

Abstract
In genome-wide association (GWA) studies (i.e., “GWAS”), hundreds of thousands of single nucleotide polymorphisms (SNPs) are tested for association with a disease trait. Typically, GWA studies give equal consideration to all SNPs tested, regardless of existing knowledge of a SNP’s functionality or biological plausibility of association. Because many tests are conducted, very low statistical significance thresholds (i.e., P<5×10−8) are required to identify true associations with confidence. By restricting GWA analyses to SNPs with enhanced prior probabilities of association, we can reduce the number of tests conducted and relax the required significance threshold, increasing power to detect association. In this analysis of existing GWA data on pancreatic cancer cases (n=1,736) and controls (n=1,802) of European descent (the PanScan study), we conduct a GWA scan restricted to SNPs that have been reported to associate with human phenotypes in prior GWA studies (with P<5×10−8). Using this method, we drastically reduce the number of tests conducted (from ~550,000 to 1,087) and test only SNPs that are known to be (or tag) variants that influence human biological processes. Of the 1,087 SNPs tested, the strongest association observed was for HNF1A SNP rs7310409 (P=3×10−5; PBonferroni=0.03), a SNP known to associate with circulating C-reactive protein. This association was replicated in an independent sample of 1,094 cases and 1,165 controls (P=0.02), producing a highly significant association in the combined datasets (P=2×10−6; PBonferroni=0.002). The HNF1A region also harbors variants that influence several human traits, including maturity-onset diabetes of the young, type 2 diabetes, LDL cholesterol, and N-glycan levels. This novel “pleiotropy scan” method may be useful for identifying susceptibility loci for other cancer phenotypes.
Keywords: pancreatic cancer, pleiotropy, HNF1A, diabetes, genome-wide association study (GWAS)
Introduction
Recently, genome-wide associations (GWA) studies have identified common single nucleotide polymorphisms (SNPs) in four genomic regions that are associated with pancreatic cancer risk (1, 2). These SNPs show associations with pancreatic cancer that are statistically significant at a “genome-wide” significance level (approximately P<5×10−8) (3). This stringent significance threshold accounts for the fact that many statistical tests are being conducted and ensures that the majority of the significant associations identified will be true positives. However, as a consequence of this threshold, variants with very weak associations may not be detectable, even in large GWA studies of pancreatic cancer, because the resulting P-values do not surpass the genome-wide significance threshold.
Reducing the number of tests conducted in an association study will relax the required significance threshold, thereby increasing statistical power to detect associations with pancreatic cancer for each SNP. One strategy for reducing the number of tests is to only test SNPs with heightened prior probabilities of association. The vast majority of the SNPs tested in a GWA study have no known consequences for human biology. However, recent GWA studies have identified thousands of loci that harbor variants that are robustly associated with human phenotypes (3, 4). By testing only these variants, we can perform a more hypothesis-based analysis, reduce number of tests conducted, relax the significance thresholds, and increase power.
In this analysis of existing GWA data on pancreatic cancer cases and controls, we conduct a GWA scan using only SNPs that have been reported to robustly associate with human phenotypes (other than pancreatic cancer) in prior GWA studies. Because these SNPs are markers of human biological processes, we argue that they are more likely, on average, to associate with pancreatic cancer risk than SNPs that have no known effects on human biology. We test these SNPs for association with pancreatic cancer risk using data on cases and controls of European descent participating in the PanScan-I GWA study. We attempt to replicate our findings in an independent dataset, the PanScan-II GWA study. By design, all observed associations will arise from SNPs that have implications for other human traits; thus, we call this approach a “pleiotropy scan”.
Materials and Methods
The Cancer Genetic Markers of Susceptibility (CGEMS) PanScan-I and PanScan-II GWA studies have been previously described (1, 2). Briefly, cases and controls were drawn from 12 cohort studies and eight case-control studies. All cases were diagnosed with primary adenocarcinoma of the exocrine pancreas. Controls were matched to cases based on birth year, sex, and race/ethnicity and were free of pancreatic cancer at the time of diagnosis of the matched case. Sample quality control and genotyping was conducted at the National Cancer Institute’s Core Genotyping Facility using Illumina HumanHap550 and HumanHap550-Duo SNP arrays (PanScan-I) and Illumina Human 610-Quad arrays (PanScan-II) (1, 2). In total, CGEMS provided high-quality genotype data for 1,895 cases and 1,937 controls from PanScan-I and for 1,478 cases and 1,534 controls from PanScan-II (after excluding duplicate samples). All data were downloaded from the database of Genotypes and Phenotypes (5).
