

Abstract
Here we report the use of a multi-genome DNA microarray to investigate the genome diversity of Bacillus cereus group members and elucidate the events associated with the emergence of B. anthracis the causative agent of anthrax–a lethal zoonotic disease. We initially performed directed genome sequencing of seven diverse B. cereus strains to identify novel sequences encoded in those genomes. The novel genes identified, combined with those publicly available, allowed the design of a “species” DNA microarray. Comparative genomic hybridization analyses of 41 strains indicates that substantial heterogeneity exists with respect to the genes comprising functional role categories. While the acquisition of the plasmid-encoded pathogenicity island (pXO1) and capsule genes (pXO2) represent a crucial landmark dictating the emergence of B. anthracis, the evolution of this species and its close relatives was associated with an overall a shift in the fraction of genes devoted to energy metabolism, cellular processes, transport, as well as virulence.
INTRODUCTION
The B. cereus sensu lato group is comprised of multiple species including B. cereus (Bc), B. thuringiensis (Bt), B. mycoides, B. pseudomycoides, B. weihenstephanensis (Bw) and B. anthracis (Ba). Based upon 16s rDNA sequence, these species share 99% sequence identity and therefore, phylogenetically they belong to one group [1-4]. The naming of species within this group has placed a historical emphasis on the distinct biological phenotypes displayed by members of the B. cereus group, most notably, the mammalian pathogen B. anthracis [5]. The reconciliation of contradictory relationships exhibited by members of this phylogenetic group of species is still ongoing. There is a growing number of complete and partial genomic sequences available in public databases that has confirmed and expanded our appreciation of the diversity displayed by the Bc group members [6]. Genome size ranges from 5-6 Mb can be attributed to high degree of plasmid heterogeneity but also to variation in the chromosomally encoded genes [6-13]. One aspect of this diversity may be explained by the dynamic repertoire of plasmids found in B. cereus group isolates [8-11,13-23]. The number and size of plasmids found in these isolates suggest that the plasmids are a significant reservoir of gene novelty enabling species fitness in a wide array of environmental niche. The specific plasmid complements encode a substantial number of genetic determinants that influence B. anthracis virulence (pXO1, pXO2), or B. thuringiensis insecticidal/pathogenic character (pBT) and Bc emetic strains (pCER270), for example [7,16,17,24]. There is evidence that mobility of plasmid encoded sequences contribute to the apparently high rate of species diversification [25-28]. One genome sequencing project, reported a B. cereus isolate recovered from a metal worker presenting symptoms consistent with inhalation anthrax [13]. This report altered the previously held belief that the virulence plasmids, pXO1, encoding the primary virulence factors, Lef, Pag and Cya were found solely in Ba [13]. These observations have been subsequently extended to other Bc isolates that encode toxin genes that cause invasive disease [17]. It remains unclear to what extent plasmid inheritance resulting in such fundamental phenotypic alterations occurs within this group.
The life cycle of B. anthracis begins with the infection of the host by the spore [24,29]. The spore germinates and become vegetative and metabolically active cells. Upon shedding or host death, the vegetative cells are often returned to the soil, where the vegetative cells go through the process of sporulation to form highly resistant spores. B. anthracis spends the majority of its life cycle as an inert spore. This may imply that Ba may have substantially reduced opportunity for gene acquisition by horizontal transfer compared to Bc counterparts that more commonly exist in the environment as vegetative cells. Comparative analysis of B. anthracis genomes indicates that Ba belong to a monomorphic group with limited diversity. This is in contrast to other Bc group genomes that display greater degrees of diversity. There is evidence that members of the Bc group undergo genetic exchange with other members of this group [14,18,20,23]. Despite several illuminating studies conducted in the last decade with regard to the genome composition and population structure of Bc group (for review see [6,10]), the evolution and emergence of Ba remains unclear. This limitation can be attributed to our lack of knowledge regarding the ecology of the Bc group members and especially Ba [5,30-33]. An additional barrier pertains to our relative inability to identify the whole set of virulence factors and accessory genes required for niche adaptation and pathogenicity [9,12,32,34,35]. Large scale comparative genomic studies while useful must be complemented with continued functional characterization of genes and their function in natural environments in order to better elucidate aspects of the evolution of ecotypes within this heterogeneous group [36].
Comparative genomic analysis of the Bc group involving complete DNA sequence and SNPs have been employed [8,9,11-13,37-39]. Here we have used comparative genome hybridization (CGH) as a means of defining genomic differences within this group, specifically to address the emergence of B. anthracis. While powerful, CGH analysis has discrete limitations related to the use of arbitrary reference genomes. As such, one may only identify genes that have been lost in query genomes, relative to that reference genome. Likewise, the DNA hybridization assay using DNA microarrays has broad, but limited capacity to detect genes that have undergone substantial divergence resulting in a relatively high frequency of false negative gene presence measurements.
In the current study we used two complementary approaches to estimate the genomic diversity among the members of the Bc group, and elucidate evolutionary aspects regarding one of the recently emerged lineages containing B. anthracis and its close relatives. Initially we implemented a directed sequencing strategy we refer to as Gene Discovery (GD) that, like subtractive hybridization, enables the identification and rapid characterization of strain-specific sequences present in microbial genomes. In order to gain insights into the unique genomic features encoded in the B. anthracis genome in the context of the B. cereus group members we applied the GD strategy to identify those genomic segments that are uniquely present in the genomes of seven diverse B. cereus strains, relative to the genome of B. anthracis. We report the analysis of genomic sequences that appear characteristic among the seven query genomes. The number of unique sequences obtained from each strain ranged significantly. Based on the unique genome sequences identified in this study and other publically available sources we developed a more comprehensive species 70-mer oligonucleotide DNA microarray for comparative genomic analyses. We carried out comparative genome hybridization (CGH) on 35 diverse Bc and six Ba strains. Due to its inclusiveness, the multi-genome microarray is able to closely approximate the gene complements of query genomes. In so doing, we were able to identify genomic events associated with the emergence of B. anthracis as a distinct lineage within the B. cereus group. We also demonstrate that the evolution of B. anthracis as a hyper-virulent, invasive phenotype is coupled with reduced energy metabolism, biosynthesis of cofactors, and transport functions compared to its predecessors. At the same time acquisition of the plasmids harboring the genes for the three toxins, and the capsule, through horizontal gene transfer, seem to have been accompanied by the acquisition of a specific set of the chromosomal virulence associated factors.
MATERIAL AND METHODS
Bacterial Isolates
A total of 48 strains, 41 B. cereus (Bc), one B. weihenstephanensis (Bw), and six B. anthracis (Ba) strains, were investigated (Table 1). Many of these strains have been previously characterized by various population genetic analysis methods such as whole genome sequencing, multi-locus enzyme electrophoresis (MLEE), multi-locus sequence typing (MLST) as well as CGH [4,12,39]. The isolates correspond to all three major groups within the B. cereus sensu lato [38,39] and represent a wide phenotypic and genotypic diversity, and geographic coverage of the species. Seven strains, were selected initially for gene discovery to complement the complete genome sequences already available in the public databases. These seven strains were selected to represent major lineages with this taxon on the basis of previously reported population genetic studies using CGH [12] and MLST [39].
