xiao L, Wang Z. Overexpression of EZH2 contributes to acquired cisplatin resistance in ovarian cancer cells in vitro and in vivo. Cancer Biol Ther. 2010:10. doi: 10.4161/cbt.10.8.12913. [DOI] [PubMed] [Google Scholar]
- 65.Dahiya N, et al. MicroRNA expression and identification of putative miRNA targets in ovarian cancer. PLoS One. 2008;3:e2436. doi: 10.1371/journal.pone.0002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wyman SK, et al. Repertoire of microRNAs in epithelial ovarian cancer as determined by next generation sequencing of small RNA cDNA libraries. PLoS One. 2009;4:e5311. doi: 10.1371/journal.pone.0005311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Larson PS, et al. CDKN1C/p57kip2 is a candidate tumor suppressor gene in human breast cancer. BMC Cancer. 2008;8:68. doi: 10.1186/1471-2407-8-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Medina R, Zaidi SK, Liu CG, Stein JL, van Wijnen AJ, Croce CM, Stein GS. MicroRNAs 221 and 222 bypass quiescence and compromise cell survival. Cancer Res. 2008;68:2773–80. doi: 10.1158/0008-5472.CAN-07-6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wurz K, Garcia RL, Goff BA, Mitchell PS, Lee JH, Tewari M, Swisher EM. MiR-221 and MiR-222 alterations in sporadic ovarian carcinoma: Relationship to CDKN1B, CDKNIC and overall survival. Genes Chromosomes Cancer. 2010;49:577–84. doi: 10.1002/gcc.20768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet. 2004;5:396–400. doi: 10.1038/nrg1328. [DOI] [PubMed] [Google Scholar]
- 71.Merritt WM, Bar-Eli M, Sood AK. The dicey role of Dicer: implications for RNAi therapy. Cancer Res. 2010;70:2571–4. doi: 10.1158/0008-5472.CAN-09-2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Merritt WM, et al. Dicer, Drosha, and outcomes in patients with ovarian cancer. N Engl J Med. 2008;359:2641–50. doi: 10.1056/NEJMoa0803785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Faggad A, et al. Prognostic significance of Dicer expression in ovarian cancer-link to global microRNA changes and oestrogen receptor expression. J Pathol. 2010;220:382–91. doi: 10.1002/path.2658. [DOI] [PubMed] [Google Scholar]
- 74.Chiosea S, Jelezcova E, Chandran U, Acquafondata M, McHale T, Sobol RW, Dhir R. Up-regulation of dicer, a component of the MicroRNA machinery, in prostate adenocarcinoma. Am J Pathol. 2006;169:1812–20. doi: 10.2353/ajpath.2006.060480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sugito N, et al. RNASEN regulates cell proliferation and affects survival in esophageal cancer patients. Clin Cancer Res. 2006;12:7322–8. doi: 10.1158/1078-0432.CCR-06-0515. [DOI] [PubMed] [Google Scholar]
- 76.Wood LD, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–13. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 77.Cooke SL, et al. Genomic analysis of genetic heterogeneity and evolution in high-grade serous ovarian carcinoma. Oncogene. 2010;29:4905–13. doi: 10.1038/onc.2010.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Baselga J, et al. Phase II study of weekly intravenous recombinant humanized anti-p185HER2 monoclonal antibody in patients with HER2/neu-overexpressing metastatic breast cancer. J Clin Oncol. 1996;14:737–44. doi: 10.1200/JCO.1996.14.3.737. [DOI] [PubMed] [Google Scholar]
- 79.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–6. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 80.Paez JG, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 81.Boyd J, et al. Clinicopathologic features of BRCA-linked and sporadic ovarian cancer. JAMA. 2000;283:2260–5. doi: 10.1001/jama.283.17.2260. [DOI] [PubMed] [Google Scholar]
- 82.Cass I, Baldwin RL, Varkey T, Moslehi R, Narod SA, Karlan BY. Improved survival in women with BRCA-associated ovarian carcinoma. Cancer. 2003;97:2187–95. doi: 10.1002/cncr.11310. [DOI] [PubMed] [Google Scholar]
- 83.Fong PC, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 84.Boettcher M, Kischkel F, Hoheisel JD. High-definition DNA methylation profiles from breast and ovarian carcinoma cell lines with differing doxorubicin resistance. PLoS One. 2010;5:e11002. doi: 10.1371/journal.pone.0011002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Farmer H, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 86.Hegi ME, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 87.Potapova A, Hoffman AM, Godwin AK, Al-Saleem T, Cairns P. Promoter hypermethylation of the PALB2 susceptibility gene in inherited and sporadic breast and ovarian cancer. Cancer Res. 2008;68:998–1002. doi: 10.1158/0008-5472.CAN-07-2418. [DOI] [PubMed] [Google Scholar]