This analysis was restricted to PanScan participants of European ancestry. We assessed population structure in both PanScan-I and PanScan-II using ~12,000 SNPs with low pair-wise linkage disequilibrium (r2<0.05 for any pair) and high call rates (<1% missing) in PanScan and HapMap3 founders (from CEU, YRI, and CHB+JPT datasets). Because PanScan-I and II contain a substantial number of individuals of non-European ancestry, the EIGENSTRAT principal components analysis (PCA) program was used to identify and exclude participants who did not cluster tightly with the CEU HapMap samples (253 in PanScan-I; 753 in PanScan-II). Based on identity-by-descent estimates, one individual from each suspected first- or second-degree relative pair was removed (14 in PanScan-I; 1 in PanScan-II). The resulting sample size for PanScan-I was 1,763 cases and 1,802 controls and PanScan-II had 1,094 cases and 1,165 controls. PCA was used to generate principal components of ancestry in the PanScan-I, PanScan-II, and combined datasets.
SNPs included in this “pleiotropy scan” were selected using the National Human Genome Research Institute’s (NHGRI) Catalog of Published GWA studies (6) which contains descriptive information on SNPs reported to associate with a human phenotype in a GWA study at a significance level of P <1.0×10−5. The catalog data was downloaded on December 20th, 2010, including SNP identifiers and P-values for the reported associations. The catalog contained 3,554 unique SNPs after excluding the four established pancreatic cancer susceptibility loci (rs9543325, rs3790844, rs401681, rs505922) and 7 SNPs in linkage disequilibrium (LD; r2>0.3) with either rs401681 (TERT region) or rs505922 (ABO region).
Approximately 57% of the 3,554 catalog SNPs (n=2,043) did not show prior evidence of association at a genome-wide significance level (P >5×10−8), and we excluded these SNPs to ensure that the vast majority of the SNPs in our analysis were truly associated with human traits. Of the remaining 1,511 catalog SNPs, 883 were present in the PanScan-I dataset. For the 628 SNPs not present, the GLIDERS program (7) was used to identify appropriate tagSNPs (r2 >0.9 in HapMap3 CEU). By incorporating an additional 211 tagSNPs into our dataset of 883 SNPs (1,094 SNPs total), we were able to tag 291 of the 628 catalog SNPs that were missing from the PanScan-I dataset (some tagSNPs tagged multiple catalog SNPs). The remaining 337 catalog SNPs were not included in this analysis; thus, we captured 78% of the 1,511 SNPs with reported P<5×10−8 in GWA studies. The 7 non-autosomal SNPs present in our dataset were excluded, resulting in a final dataset of 1,087 SNPs eligible for analysis. No individual was missing >5% of SNP data and no SNP was missing >5% of genotype data.
Our general analytic strategy was to test all 1,087 SNPs for association with pancreatic cancer in the larger PanScan-I dataset, attempt to replicate the top 10 hits in the PanScan-II dataset, and assess their overall statistical significance based on the combined PanScan-I and PanScan-II dataset. Logistic regression adjusted for five principal components, sex, and 10-year age groups (categorical) was used to generate odds ratios (ORs), 95% confidence intervals (CIs), and P-values for each of the 1,087 SNPs selected from the PanScan-I GWA dataset. All SNPs were coded as 0, 1, or 2 minor alleles (a log-additive model).
In addition to Bonferroni-corrected P-values, permutation-based P-values were calculated to obtain less-conservative, empirical significance measures that account for the LD structure of the SNPs in the GWA catalog. Permutation-based P-values were generated for the combined PanScan-I/II dataset by performing the pleiotropy scan on 10,000 datasets in which each subject’s phenotype data was randomly re-assigned to another subject’s genotype data. The test statistics observed in the original analysis were compared to the distribution of maximum test statistics from each simulated dataset to determine how often the observed P-value occurred by chance.
To characterize the statistically significant association signals, we examined associations for all SNPs in the region of interest, using all SNPs present in the combined PanScan datasets. Statistical analyses were performed using PLINK (8). Figures were generated using R (9), and LocusZoom (10).