Table 1.
Strains used in the analysis.
| Species | Strain | Alternative Designations | Year of isolation | Geographic Origin | Source | Type of infection (human isolates) | MLST sequence type (ST)& | Plasmid Profile | Main phylogenetic cluster$ |
|---|---|---|---|---|---|---|---|---|---|
| B. cereus | AH259* | 6A1 in BGSC | Not typed | II | |||||
| B. cereus | AH607* | Norway | Dairy | 17 | I | ||||
| B. cereus | AH535* | 1994 | Norway | Soil (strawberry field) | 77 | III | |||
| B. cereus | AH819* | 1995 | Norway | Human | Periodontitis | 40 | I | ||
| B. cereus | AH812* | 1995 | Norway | Human | Periodontitis | 38 | I | ||
| B. cereus | AH1123* | France | Human | Blood | 45 | I | |||
| B.weihenstephanensis | AH1143* | Germany | Dairy (milk) | 68 | III | ||||
| B. cereus | ATCC 10987* | 1930 | Canada | Spoiled cheese | 2 | I | |||
| B. cereus | ATCC 14579* | 1916 | USA | Air, farmhouse | 33 | II | |||
| B. cereus | AH404 | Finland | Dairy | Not typed | III | ||||
| B. cereus | AH405 | Norway | Dairy | Not typed | II | ||||
| B. cereus | AH407 | Finland | Dairy | 9 | III | ||||
| B. cereus | AH533 | 1993 | Norway | Soil (strawberry field) | Not typed | IV§ | |||
| B. cereus | AH540 | 1993 | Norway | Soil | Not typed | I | |||
| B. cereus | AH557 | 1993 | Norway | Soil | Not typed | I | |||
| B. cereus | AH564 | 1993 | Norway | Soil | Not typed | I | |||
| B. cereus | AH568 | 1993 | Norway | Soil | Not typed | I | |||
| B. cereus | AH597 | Norway | Dairy | Not typed | III | ||||
| B. cereus | AH598 | Norway | Dairy | Not typed | I | ||||
| B. cereus | AH599 | Norway | Dairy | Not typed | I | ||||
| B. cereus | AH601 | Norway | Dairy | Not typed | II | ||||
| B. cereus | AH602 | Norway | Dairy | Not typed | III | ||||
| B. cereus | AH603 | Norway | Dairy | Not typed | III | ||||
| B. cereus | AH604 | Norway | Dairy | Not typed | I | ||||
| B. cereus | AH608 | Norway | Dairy | Not typed | I | ||||
| B. cereus | AH648 | 1994 | Norway | Soil | Not typed | III | |||
| B. cereus | AH810 | 1994 | Norway | Human | Periodontitis | 36 | I | ||
| B. cereus | AH811 | 1995 | Brazil | Human | Periodontitis | 37 | II | ||
| B. cereus | AH813 | 1995 | Brazil | Human | Periodontitis | 39 | I | ||
| B. cereus | AH814 | 1995 | Norway | Human | Periodontitis | 84 | II | ||
| B. cereus | AH815 | 1995 | Norway | Human | Periodontitis | 85 | II | ||
| B. cereus | AH816 | 1995 | Norway | Human | Periodontitis | 39 | pPER272 | I | |
| B. cereus | AH817 | 1995 | Norway | Human | Periodontitis | 3 | pPER272 | I | |
| B. cereus | AH818 | 1995 | Brazil | Human | Periodontitis | 39 | I | ||
| B. cereus | AH820 | 1995 | Norway | Human | Periodontitis | 39 | I | ||
| B. cereus | AH823 | 1996 | Norway | Human | Periodontitis | 40 | I | ||
| B. cereus | AH825 | 1996 | Norway | Human | Periodontitis | 40 | I | ||
| B. cereus | AH826 | 1996 | Norway | Human | Periodontitis | 40 | I | ||
| B. cereus | AH827 | 1996 | Norway | Human | Periodontitis | 40 | I | ||
| B. cereus | AH828 | 1996 | Norway | Human | Periodontitis | 40 | I | ||
| B. cereus | AH830 | 1995 | Norway | Human | Periodontitis | 41 | I | ||
| B. cereus | AH831 | 1996 | Norway | Human | Periodontitis | 42 | I | ||
| B. anthracis | Ames | 1981 | USA | Cow | 1 | pXO+ /pXO2+ | I | ||
| B. anthracis | Australia94 | A0039 | 1994 | Australia | Cattle | 1 | pXO+ /pXO2+ | I | |
| B. anthracis | Tsiankovskii-I | Former USSR | Livestock vaccine strain | 1 | pXO+ /pXO2+ | I | |||
| B. anthracis | Vollum | A4088 | 1935 | UK | Cow | 1 | pXO+ /pXO2+ | I | |
| B. anthracis | Sterne | 34F2 | 1937 | South Africa | Cow (animal vaccine strain) | 1 | pXO+ /pXO2- | I | |
| B. anthracis | Pasteur | 1 | pXO- /pXO2+ | I |
Strains used for gene discovery
According to the optimized scheme of Tourasse, Helgason et al., (2006)
As determined by MLST, MLEE, and/or AFLP typing data (HyperCAT database, http://mlstoslo.uio.no)
Preparation of Genomic DNA
For each strain in Table 1, a single colony from a plate of LB growth was inoculated into 10 mL LB and grown overnight at 30°C. Fresh LB medium (250 mL) was inoculated from the overnight culture at a 1:50 dilution and grown for 4 hours at 30°C with shaking (250 rpm). The culture was harvested by centrifugation at 8500 rpm for 10 min at 20°C. Genomic DNA was extracted and purified from the pellets as previously described [39]. All genomic DNA (gDNA) samples were assessed for DNA integrity by gel electrophoresis before further analysis.