- 88.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4:814–9. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 89.Collins N, Wooster R, Stratton MR. Absence of methylation of CpG dinucleotides within the promoter of the breast cancer susceptibility gene BRCA2 in normal tissues and in breast and ovarian cancers. Br J Cancer. 1997;76:1150–6. doi: 10.1038/bjc.1997.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gras E, Cortes J, Diez O, Alonso C, Matias-Guiu X, Baiget M, Prat J. Loss of heterozygosity on chromosome 13q12-q14, BRCA-2 mutations and lack of BRCA-2 promoter hypermethylation in sporadic epithelial ovarian tumors. Cancer. 2001;92:787–95. doi: 10.1002/1097-0142(20010815)92:4<787::aid-cncr1384>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 91.Hilton JL, Geisler JP, Rathe JA, Hattermann-Zogg MA, DeYoung B, Buller RE. Inactivation of BRCA1 and BRCA2 in ovarian cancer. J Natl Cancer Inst. 2002;94:1396–406. doi: 10.1093/jnci/94.18.1396. [DOI] [PubMed] [Google Scholar]
- 92.Taniguchi T, Tischkowitz M, Ameziane N, Hodgson SV, Mathew CG, Joenje H, Mok SC, D’Andrea AD. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med. 2003;9:568–74. doi: 10.1038/nm852. [DOI] [PubMed] [Google Scholar]
- 93.Tokunaga E, et al. Low incidence of methylation of the promoter region of the FANCF gene in Japanese primary breast cancer. Breast Cancer. 2009 doi: 10.1007/s12282-009-0175-z. [DOI] [PubMed] [Google Scholar]
- 94.Lim SL, et al. Promoter hypermethylation of FANCF and outcome in advanced ovarian cancer. Br J Cancer. 2008;98:1452–6. doi: 10.1038/sj.bjc.6604325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Xia B, et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. 2006;22:719–29. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 96.van Jaarsveld MT, Helleman J, Berns EM, Wiemer EA. MicroRNAs in ovarian cancer biology and therapy resistance. Int J Biochem Cell Biol. 2010;42:1282–90. doi: 10.1016/j.biocel.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 97.Eitan R, et al. Tumor microRNA expression patterns associated with resistance to platinum based chemotherapy and survival in ovarian cancer patients. Gynecol Oncol. 2009;114:253–9. doi: 10.1016/j.ygyno.2009.04.024. [DOI] [PubMed] [Google Scholar]
- 98.Yang N, et al. MicroRNA microarray identifies Let-7i as a novel biomarker and therapeutic target in human epithelial ovarian cancer. Cancer Res. 2008;68:10307–14. doi: 10.1158/0008-5472.CAN-08-1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Huang KH, Huang SF, Chen IH, Liao CT, Wang HM, Hsieh LL. Methylation of RASSF1A, RASSF2A, and HIN-1 is associated with poor outcome after radiotherapy, but not surgery, in oral squamous cell carcinoma. Clin Cancer Res. 2009;15:4174–80. doi: 10.1158/1078-0432.CCR-08-2929. [DOI] [PubMed] [Google Scholar]
- 100.Clarke MF, et al. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–44. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 101.Eramo A, et al. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006;13:1238–41. doi: 10.1038/sj.cdd.4401872. [DOI] [PubMed] [Google Scholar]
- 102.Ghods AJ, et al. Spheres isolated from 9L gliosarcoma rat cell line possess chemoresistant and aggressive cancer stem-like cells. Stem Cells. 2007;25:1645–53. doi: 10.1634/stemcells.2006-0624. [DOI] [PubMed] [Google Scholar]
- 103.Kang MK, Kang SK. Tumorigenesis of chemotherapeutic drug-resistant cancer stem-like cells in brain glioma. Stem Cells Dev. 2007;16:837–47. doi: 10.1089/scd.2007.0006. [DOI] [PubMed] [Google Scholar]
- 104.Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008;27:1749–58. doi: 10.1038/sj.onc.1210811. [DOI] [PubMed] [Google Scholar]
- 105.Mimeault M, Hauke R, Batra SK. Recent advances on the molecular mechanisms involved in the drug resistance of cancer cells and novel targeting therapies. Clin Pharmacol Ther. 2008;83:673–91. doi: 10.1038/sj.clpt.6100296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ibanez de Caceres I, Cairns P. Methylated DNA sequences for early cancer detection, molecular classification and chemotherapy response prediction. Clin Transl Oncol. 2007;9:429–37. doi: 10.1007/s12094-007-0081-9. [DOI] [PubMed] [Google Scholar]