Results
After PCA-based exclusions, PanScan-I had a slightly higher percentage of females (49%) than PanScan-II (46%). The age distributions categorized in <51, 51–60, 61–70, 74–80, and >80 age groups were 3%,14%, 39%, 37%, 7% for PanScan-I and 11%, 25%, 34%, 25%, and 4% for PanScan-II.
The vast majority of the p-values for the 1,087 SNPs included in the PanScan-I pleiotropy scan did not show systematic departure from the expected uniform distribution (lambda=1.01), suggesting that population stratification had been adequately accounted for using principal component methods. The ten most significantly associated SNPs from PanScan-I are shown in Table 1. Only rs7310409 showed a significant association with pancreatic cancer after Bonferroni correction (PBonferronni = 0.03). Two additional HNF1A SNPs, rs735396 and rs2650000, were ranked 3 and 5 among the top 10 associations and were in LD with lead SNP rs7310409 (r2= 0.77 and 0.55, respectively).
Table 1.
Association estimates for the ten most significant associations observed in PanScan pleiotropy scan
| SNP | Chrom. | Gene Region | MAF (MA) | PanScan I (1,763 cases, 1,802 controls)
|
PanScan II (1,094 cases, 1,165 controls)
|
PanScan I+II (2,857 cases, 2,967 controls)
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR | 95% CI | P | PBonferroni | OR | 95% CI | P | P | PBonf | Pperm | ||||
| rs7310409 | 12 | HNF1A | 0.40 (A) | 1.22 | 1.11–1.35 | 0.00003 | 0.03 | 1.16 | 1.02–1.30 | 0.02 | 2×10−6 | 0.002 | 0.0008 |
| rs3748816 | 1 | MMEL1 | 0.33 (C) | 1.20 | 1.09–1.33 | 0.0002 | 0.21 | 0.88 | 0.78–1.00 | 0.05 | 0.66 | 1.00 | 1.00 |
| rs735396 | 12 | HNF1A | 0.38 (G) | 1.19 | 1.08–1.31 | 0.0005 | 0.51 | 1.10 | 0.97–1.24 | 0.14 | 0.0003 | 0.33 | 0.07 |
| rs7202877 | 16 | Intergenic | 0.11 (G) | 1.28 | 1.11–1.48 | 0.0009 | 1.00 | 1.19 | 0.99–1.43 | 0.07 | 0.0003 | 0.32 | 0.19 |
| rs2650000 | 12 | HNF1A | 0.37 (T) | 1.17 | 1.06–1.28 | 0.002 | 1.00 | 1.15 | 1.02–1.30 | 0.03 | 0.0001 | 0.11 | 0.04 |
| rs2736990 | 4 | SNCA | 0.45 (C) | 0.86 | 0.79–0.95 | 0.002 | 1.00 | 0.99 | 0.88–1.11 | 0.86 | 0.01 | 1.00 | 1.00 |
| rs727957 | 21 | KCNE1 | 0.18 (T) | 1.20 | 1.07–0.36 | 0.003 | 1.00 | 1.10 | 0.94–1.28 | 0.23 | 0.001 | 1.00 | 0.95 |
| rs2126259 | 8 | PPP1R3B | 0.09 (A) | 0.79 | 0.67–0.92 | 0.003 | 1.00 | 0.92 | 0.75–1.14 | 0.46 | 0.005 | 1.00 | 0.98 |
| rs9989419 | 16 | CETP | 0.40 (A) | 0.87 | 0.79–0.96 | 0.004 | 1.00 | 1.00 | 0.89–1.13 | 0.99 | 0.03 | 1.00 | 1.00 |
| rs9368699 | 6 | SNORD52 | 0.04 (C) | 0.69 | 0.53–0.88 | 0.004 | 1.00 | 1.10 | 0.82–1.48 | 0.53 | 0.07 | 1.00 | 1.00 |
MAF, minor allele frequency; MA, minor allele; OR, odds ratio; 95% CI, 95% confidence interval
Interestingly, there were more SNPs contained in the low end of the P-value distribution than expected under null hypothesis of a uniform P-value distribution. In other words, the −log10(P-values) at the high end of the distribution were higher than expected according to the 95% confidence envelope of the Q-Q plot (Figure 1). After exclusion of SNPs in the HNF1A region, there was still suggestive evidence of departure from the null distribution, indicating additional trait-associated SNPs may have true associations with pancreatic cancer.
Figure 1. A quantile-quantile plot of −log10(P-values) for associations of 1,087 trait-associated SNPs with pancreatic cancer (PanScan-I data).