Gene Discovery (GD): Genomic Library Preparation, Screening and Sequencing
Gene Discovery strategy was applied as previously described [40]. Library generation and screening for GD are summarized in Figure 1s. Briefly, a partial Tsp509I digestion was performed with 3 μg each of the seven selected Bc strains to generate a mass peak centered at ~500 base pair fragments. Digested DNA was electrophoresed on 1% ultra pure agarose gel (Invitrogen, Carlsbad, CA), and fragments of an apparent MW ~300-800bp. were excised and recovered using the QIAquick gel extraction kit as per manufacturer’s instruction (QIAgen, Valencia, CA). These fragments were cloned into the EcoRI site of pUC19 [41] and transformed into ElectroMAX DH10B cells (Invitrogen, Calsbad, CA) by electroporation. High-throughput plasmid purification was performed at the J. Craig Venter Science Foundation, Joint Technology Center (Rockville, MD). To determine how many plasmids to screen from each genomic library we used Moore’s formula [42] assuming an average insert size of base pairs. For each strain, 37,632 plasmids were produced representing greater than 5X coverage of each genome. Slides for genomic library screening (GD) were generated by printing plasmid DNA templates at a concentration of ca. 200ng/μL. In order to identify sequences uniquely present in query strains, each library was screened through flip-dye competitive hybridizations using differentially labeled probes from the genomic DNA of the strain that the library was generated from, and B. anthracis Ames strain [12]. Plasmids were printed and screened using Bacillus spp./ and Ba genomic probes labeled with either Cy3 or Cy5. Replicate hybridizations were conducted with flip-dye replicates. Genomic DNA hybridizations were performed essentially as described at http://pfgrc.jcvi.org/index.php/microarray/protocols.html [40,43]. The slides were dried and then scanned using the GenePiX 4000B scanner (Axon Instruments, Union City, CA). The signal intensity ratios were used to indicate those plasmids that contained inserts that were divergent or uniquely present in the query Bacillus spp. genome relative to Ba. Hybridization signals displaying a query Bacillus spp./reference Ba. log-ratio of 5.0 or greater were identified and the corresponding plasmid DNAs were subjected to DNA sequencing in both directions using vector-based primers. All sequence reads were assembled using the Celera Assembler [44]. Automatic annotation was performed to identify entire or partial open reading frames (ORFs) as previously described [45,46]. Annotation of the contigs produced putative ORFs or “features.” These features represent both complete and partial genes and are referred to as “pORFs” hereafter.
Gene Discovery Sequence Analysis
Figure 2s summarizes the bioinformatic approach used to characterize the sequences from B. cereus strains subjected to gene discovery. Briefly, template sequences obtained from the GD process were compared to publicly available genome sequences from Bacillus as well as non-Bacillus species to identify unique sequences. Obtained sequences were initially filtered for uniqueness against B. anthracis, and B. cereus genomes, using the blastn algorithm [47]. Nucleotide matches of <100 nucleotides and e-values >10-5 were preliminarily considered novel. Translated sequences were then compared against the non-redundant amino acid (NRAA) database using blastp [47]. ORFs showing BLAST hits with homology e-value <10-5 were considered homologous to previously characterized proteins, while those with no match or with e-values >10-5, were considered unique.
Species Microarray Design, Preparation, and Hybridization
ArrayOligoSelector [48] was used to design seventy base oligonucleotides (70-mer genomic markers) for the multi-genome species microarray. A total of 29,682 ORFs representing unique features from GD and from other partial and complete genome sequencing projects (NCBI release 148) were printed on the species microarray. Oligonuceotide preparation, printing and hybridizations were performed as previously described [12,40]. Ba Ames gDNA was used as reference in all CGH experiments.
DNA Microarray Data Analysis
Microarray data were analyzed as previously described [40,49]. Raw microarray images were analyzed using SpotFinder version 2.2.2 from the TM4 Microarray Software Suite. Fluorescence intensities were normalized using Histogram Mode Centering (HMC) algorithm with spots less than 20,000 relative fluorescence units filtered from subsequent analysis.
CGH Data Clustering
In order to minimize bias and noise due to the individual log-ratio values when investigating global gene presence/absence patterns among the query strains, the final data set were assigned one of three different values: 0 = absent, 0.5 = divergent and 1 = present. Hierarchical clustering of the query genomes based on CGH data was performed as previously described [40,49-53] using the Multiple Experiment Viewer (MeV) of TM4 Software as well as MrBayes.
Allelic Grouping and Gene Calling
Our oligonucleotide design approach allowed us to distinguish subtle sequence differences among variants of orthologous and paralogous genes. In order not to confound the final gene calls or the total gene estimates, we identified and clustered related gene variants into “allelic groups” as previously described [40]. Each group includes putative variants of the same coding DNA sequence (CDS). Clustering of the orthologous sequences into allelic groups was primarily based on oligo-to-gene, gene-to-gene, and protein-to-protein relationships. Allelic groups containing multiple paralogs were sub-divided when possible such that each paralogous variant had its own sub-group.
Statistical Analysis of the CGH Data
The Fisher exact test [54] was used to perform genomic marker association analyses (MAA) between the genomic attributes i.e. gene presence or absence, and clades or groups of strains displaying a particular phenotype. Markers, and consequently their corresponding ORFs, were considered “characteristic for” or “prevalent in” a particular group as inferred by MAA using p<0.05 as a cut-off value for statistical significance [40].
RESULTS
Genomic Library Screening
We have developed a directed sequencing strategy referred to as Gene Discovery (GD) that enables the identification and rapid characterization of strain-specific sequences present in microbial genomes (Figure 1s). We applied GD analysis to nine strains belonging to the B. cereus sensu lato group. They consisted of two fully sequenced strains (ATCC10987 and ATCC14579), and seven uncharacterized strains. Random genomic libraries from each strain consisting of nearly 38,000 recombinant pUC plasmids with inserts of 300-800 bp were printed onto glass slides. Each library was screened through flip-dye hybridization using two differentially labeled probes. The reference channel in each case was B. anthracis Ames [12] paired with the strain that was used for library construction. Based on hybridization intensity, plasmids that hybridized to the query strain (library) but not B. anthracis, were considered candidate strain-specific sequences. Fully sequenced strains ATCC10987 and ATCC14579 were included to evaluate the efficiency of the novel gene identification. None of the sequence reads obtained from either library – ATCC10987 and ATCC14579, aligned significantly with any identified Ba sequences. The fraction of strain-specific genes encoded in the ATCC10987 and ATCC14579 recovered in the screen was 100% for both control strains (e-value <10-5 over >100 nucleotides, Table 1s, [9,11]). These findings suggest that both the experimental and bioinformatic thresholds were appropriate. Over 75% of the recovered sequences displayed significant similarity at the nucleotide and protein levels to sequences derived from previously sequenced Bacillus spp. or other gram-positive bacteria. The number of unique and orthologous reads varied for each query genome screened (7%-35%). Strains AH1123 and AH1143 had the largest numbers of unique reads identified (614 and 913 respectively). Strains AH812 and AH259 had the smallest number of novel sequences identified (46 and 48 respectively).
Analysis of Unique Genomic Features
A total of 4,630 contigs and 4,008 singletons were obtained from the seven query strains. Contig size ranged from 270 – 4,800 bp with an average length and sequence coverage of 740 bp and 1.9 reads per contig respectively. The amount of novel sequence information in the query B. cereus strains (the total length of contigs and singletons) relative to B. anthracis ranged from 104.4 Kb (AH812) to 1.5 Mbp (AH535) (Table 2). Genes encoding hypothetical proteins constituted the largest group of features (35-76%, Figures 2 and 3s). Among pORFs with a match in the NRAA database, the vast majority (87%) shared sequence identity to Gram-positive bacteria and bacteriophage. While 71 % of the novel sequence displayed the strongest similarities to other Bacillus spp., pORFs with identity to other non-Bacillus spp., low G+C Gram-positives such as Clostridium spp. and Listeria spp. were also prevalent (Figure 1).