- 107.Issa JP, Kantarjian HM. Targeting DNA methylation. Clin Cancer Res. 2009;15:3938–46. doi: 10.1158/1078-0432.CCR-08-2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Silverman LR, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002;20:2429–40. doi: 10.1200/JCO.2002.04.117. [DOI] [PubMed] [Google Scholar]
- 109.Wijermans P, Lubbert M, Verhoef G, Bosly A, Ravoet C, Andre M, Ferrant A. Low-dose 5-aza-2′-deoxycytidine, a DNA hypomethylating agent, for the treatment of high-risk myelodysplastic syndrome: a multicenter phase II study in elderly patients. J Clin Oncol. 2000;18:956–62. doi: 10.1200/JCO.2000.18.5.956. [DOI] [PubMed] [Google Scholar]
- 110.Glasspool RM, Teodoridis JM, Brown R. Epigenetics as a mechanism driving polygenic clinical drug resistance. Br J Cancer. 2006;94:1087–92. doi: 10.1038/sj.bjc.6603024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Agathanggelou A, Cooper WN, Latif F. Role of the Ras-association domain family 1 tumor suppressor gene in human cancers. Cancer Res. 2005;65:3497–508. doi: 10.1158/0008-5472.CAN-04-4088. [DOI] [PubMed] [Google Scholar]
- 112.Fu S, et al. Phase 1b-2a study to reverse platinum resistance through use of a hypomethylating agent, azacitidine, in patients with platinum-resistant or platinum-refractory epithelial ovarian cancer. Cancer. 2010 doi: 10.1002/cncr.25701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fang F, et al. A phase 1 and pharmacodynamic study of decitabine in combination with carboplatin in patients with recurrent, platinum-resistant, epithelial ovarian cancer. Cancer. 2010;116:4043–53. doi: 10.1002/cncr.25204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Frost JM, Moore GE. The importance of imprinting in the human placenta. PLoS Genet. 2010;6:e1001015. doi: 10.1371/journal.pgen.1001015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- 117.Ishibashi T, Dryhurst D, Rose KL, Shabanowitz J, Hunt DF, Ausio J. Acetylation of vertebrate H2A.Z and its effect on the structure of the nucleosome. Biochemistry. 2009;48:5007–17. doi: 10.1021/bi900196c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gore SD, et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res. 2006;66:6361–9. doi: 10.1158/0008-5472.CAN-06-0080. [DOI] [PubMed] [Google Scholar]
- 119.Matei DE, Nephew KP. Epigenetic therapies for chemoresensitization of epithelial ovarian cancer. Gynecol Oncol. 2010;116:195–201. doi: 10.1016/j.ygyno.2009.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Volinia S, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–61. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yanaihara N, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9:189–98. doi: 10.1016/j.ccr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 122.Weiler J, Hunziker J, Hall J. Anti-miRNA oligonucleotides (AMOs): ammunition to target miRNAs implicated in human disease? Gene Ther. 2006;13:496–502. doi: 10.1038/sj.gt.3302654. [DOI] [PubMed] [Google Scholar]
- 123.Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov. 2010;9:775–89. doi: 10.1038/nrd3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xiao J, Yang B, Lin H, Lu Y, Luo X, Wang Z. Novel approaches for gene-specific interference via manipulating actions of microRNAs: examination on the pacemaker channel genes HCN2 and HCN4. J Cell Physiol. 2007;212:285–92. doi: 10.1002/jcp.21062. [DOI] [PubMed] [Google Scholar]
- 125.Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4:721–6. doi: 10.1038/nmeth1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gumireddy K, Young DD, Xiong X, Hogenesch JB, Huang Q, Deiters A. Small-molecule inhibitors of microrna miR-21 function. Angew Chem Int Ed Engl. 2008;47:7482–4. doi: 10.1002/anie.200801555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wahid F, Shehzad A, Khan T, Kim YY. MicroRNAs: synthesis, mechanism, function, and recent clinical trials. Biochim Biophys Acta. 2010;1803:1231–43. doi: 10.1016/j.bbamcr.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 128.Akinc A, et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat Biotechnol. 2008;26:561–9. doi: 10.1038/nbt1402. [DOI] [PMC free article] [PubMed] [Google Scholar]