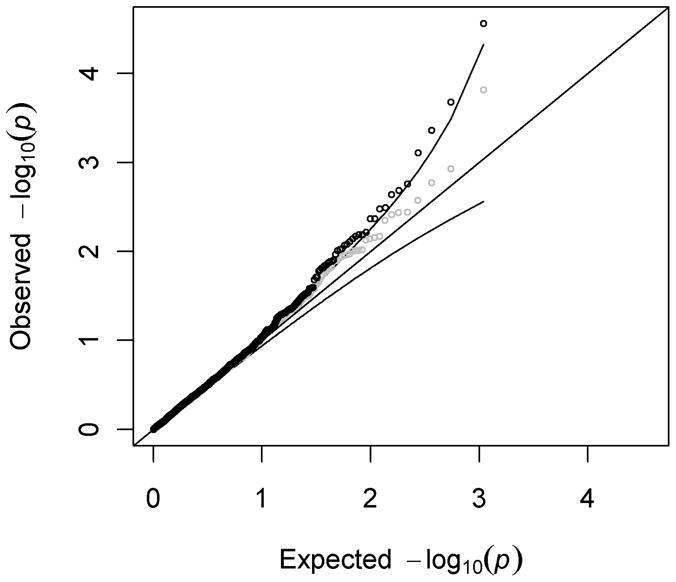
Data is shown both before (black) and after (grey) exclusion of all SNPs in the HNF1A region of association. The curved lines represent the 95% confidence envelope.
In the PanScan-II replication analysis of the ten most significant associations from PanScan-I, only the lead SNP from PanScan-I (HNF1A SNP rs7310409) and a correlated HNF1A SNP rs2650000 showed a nominally significant association with pancreatic cancer risk (P=0.02 and P=0.03, respectively). When the PanScan-I and PanScan-II datasets were combined, the association for rs7310409 reached a P-value 2×10−6 (PBonferroni = 0.002; Table 1). The other two HNF1A SNPs showing associations with pancreatic cancer (rs735396 and rs2650000) showed P-values of 0.0002 (PBonferroni = 0.22) and 0.0001 (PBonferroni = 0.11) in the combined dataset.
When all SNPs in the HNF1A region were analyzed in the combined PanScan-I and PanScan-II dataset, including all non-catalog SNPs included on the Illumina platforms, the strongest association was still rs7310409 (Figure 2A), with several nearby correlated SNPs showing weaker associations. When rs7310409 was included as a covariate in the logistic regression analyses, all associations arising from the HNF1A region were essentially eliminated (Figure 2B), indicating that there is most likely a singular association signal arising from the HNF1A region in this dataset.
Figure 2. Association results for all genotyped SNPs in the HNF1A region (PanScan I+II data).

P-values for each SNP are plotted both before (Panel A) and after (Panel B) adjusting each logistic regression for lead SNP rs7310409. Color coding of plotted P-values represents correlation with rs7310409 (in HapMap CEU population). This figure includes all genotyped SNPs, including SNPs not contained in the NHGRI’s GWA study catalog.
In the PanScanI+II combined dataset of 900 independent catalog SNPs (excluding NHF1A SNPs and SNPs with pair-wise r2>0.5 with other SNPs), 5.3% of SNPs had a P<0.05 and 1.6% of SNPs with P<0.01. These results provide evidence for mild enrichment of non-HNF1A catalog SNPs for association with pancreatic cancer.
Discussion
In this study we have tested 1,087 SNPs with known implications for human phenotypes for association with pancreatic cancer risk using data from a large two-stage GWA study of pancreatic cancer. By focusing on SNPs that represent genetic variation with effects on human biology, we have drastically reduced the number of tests typically conducted in a GWA study, while focusing on SNPs that are likely to have increased prior probabilities of association with human phenotypes, including pancreatic cancer. The 1,087 SNPs we interrogated were selected based on their strong evidence of association with human phenotypes in prior GWA studies.
We have identified an association signal in the HNF1A region. SNP rs7310409, an intronic SNP that lies <2kb upstream of exon 2 of the HNF1A gene, showed convincing evidence of association with pancreatic cancer in the PanScan-I study, even after a conservative Bonferroni correction for multiple testing. This finding was replicated in PanScan-II, producing combined Bonferroni P-value of 0.002. All associated SNPs in this region are in substantial LD with rs7310409, indicating that there is a singular association signal for pancreatic cancer in this region. Evidence for additional associations outside of the HNF1A regions was suggestive.