Table 2.
Comparative sequence analysis summary of B. cereus novel sequences found from gene discovery.
| B. cereus strains: | AH819 | AH607 | AH535 | AH1123 | AH812 | AH1143 | AH259 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total bases sequenced / strain: | 1,076,892 | bp | 1,237,775 | bp | 1,500,190 | bp | 443,218 | bp | 104,276 | bp | 1,422,879 | bp | 177,983 | bp |
| Fraction relative to the size of an average B. cereus genome: | 20% | 23% | 28% | 8% | 2% | 27% | 3% | |||||||
| Total number of annotated features: | 2,041 | 2,204 | 3,138 | 871 | 200 | 2,874 | 383 | |||||||
| Species matches | ||||||||||||||
| B. anthracis | 43 | 2% a | 122 | 6% a | 132 | 4% a | 52 | 6% a | 7 | 4% a | 92 | 3% a | 11 | 3% a |
| B. anthracis pXO1 & pXO2 | 4 | 0% a | 0 | 0% a | 0 | 0% a | 0 | 0% a | 1 | 1% | 2 | 0% a | 0 | 0% a |
| B. cereus 10987 | 503 | 25% a | 510 | 23% a | 431 | 14% a | 85 | 10% a | 34 | 17% | 373 | 13% a | 4 | 1% a |
| pBc10987 | 121 | 6% a | 3 | 0% a | 1 | 0% a | 1 | 0% a | 17 | 9% | 0 | 0% a | 0 | 0% a |
| B. cereus ATCC14579 | 143 | 7% a | 183 | 8% a | 427 | 14% a | 64 | 7% a | 15 | 8% | 394 | 14% a | 271 | 71% a |
| B. cereus (other strain genomes) | 200 | 10% a | 275 | 12% a | 284 | 9% a | 192 | 22% a | 21 | 11% | 227 | 8% a | 12 | 3% a |
| B. cereus (other strain plasmids) | 49 | 2% a | 70 | 3% a | 112 | 4% a | 18 | 2% a | 12 | 6% | 73 | 3% a | 0 | 0% a |
| B. thuringiensis | 97 | 5% a | 245 | 11% a | 490 | 16% a | 52 | 6% a | 7 | 4% | 474 | 16% a | 40 | 10% a |
| B. thuringiensis plasmids | 0 | 0% a | 6 | 0% a | 3 | 0% a | 0 | 0% a | 0 | 0% | 0 | 0% a | 0 | 0% a |
| Bacillus spp. | 1 | 0.05% a | 1 | 0.05% a | 5 | 0.16% a | 0 | 0.00% a | 0 | 0.00% | 6 | 0.21% a | 0 | 0.00% a |
| Bacillus spp. plasmids | 0 | 0% a | 2 | 0% a | 0 | 0% a | 0 | 0% a | 0 | 0% | 1 | 0% a | 0 | 0% a |
| Bacillus spp. phages | 0 | 0% a | 0 | 0% a | 0 | 0% | 0 | 0% a | 0 | 0% | 0 | 0% a | 0 | 0% a |
| Overall Number of features similar to Bacillus species by BlastN: | 1,161 | 57% a | 1,417 | 64% a | 1,885 | 60% a | 464 | 53% a | 114 | 57% a | 1,642 | 57% a | 338 | 88% a |
| Remaining relatively novel features (by BlastN): | 880 | 43% a | 787 | 36% a | 1,253 | 40% a | 407 | 47% a | 86 | 43% a | 1,232 | 43% a | 45 | 12% a |
| Truly unique features from the relatively novel gene set c: | 452 | 22% a | 312 | 14% a | 887 | 28% a | 324 | 37% a | 71 | 36% a | 1,054 | 37% a | 19 | 5% a |
Fraction from the total number of annotated features in the corresponding strain.
Remaining pORFs whose amino acid sequence shows no significant homology to proteins in databases by BlastP.
Figure 2.
Comparative summary of genes considered missing or novel among the seven query B. cereus strains relative to B. anthracis. Absent genes are identified from a previous CGH study [12] whereas the novel genes are those discovered through gene discovery (GD).
Figure 1.
Sequence analysis summary of the novel genomic content present in B. cereus strains based on blast [47]. Species hit summary results are based on feature (partial ORF) sequence analysis resulting from blastp.
We observed differences among the unique features obtained from each query genome with respect to predicted functions (Figure 3s, Table 2s). The majority of the novel features (67%) encoding proteins with predicted functions defined only seven major role categories including: cell envelope (14%), transport (14%), regulatory/signal transduction (13%), cellular processes (10%), energy metabolism (9%) and mobile elements (7%). Among transporters, proteins predicted to be responsible for the uptake of glycopeptides, amino acids and their derivatives, trace elements, calcium, sodium, phosphates as well as sulfates were commonly found. In addition to some S-layer proteins, the novel cell envelope fraction was dominated by proteins involved in various steps of peptidoglycan or capsule synthesis; most of them were sugar transferases, epimerases, amidases, transfereases, D-alanine-D-alanine ligases N-acetylglucosaminyltransferases, or murein hydrolases. Novel features related to regulatory functions included various members of two-component regulatory systems, transcriptional regulators belonging to MarR, TerRAcrC, LysR, LuxR, LytR, AraC/XylS families, σ54-dependent transcriptional activator, BglG family anti-terminators, as well as kinases involved in sprorulation. Among the unique pORFs, we identified several that are predicted to perform important house-keeping functions within the cell such as protein synthesis and fate, and DNA metabolism. This group of features was enriched for variants of tRNA-synthetases. Among non-essential genes there appeared to be significant variation in genes encoding chaperones, proteases, and restriction modification enzymes (Figures 3 and 3s, and Table 2s).
Figure 3.
Simulation model for predicting the effect of additional strain sampling to estimate (A) the number of novel pORFs, and (B) the limits of unique genomic DNA expected among B. cereus group members.
Distribution of Virulence-associated and Anti-microbial Resistance Genes
We mined the novel sequence data to identify genes with similarity to well characterized penicillin binding proteins (pbp- or fmt-like) and other antibiotics, heavy metals, multi-drug efflux pump regulators and lantibiotic transporters (Table 2s and 3s). Virulence associated genes (VAGs) included orthologs of Listeria spp. internalin, enterotoxins, putative cereolysin O (a thiol-activated cytolysin variant from B. weihenstephanensis), collagen adhesion protein (B. cereus ATCC10987), mouse virulence factor mviN, and a perfringolysin O precursor; almost all occurrences of these genes were found in non-clinical (environmental) isolates. Interestingly, we found a gene derived from the strain Bw AH1143 annotated as “protective antigen.” This CDS (2,127 bp) displayed significant sequence identity (61 % nt, 47 % aa) to the B. anthracis protective antigen. Interestingly, the Bw AH1143 pag-ortholog had 97 % nucleotide identity over its length to a 1846 bp contig, encoding a partial CDS also annotated as protective antigen, in the recently sequenced genome of B. cereus BDRD-ST196 (NCBI Accession Number: ACMD01000248).
B. cereus Pan-genome Predictions
To evaluate the comprehensiveness of gene discovery as it relates to the complete gene pool encoded by B. cereus sensu lato, we performed statistical modeling based on the novel genomic content observed among the query genomes (Figure 3). The number of novel genes obtained from each query strain follow a strong regression pattern approximating the Boltzmann equation (R2=0.96). This analysis predicts that members of the Bc group may vary in genome size by as much as 1.5 Mb, nearly 30% of an average genomes coding capacity. It is interesting to note that this value is quite close to traditional threshold for species differentiation (≥ 30 % genome difference by DNA-DNA hybridization) [55,56]. Following the identification of novel genes from seven Bc strains, we estimated that an asymptote was reached predicting that the number of the new genes that may be discovered through additional screening of genomes may not exceed 250. The fact that the number does not drop to zero is consistent with the existing knowledge that B. cereus sensu lato is part of an open genome [45].
B. cereus/B. anthracis Multi-Genome DNA Microarray Design
Annotated genes derived from novel gene sequences discovered in this study and previous genome and plasmid sequencing projects were used to create a database of B. cereus group DNA sequence to enable 70-mer oligonucleotide design using a modified version of the Pick-70 tool [48]. Table 4s presents a summary of the 29,977 unique 70-mer oligonucleotides that together, represent 29,682 putative ORFs. We were able to assign these ORFs to a smaller number of 15,548 orthologous groups including sequence variants of orthologous coding DNA sequences (CDSs). It should be noted that many singleton reads annotated as unknowns, may represent unlinked sequence belonging to the same ORF or orthologous sequence. Therefore, the frequency and identity of feature redundancy for these ORFs is unknown. Given these uncertainties, the estimated number of oligonucleotides per allelic group is 1.93. There were 9,909 (59%) CDS represented by single oligonucleotides. Among the orthologous groups with two or more CDSs, 3,117 (48%), 1,823 (28%), 580 (9%), 344 (5%), and 225 (3%) are represented by two, three, four, five and six oligonucleotides respectively. Allelic grouping was then used as the basis for estimating the genomic content of each query strain based on hybridization results.
Genome Size and Conservation Estimates
We interrogated the B. cereus/B. anthracis species DNA microarray with a set of 35 diverse Bacillus isolates including soil, dairy and periodontal isolates and six B. anthracis strains (Table 1). CGH profiles of the strains are summarized in Table 5s. In order to assess the accuracy of using CGH data to estimate gene content and genome size, CGH-derived total gene estimates for four B. anthracis strains (Ames, Sterne 34F2, Vollum, Australia 94, Tsiankovskii-I) and three B. cereus 14579, 10987 and AH820) were compared to the annotated genes from their published genome sequences. The margin of error of the CGH-based method ranged from -7.9 to 13% (median 2%, average 4%). Using this procedure to estimate genome size, we observed that genome sizes are generally conserved within the members of this group (Figure 4s). For the majority of query strains, total gene counts ranged from 5,000 to 6,000. These results are comparable to genome size estimates determined by complete genome sequencing (average genome size among B. cereus group members varies between 5.3-5.6 Mb). Some notable exceptions to the overall genome size conservation included three strains (AH533, AH815 and AH830) that had gene counts below 4,000 and five others (AH404, AH597, AH601 AH604 and AH648) that had total gene estimates exceeding 6,000.
CGH Profiling of Mobile Elements
Plasmid profiles inferred from CGH patterns appeared complex (Table 6s, Figure 4). Therefore, we focused our analysis on the best-known and most clinically relevant plasmids such as pXO1, pXO2, and pBC218 [13,27]. Plasmid pXO1 has been shown to share a high degree of sequence similarity, with three completely sequenced plasmids i.e. pBC10987, pCER, and pPER272 [16]. Here we refer to the presence of a plasmid or a plasmid backbone if CGH results indicate that ≥ 20% of genes normally associated with a plasmid are present. In no case have we confirmed that the detected genes are indeed plasmid-based. Among the 35 Bc strains investigated, 19 (54 %) appeared to harbor pXO1-like plasmids. These findings confirm previous observations that pXO1-like plasmid variants are common among B. cereus group members [6,8,11-14,16].
Figure 4.
Summary of comparative clade analysis based on the marker association analysis. The dendrogram on the left represents strain clustering based on the global CGH patterns. Comparative clade analyses are color-coded with square brackets accordingly. Markers representing clade-characteristic CDSs are summarized in the form of predicted functional role categories of the genes they represent. Stars next to the role category sections on each bar indicate that there is a statistical difference with respect to their corresponding fractions between groups.
CGH profile analysis indicated that variants of capsule carrying plasmids such as pXO2 or pBC218, may also be harbored by some Bc strains but at a significantly reduced frequency. pXO2 variants were found in four Bc strains (AH813, AH815, AH816, and AH818). With one exception (AH813), all these strains also appeared to harbor pXO1-like plasmids. Traces of pBC218 plasmid (10-19% of its genes) were observed in eight Bc strains, four of which (AH817, AH598, AH599, AH601), also seemed to harbor pXO1-like plasmid variants. Despite the prevalence of the virulence plasmid variants among Bc strains investigated, the presence of B. anthracis toxin genes was very rare. Among the B. cereus strains examined, we observed a positive signal for the regulator of tripartite toxins gene expression, atxA (pXO1) only in strain AH568. Five other strains, i.e. AH601, AH597, AH648, AH404 and AH818, generated a positive signals for pXO2-borne capA gene. Similarly, strain AH608 generated a positive signal for capC. Only two additional B. cereus strains, AH815, AH811, generated positive signals for both capA and capC, however both were negative for capB as well as the regulatory genes controlling cap gene expression (Table 7s).
Phylogenomic Relationships of Bacillus Strains
We performed hierarchical clustering to reveal strain relatedness on a genomic scale. The dendrogram shown in Figure 4 represents a summary of phylogenomic relationships based on the data for all 29,977 oligonucleotides using the trinary designations (“absent”, “divergent” and “present”). Strain relationships based on CGH patterns (trinary values) of plasmid marker sets are summarized in Figure 5s. Overall, phylogenomic clustering resulted in similar strain relationships as those generated by other approaches such as MLEE, MLST, AFLP or sequencing [4,6,8,38,39,57,58]. The finding that four periodontal isolates, strains AH820, AH813, AH816 and AH817, clustered tightly with the B. anthracis clade (Clade 6) was of interest. Nearly all of these isolates harbor pXO1- and/or pXO2-like plasmids. These isolates were distinct from six other B. cereus strains of clinical origin i.e. AH823, AH825, AH826, AH827, AH828 and AH831 (Clade 2) that constitute a separate clade, somewhat distant from B. anthracis.