Several prior studies have considered integrating biological information, such as gene expression data (11), into GWA studies to improve gene-discovery. However, to our knowledge, this is the first study to leverage biological information drawn from prior GWA studies. A recent study demonstrated that trait-associated SNPs are more likely to be expression quantitative trait loci (eQTLs) than other SNPs on GWA platforms (12), suggesting that incorporating eQTL information into GWA studies may facilitate the discovery of new associations and provide a better understanding of disease mechanisms. In a similar fashion, our results suggest that SNPs identified in GWA studies may be more likely to show associations with other human traits than SNPs with no prior evidence of association. While this theory warrants further evaluation in independent datasets, there is emerging evidence for pleiotropy in the cancer GWA literature, as several susceptibility loci (e.g., 8q24 (13), TERT (14), CDKN2A/2B (15–17)) have been linked to multiple cancers and other complex traits.
HNF1A (12q24.31) codes for hepatocyte nuclear factor 1 homeobox A (TCF1), a transcription factor expressed in the human liver, pancreas, kidney, and gut (18). HNF1A is known to be a critical member of a regulatory transcription factor circuit in the developing and mature pancreas (19). Common variants in the HNF1A region have been implicated by GWA studies in a several human phenotypes, including circulating levels of the acute-phase inflammatory marker C-reactive protein (CRP) (20–22), liver enzyme gamma-glutamyl transferase (GGT) (23), LDL cholesterol (24), coronary heart disease (25), type 2 diabetes (26), and N-glycan levels in plasma (27) (Table 2). However, this is the first study to link HNF1A variants to risk for any type of cancer. While the lead SNP from this analysis (rs7310409) is in the NHGRI catalog due to its reported associations with CRP, based on HapMap3 CEU LD estimates for nearby SNPs, rs7310409 would also be expected to associate with GGT levels, LDL cholesterol, coronary heart disease, and N-glycan levels in European populations (Table 2).
Table 2.
A description of SNPs in the HNF1A region with associations reported in the NHGRI GWA study catalog
| SNP | r2 with rs7310409a | Associations reported in GWA catalog
|
PanScan I+II
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| Associated Trait | Risk (or increaser) Allele | Reported P-value | Ref. | Best tagSNP (r2) | OR | CI | P | ||
| rs7310409 | N/A | C-reactive protein | A | 7 × 10−17 | (22, 31) | SNP present | 1.19 | 1.11–1.19 | 2×10−6 |
| rs1169310 | 0.71 | C-reactive protein | A | 2×10−8 | (21) | rs735396 (1.00) | 1.15 | 1.07–1.24 | 0.0003 |
| rs1183910 | 0.64 | C-reactive protein | T | 1×10−30 | (20) | rs2650000 (0.88) | 0.16 | 1.07–1.25 | 0.0001 |
| rs1169313 | 0.64 | Gamma-glutamyl transferase | C | 2×10−10 | (23) | rs735396 (1.00) | 1.15 | 1.07–1.24 | 0.0003 |
| rs2650000 | 0.45 | LDL cholesterol | A | 2×10−8 | (24) | SNP present | 0.16 | 1.07–1.25 | 0.0001 |
| rs7957197 | 0.11 | Type 2 diabetes | T | 2×10−8 | (26) | rs7965349 (0.82) | 0.97 | 0.88–1.07 | 0.52 |
| rs2259816 | 0.64 | Coronary heart disease | T | 5×10−7 | (25) | rs735396 (1.00) | 1.15 | 1.07–1.24 | 0.0003 |
| rs7953249 | 0.79 | N-glycan levels | G | 2×10−8 | (27) | rs7310409 (0.89) | 1.19 | 1.11–1.19 | 2×10−6 |
| rs735396 | 0.64 | N-glycan levels | G | 4×10−8 | (27) | SNP present | 1.15 | 1.07–1.24 | 0.0003 |
All r values are derived from HapMap3 CEU data
Interestingly, the lead SNP rs7310409 is not strongly correlated with the type 2 diabetes risk variant rs7957197 (r2=0.11). Although rs7957197 is not present in our dataset, its best tagSNP, rs7965349 (r2=0.82), is not associated with pancreatic cancer (P=0.53, Table 2), suggesting that the diabetes and pancreatic cancer association signals are independent. Type 2 diabetes is a well-established risk factor for pancreatic cancer (28), although the potential causality of this association is not well understood. Rare variants HNF1A are also known to cause maturity-onset diabetes of the young (MODY) (29). It is also worth noting that in the combined PanScan-I+II dataset, the strongest non-HNF1A association observed was for type 1 diabetes-associated SNP rs7202877 (P=0.0003, PBonferroni=0.32) (30). Further investigation of this result is warranted.