Phylogenomic cluster analysis further support previous observation that the B. anthracis clade appears to be closely related to a distinct group of B. cereus (Clade 3) [4,39]. Based on the data generated, seven Bc isolates, together with the B. anthracis strains, define a major clade (Clade 5). It is also of interest to note that the three strains in this clade are of environmental origin (Clade 5). The pathogenic potential of these isolates is unknown. Strains constituting Clade 2 were the closest relatives of Clade 4. Clades 2 and 4 appear to have originated from a common B. cereus ancestral lineage that correspond to the previously defined B. cereus group I [38,39]. Most of the strains within this group appear to harbor pXO1-like plasmid variants. The remaining major Clade 1 was a diverse group comprised almost exclusively of environmental or dairy isolates. Four of the seven members of this clade appeared to harbor pBC218-like plasmids.
Comparative Clade Analysis Reveals Genomic Flux and Alteration Events
Based on the identified phylogenomic relationships, we next conducted a comparative analysis of the gene complements of isolates within each distinct clade. The focus of our analyses was the identification of genomic patterns (gene content) associated with the emergence of B. anthracis using the Fisher’s Exact Test (Figure 4, Table 5s). We investigated the main role category profiles that provided an overview of the differences. We then conducted a more detailed examination of sub-role categories and KEGG pathways. We considered that the function(s) or pathways may be impaired if the number of missing genes classified under such functions or pathways was ≥ 2 [40].
We noticed several differences with respect to the main functional categories across the clades. For example, the fraction of genes involved in energy utilization, cellular processes, transport, and in some house-keeping functions such as amino acid biosynthesis, protein synthesis and protein fate, was relatively smaller among B. anthracis genomes and their neighbors compared to their distant relatives (e.g. Clade 8 vs. Clade 6 and/or Clade 4 vs. Clade 2), (Figure 4). This observation was also true when comparing clinical isolates with those of limited or no virulence potential. Furthermore, the fraction of transport, energy metabolism, biosynthesis of cofactors cellular processes, amino acids metabolism, and protein synthesis and fate functions as a whole in Clade 4 members is half of the corresponding group compared to the remainder of B. cereus (18% vs. 35%, Figure 4). The evolution of Clade 4, and especially B. anthracis, have been associated with the acquisition of mobile elements and most dominantly, genes of unknown function. The expansion of mobile elements in B. anthracis compared to other clades suggests that the primary mechanism for the numerous gene acquisition events occurring throughout the B. cereus group are mediated by mobile element movement.
We found a significantly smaller number of genes representing ORFs predicted to be involved in protein production i.e. amino acid biosynthesis, protein fate (post-translational modification) and synthesis among Clade 3 members compared to those belonging to Clade 1 (29 vs. 316). Likewise, fewer ORFs predicted to be involved in protein production were observed when comparing Clade 4 vs. Clade 2. (19 vs. 90) or Clade 4 vs. the remainder of all other Bc genomes (29 vs. 158). We also note a smaller number of genes devoted to the metabolism of n-acetylglucosamine, n-acetylgalactosamine or n-acetylmuramic acid derivatives (classified under cell envelope functions) among Clade 3 genomes compared to those belonging to Clade 1 (10 vs. 40) as well as among Clade 4 vs. the remainder of all other Bc genomes (11 vs. 17). Similar patterns were also observed when investigating genes involved in sugar metabolism, which is classified under energy metabolism. For example, genes involved in the uptake and metabolism of sugars in general, and mannose, fructose, ribose/ribulose or glucose in particular, were more prevalent among Clade 1 members compared to those in Clade 3 (48 vs. 11). Similarly, more genes involved in sugar metabolism were found when comparing Clade 2 members with to those belonging to Clade 4 (31 vs. 8). Clade 4 genomes also appeared to possess fewer genes involved in sugar metabolism when compared the remainder of all other Bc genomes (3 vs. 12).
Results of sub-role functions or KEGG pathways survey indicated that seven functions or pathways may be incomplete or impaired in Clade 3 members compared to those of Clade 1 affecting the metabolism of: arginine and proline, aspartate derivatives involved in amino acid biosynthesis, metabolism of butanoate, starch and sucrose, biosynthesis of secondary metabolites as well as metabolism in diverse environments. Likewise, when compared to the rest of B. cereus, Clade 4 members appeared to have incomplete or impaired functions or pathways affecting the biosynthesis of glutamate-, aspartate- and pyruvate derivatives involved in amino acid biosynthesis, degradation of proteins, peptides, and glycopeptides, degradation of amino acids and amines, biosynthesis of thiamine, adaptation to atypical conditions as well as DNA recombination and repair. On the other hand, Clade 4 members appeared to possess significantly large numbers of features representing mobile elements and enzymes of unknown functions. In addition, we discovered that Clade 4 members seemed to have four genes belonging to a segment of the inositol phosphate metabolism which enable the catabolism of myo-inositol to Acetyl –CoA or Glyceraldehyde-3P, which in turn may enter the glycolysis/glyconeogenesis pathways.
Transporters were another functional group of interest. We found that Clade 1, which contains more environmental isolates, has more transporters than Clade 3, which moslty contains strains of clinical origin. Similarly, both Clade 4 and Clade 8 members, compared to the rest of B. cereus genomes, seem to possess fewer transporters in general, especially sugar transporters. On the other hand, we found more urea/amide transporters among Clade 3 members compared to those of Clade1, and more iron transporters in B. anthracis (Clade 8) than in B. cereus genomes.
The evolution of B. anthracis (comparing Clade 3 to Clade 4) appear to have involved an overall increase in genome size and coding capacity. For example the average gene number estimates for Clade 1, and Clades 3 and Clade 4 (excluding B.a.) are 5,600, 5,200 and 5,000 respectively. By contrast, the average number of genes encoded by human-associated Clade 4 members is 460 genes larger than those of environmental origin. B. anthracis genomes also contain on average 480 more genes than their four nearest neighbors. A large fraction of the additional genes are the result of B. anthracis-specific pXO1 and pXO2 CDS and the four Ba chromosomal phage insertions. In addition, we conducted a more detailed comparative gene content analysis among Ba (Clade 8, excluding plasmid deficient mutant strains) and Clade 6 members. The results of this analysis have been summarized in Figure 5. Overall, we found 1,459 CDSs that were shared by at least two strains in Clade 7 beyond the conserved gene set. Among this group of 1,459 partially conserved genes, 475 were found in common between B. anthracis and at least another member of Clade 6. From this analysis, we also identified 645 ORFs unique to B. anthracis with respect to Clade 6 members. Besides of consisting of a significantly large number of genes, the Ba unique gene set also displayed a characteristic functional role profile. Mobile elements (34%) and genes encoding for proteins of unknown functions (45%) constituted 79% the B. anthracis unique gene set with respect to Clade 6 genomes. Interestingly, the remainder of the unique genes consisted of almost equal shares of ORFs coding for proteins involved in cell envelope (5%), cellular processes (5%), transport (4%), and regulation of gene expression (4%). It is worth noting here that of the 27 CDSs constituting the latter group of genes involved in regulatory functions only six were found within phage features. This finding suggests that, aside from several phage- and plasmid-based genes that could be just hitch-hikers within phages or plasmids, Ba seems to have acquired at least 21 single genes or operons that may contribute to its distinct pathobiology. While two of them – atxA and capR are well-characterized, the role of the other putative regulatory genes remains yet to be discovered. On the other hand, 45% (13/29) of the genes predicted to participate in cell envelope functions consisted of those encoding proteins involved in LPS biosynthesis, lipoproteins, and S-layer proteins. Finally over half (54%, 13/24) of the genes involved in transport functions consisted of efflux pumps and protein involved in transport of amino acids and trace elements.
Figure 5.
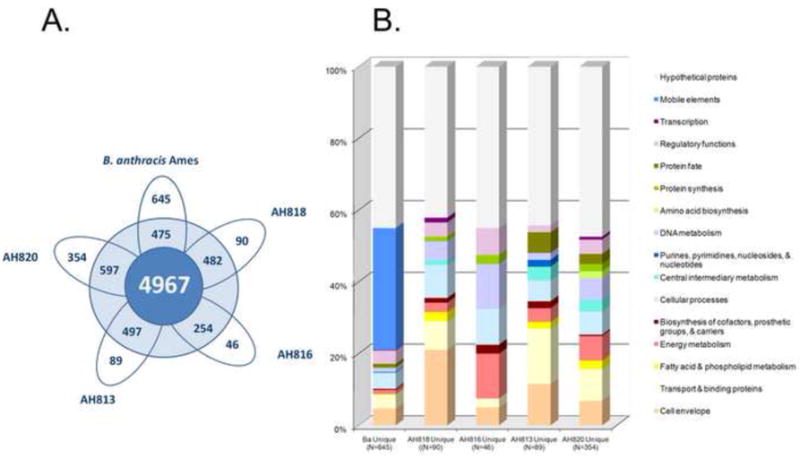
Comparison of gene content among B. anthracis (Clade 8) and four B. cereus genomes constituting Clade 6 (Figure 4, Table 5s). A. Unique and shared genes. We defined the conserved gene set shared by Clade 7 members as the group of ORFs that are found among ≥75% of both Clade 6 and Clade 8. The remaining genes from each genome were either unique or shared by at least one other genome in Clade 7. We used B. anthracis Ames as a representative of Clade 8. The number in the inner circle represent the gene set commonly found among members of both Clade 6 and Clade 8 (present in >= 75% of the genomes of Clade 7). Going outward, numbers in the second circle represent genes of a particular strain that are shared with at least one other member of the Clade 7. The remainder of the numbers represents the number of unique genes for each individual strain. B. Role category profiling of genes that are unique for B. anthracis Ames and Clade 6 B. cereus genomes.
Acquisition of Virulence Associated Genes in B. anthracis
We conducted a specific mining of the dataset to identify the characteristic genomic events associated with the evolution and the emergence of B. anthracis. Initially we identified a group of 1,512 genes that were characteristic for B. anthracis compared to the all B. cereus group genomes analyzed, (which appeared to have 302 genes in common). It should be noted that not all the genes that were characteristic for B. anthracis by marker association analysis were unique. Some of them, despite being present in all B. anthracis genomes, were occasionally present in other B. cereus genomes [6]. The picture that emerges from this analysis supports the idea that the evolution of the B. anthracis lineage from its predecessors is complex involving more than the acquisition of the two virulence plasmids. Therefore, we expanded our analysis to additional genes that may have contributed to the enhanced B. anthracis virulence, beyond those encoding tripartite toxins and the capsule.
Among the oligonucleotides represented on the DNA microarray, are 772 markers that were annotated to represent putative virulence associated genes (VAGs). This VAG set may be divided into two sub-groups. The first one is comprised primarily of toxins, capsular genes, cytadhesins, invasins, hemolysins, collagenases, and phospholipases. The second sub-group includes genes whose products are predicted to contain, or are in the involved acquisition and utilization of trace elements such as iron, cobalt, zinc, and copper. With the exception of zinc, which is a component of metallo-proteases, other elements function as co-factors primarily involved in redox pathways. While the proteins of the first group are considered primary virulence factors, those belonging to the second group may be considered as accessory genes that play a role in adaptation to the host environment, especially under stress conditions imposed by the human immune response [59-71].
Marker association analysis identified a small group of 31 VAGs (Ba-VAGs) that are characteristic for B. anthracis compared to the remainder of B. cereus genomes analyzed (Table 3). With the exception of toxin and capsule encoding genes, many Ba-VAGs are shared among other Clade 4 members.
Table 3.
Characteristic virulence associated genes for the clades of B. anthracis and its near neighbors.
| Locus | Common Name | Characteristic for | |
|---|---|---|---|
| Clade 4 genomes vs. other B. cereus | B. anthracis vs. B. cereus | ||
| BA0552 | internalin, putative | (+)a | + |
| BA1489 | superoxide dismutase | + | |
| BA1760 | cobalamin synthesis protein, putative | + | + |
| BA1902 | multicopper oxidase family protein | + | |
| BA1925 | cobalamin synthesis protein/P47K family protein | + | + |
| BA1981 o | siderophore biosynthesis protein, putative | + | + |
| BA1982 o | siderophore biosynthesis protein, putative | + | + |
| BA1983 o | AMP-binding protein | + | + |
| BA1984 o | hypothetical protein | + | + |
| BA1985 o | hypothetical protein | + | + |
| BA1986 o | conserved hypothetical protein | + | + |
| BA1992 | phospholipase, putative | + | |
| BA2222 | alcohol dehydrogenase, iron-containing | + | + |
| BA2372 o | nonribosomal peptide synthetase DhbF | + | + |
| BA2588 | alcohol dehydrogenase, zinc-containing | + | + |
| BA2642 | cobalt transport protein | + | + |
| BA2730 | neutral protease | + | |
| BA3100 | copper homeostasis protein CutC, putative | + | |
| BA3131 | alcohol dehydrogenase, zinc-containing | + | |
| BA3189 | manganese ABC transporter, substrate-binding protein/adhesin | (+)a | + |
| BA3269 | iron-sulfur cluster-binding protein | + | + |
| BA3299 | microbial collagenase, putative | + | |
| BA3307 o | L-serine dehydratase, iron-sulfur-dependent, alpha subunit | + | + |
| BA3308 o | L-serine dehydratase, iron-sulfur-dependent, beta subunit | + | + |
| BA3435 o | alcohol dehydrogenase, zinc-containing | + | |
| BA3442 | neutral protease | + | + |
| BA3515 | alcohol dehydrogenase, zinc-containing, authentic point mutation | + | |
| BA3584 | microbial collagenase, putative | + | |
| BA3703 | phospholipase/carboxylesterase family protein | + | |
| BA3922 | zinc protease, insulinase family | + | |
| BA4766 o | iron compound ABC transporter, iron compound-binding protein | + | + |
| BA4767 o | iron compound ABC transporter, permease protein | + | + |
| BA4784 o | iron compound ABC transporter, ATP-binding protein | + | |
| BA5701 | channel protein, hemolysin III family | + | |
| BXA0142 | calmodulin-sensitive adenylate cyclase, Cya | + b | |
| BXA0146 | transcriptional activator AtxA, (pXO1-119) | + b | |
| BXA0163 | protective antigen-related protein, (pXO1-111) | + b | |
| BXA0164 | protective antigen, PagA | + b | |
| BXA0172 | lethal factor, Lef | + b | |
| BXA0197 | zinc-binding lipoprotein AdcA domain protein, (pXO1-130) | + b | |
| BXB0060 | capsule synthesis trans-acting positive regulator, putative, (pXO2-53) | + | |
| BXB0062 o | hypothetical protein, CapE (pXO2-54) | + b | |
| BXB0063 o | gamma-glutamyltranspeptidase, capD (pXO2-55) | + b | |
| BXB0064 o | capsule biosynthesis protein CapA, (pXO2-56) | + | |
| BXB0065 o | capsule biosynthesis protein CapC, (pXO2-57) | + b | |
| BXB0066 o | capsule biosynthesis protein CapB, (pXO2-58) | + b | |
| BXB0084 | capsule synthesis trans-acting positive regulator, CapR (pXO2-64) | + b | |
Present in all B. anthracis strains and three B. cereus of Cl.4, Clade2, and Clade1.