Additional research is needed to identify the causal variant for pancreatic cancer in this region. Lead SNP rs7310409 is intronic and is in moderate LD with non-synonymous coding HNF1A SNPs rs1169288 (r2=0.62) and rs2464196 (r2=0.55) and HNF1A 5′ UTR SNPs rs1169310 (r2=0.71) and rs1169312 (r2=0.73). It is also possible that the causal variant is untyped, potentially with a low minor allele frequency. Additional genes within approximately 100kb of the HNF1A signal include C12orf27, C12org43, OASL, SPPL3, and P2RX7.
This study was limited by our inability to test all SNPs of interest for association with pancreatic cancer. A total of 337 out of 1,511 eligible SNPs in the GWA catalog (~22%) were neither present in our dataset nor strongly correlated with any SNP in our dataset. Thus, we may have missed additional regions with pleiotropic effects on pancreatic cancer risk. We also acknowledge that secondary analyses of GWA data have the potential to generate false positive associations. Thus, for secondary GWA analyses that integrate biological information, we stress the importance of clear hypotheses, careful treatment of multiple testing issues, and ideally, a strategy for replication. Multiple testing issues are particularly important when multiple hypotheses are evaluated and/or multiple analyses are carried out in relation to multiple traits.
In summary, this study has used a novel “pleiotropy scan” approach to identify HNF1A as a pancreatic cancer risk locus. In this GWA-based approach, we have interrogated only SNPs that have been shown in previous GWA studies to associate with human traits. This approach increases the statistical power of GWA approaches by reducing the number of tests conducted and focusing on SNPs with heightened prior probabilities of association with disease risk. The central findings of this study further emphasize the importance of variation in the HNF1A gene region in relation to pancreas function and related disease phenotypes, including type 2 diabetes, MODY, and pancreatic cancer.
Acknowledgments
Grant Support: This work was supported by National Institutes of Health [CA122171 and CA102484 to H.A.] and Department of Defense [W81XWH-10-1-0499 to B.P.]
The authors would like to thank all researchers and study participants for contributing to the PanScan study and making this genetic data available to the research community. We would also like to thank Lin Tong for her assistance preparing the datasets.
Footnotes
Conflicts of interest: None
References
- 1.Amundadottir L, Kraft P, Stolzenberg-Solomon RZ, Fuchs CS, Petersen GM, Arslan AA, et al. Genome-wide association study identifies variants in the ABO locus associated with susceptibility to pancreatic cancer. Nat Genet. 2009;41:986–90. doi: 10.1038/ng.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petersen GM, Amundadottir L, Fuchs CS, Kraft P, Stolzenberg-Solomon RZ, Jacobs KB, et al. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nat Genet. 2010;42:224–8. doi: 10.1038/ng.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JP, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–69. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 4.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106:9362–7. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mailman MD, Feolo M, Jin Y, Kimura M, Tryka K, Bagoutdinov R, et al. The NCBI dbGaP database of genotypes and phenotypes. Nat Genet. 2007;39:1181–6. doi: 10.1038/ng1007-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hindorff L, Junkins H, Hall P, Mehta J, Manolio T. [Accessed [December 20, 2010]];A Catalog of Published Genome-Wide Association Studies. Available at: www.genome.gov/gwastudies.
- 7.Lawrence R, Day-Williams AG, Mott R, Broxholme J, Cardon LR, Zeggini E. GLIDERS--a web-based search engine for genome-wide linkage disequilibrium between HapMap SNPs. BMC Bioinformatics. 2009;10:367. doi: 10.1186/1471-2105-10-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundataion for Statistical Computing; 2010. [Google Scholar]
- 10.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. LocusZoom: Regional visualization of genome-wide association scan results. Bioinformatics. 2010 doi: 10.1093/bioinformatics/btq419. (In Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhong H, Yang X, Kaplan LM, Molony C, Schadt EE. Integrating pathway analysis and genetics of gene expression for genome-wide association studies. Am J Hum Genet. 86:581–91. doi: 10.1016/j.ajhg.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ioannidis JP, Thomas G, Daly MJ. Validating, augmenting and refining genome-wide association signals. Nat Rev Genet. 2009;10:318–29. doi: 10.1038/nrg2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rafnar T, Sulem P, Stacey SN, Geller F, Gudmundsson J, Sigurdsson A, et al. Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat Genet. 2009;41:221–7. doi: 10.1038/ng.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bishop DT, Demenais F, Iles MM, Harland M, Taylor JC, Corda E, et al. Genome-wide association study identifies three loci associated with melanoma risk. Nat Genet. 2009;41:920–5. doi: 10.1038/ng.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shete S, Hosking FJ, Robertson LB, Dobbins SE, Sanson M, Malmer B, et al. Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet. 2009;41:899–904. doi: 10.1038/ng.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turnbull C, Ahmed S, Morrison J, Pernet D, Renwick A, Maranian M, et al. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat Genet. 2010;42:504–7. doi: 10.1038/ng.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harries LW, Ellard S, Stride A, Morgan NG, Hattersley AT. Isomers of the TCF1 gene encoding hepatocyte nuclear factor-1 alpha show differential expression in the pancreas and define the relationship between mutation position and clinical phenotype in monogenic diabetes. Hum Mol Genet. 2006;15:2216–24. doi: 10.1093/hmg/ddl147. [DOI] [PubMed] [Google Scholar]
- 19.Boj SF, Parrizas M, Maestro MA, Ferrer J. A transcription factor regulatory circuit in differentiated pancreatic cells. Proc Natl Acad Sci U S A. 2001;98:14481–6. doi: 10.1073/pnas.241349398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elliott P, Chambers JC, Zhang W, Clarke R, Hopewell JC, Peden JF, et al. Genetic Loci associated with C-reactive protein levels and risk of coronary heart disease. JAMA. 2009;302:37–48. doi: 10.1001/jama.2009.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reiner AP, Barber MJ, Guan Y, Ridker PM, Lange LA, Chasman DI, et al. Polymorphisms of the HNF1A gene encoding hepatocyte nuclear factor-1 alpha are associated with C-reactive protein. Am J Hum Genet. 2008;82:1193–201. doi: 10.1016/j.ajhg.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ridker PM, Pare G, Parker A, Zee RY, Danik JS, Buring JE, et al. Loci related to metabolic-syndrome pathways including LEPR, HNF1A, IL6R, and GCKR associate with plasma C-reactive protein: the Women’s Genome Health Study. Am J Hum Genet. 2008;82:1185–92. doi: 10.1016/j.ajhg.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuan X, Waterworth D, Perry JR, Lim N, Song K, Chambers JC, et al. Population-based genome-wide association studies reveal six loci influencing plasma levels of liver enzymes. Am J Hum Genet. 2008;83:520–8. doi: 10.1016/j.ajhg.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kathiresan S, Willer CJ, Peloso GM, Demissie S, Musunuru K, Schadt EE, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009;41:56–65. doi: 10.1038/ng.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erdmann J, Grosshennig A, Braund PS, Konig IR, Hengstenberg C, Hall AS, et al. New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet. 2009;41:280–2. doi: 10.1038/ng.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42:579–89. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lauc G, Essafi A, Huffman JE, Hayward C, Knezevic A, Kattla JJ, et al. Genomics meets glycomics-the first GWAS study of human N-Glycome identifies HNF1alpha as a master regulator of plasma protein fucosylation. PLoS Genet. 2010;6:e1001256. doi: 10.1371/journal.pgen.1001256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huxley R, Ansary-Moghaddam A, Berrington de Gonzalez A, Barzi F, Woodward M. Type-II diabetes and pancreatic cancer: a meta-analysis of 36 studies. Br J Cancer. 2005;92:2076–83. doi: 10.1038/sj.bjc.6602619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winter WE, Silverstein JH. Molecular and genetic bases for maturity onset diabetes of youth. Curr Opin Pediatr. 2000;12:388–93. doi: 10.1097/00008480-200008000-00019. [DOI] [PubMed] [Google Scholar]
- 30.Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41:703–7. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okada Y, Takahashi A, Ohmiya H, Kumasaka N, Kamatani Y, Hosono N, et al. Genome-wide association study for C-reactive protein levels identified pleiotropic associations in the IL6 locus. Hum Mol Genet. 2011;20:1224–31. doi: 10.1093/hmg/ddq551. [DOI] [PubMed] [Google Scholar]