Present in all B. anthracis strain and three B. cereus one of which belong to Clade 4.
Gene is part of an operon.
In our analysis of VAGs in B. cereus lineages we noted that clades comprised of human associated strains do not necessarily encode more VAGs than strains of environmental origin. For example, we identified 29 VAGs that were characteristic for Clade 3 compared to 103 that were characteristic for Clade 1 strains. Likewise, Clade 4 genomes appear to have fewer characteristic VAGs when compared to the other Bc strains — 37 vs. 47. We also conducted a global clade distribution analysis with respect to VAGs and found that that B. anthracis and its near neighbors have indeed fewer VAGs than their distant relatives (Figure 6). Taken together, these findings may indicate that the evolution of B. anthracis was associated with the acquisition and/or maintenance of a limited but specific set of chromosomally encoded VAGs in addition to those encoded on the virulence plasmids.
Figure 6.
Summary of VAG content distribution analysis among various groups of genomes.
DISCUSSION
The generation of diversity is of fundamental importance for most microbial populations. These diversification processes, together with environmental selection, drive cell adaptation [72-74]. In this study, we attempted to elucidate evolutionary aspects or trends associated with the emergence of one of the hyper-virulent variants of this taxon, B. anthracis. Genes identified through targeted genome sequencing of diverse isolates were complemented with sequences from publicly available sources to design a comprehensive oligonucleotide-based species microarray. This microarray was then used to screen a diverse group of strains for their genomic content by CGH.
Total gene estimates for the majority of the query strains varied from 5,000 to 6,000, which is consistent with previous findings regarding genome size of spore forming members of the B. cereus group (NCBI (http://www.ncbi.nlm.nih.gov ) and [4-6,10,11,13,39]). Some exceptions were noted as some strains displayed markedly reduced coding capacity and total gene estimates as low as 3,400 – 4,500, or as high as 6,500. These findings are consistent with a recent report on a highly cytotoxic B. cereus strain with genome size of 4.2 Mbp [75].
The profiling of putative role categories associated with both novel sequence identification and CGH data provided a global perspective with regard to the diversity of genome complements defining Bc group members. Based on all data generated, we conclude that 67% of the strain-variable CDS identified in the seven Bc strains analyzed correspond to seven role categories including genes of unknown function, mobile elements, transport, regulatory functions, cellular processes, energy metabolism, and cell envelope. Interestingly, the same role categories constitute the majority of functions that were absent in these seven query strains as compared to the reference genome. Hence, we infer that the heterogeneity of gene complements throughout this lineage has been driven by significant horizontal gene acquisition, and orthologous and non-orthologous gene displacements events.
Plasmids play an important role in the biology of Bacillus species. Together with other mobile elements such as bacteriophages, they enable horizontal gene transfer among members of this genus, a major force driving genetic diversity. Plasmids vary considerably in size and gene content among Bacillus spp. and contribute strongly to genome variability. pXO1-like plasmids are common among Clade 3 members, and together with pXO2 variants, pXO1-like plasmids are prevalent among isolates of human origin. In contrast, pBC218 variants are frequently found among non-clinical strains, or strains outside of the Clade 3 lineage. Our data do not allow us to define gene content as plasmid based or chromosomal. However, the presence of previously identified virulence factors and capsular genes among the gene complements of B. cereus isolates is of interest and potential importance. Two of the four Clade 6 genomes appear to harbor parts of both pXO1 and pXO2. The relevance these plasmid sequences in the pathobiology of the periodontal-associated strains constituting Clade 6 is not yet clear. Clade 6 strains in this study, together with isolates obtained from metal workers in Texas, US, [17] and wild great apes from Coté d’Ivoire, Cameroon, [15] causing anthrax-like disease are the only B. cereus strains to date that appear to harbor both pXO1- and pXO2-like plasmids. These periodontal strains, however, are missing both the B. anthracis pathogenicity island and the cap gene locus, further emphasizing the major role of these two loci in the emergence and pathogenicity of B. anthracis. Taken together, these observations suggest that the evolution of both B. anthracis plasmids appears to have been a complex process involving multiple gene acquisition events.
The phylogenomic relationships inferred by complete gene complement-based clustering do not in general, reproduce the phylogeny of a species in terms of vertical evolution, but rather depict the overall relatedness of genomes to one another. We compared the B. cereus var. anthracis CI genome [76] content and its relatedness with other genomes we investigated in this study by simulating CGH as previously reported [40]. From both marker association as well as phylogenomic analyses B. cereus var. anthracis CI appears to represent a distinct lineage within Clade 3 that is closely related to Clade 4. This finding further suggests that acquisition of both pXO1 and pXO2—as well as the full expression of the virulence genes they carry—by B. cereus var. anthracis CI may have taken place in a particular genomic (chromosomal) background.
A systematic comparative clade analysis indicates that gene acquisition and fixation appear to have driven early Clade 3 members and subsequently those constituting the Clade 4 lineage toward an altered interaction with the (mammalian) host, which eventually resulted in variants with a distinct pathobiology. The observed lineage-specific gene gain and loss events may reflect step-wise adaptation to a new selective host environment or may reflect fitness adaptation in response to the emergence of a lineage with a hyper-virulent phenotype. Although Clade 5 and Clade 6 represent near-neighbors of B. anthracis, neither the gene acquisition and gene loss events, nor the specific relevance of all these events to anthrax disease can be deduced with absolute clarity. However, it does appear that the emergence of B. anthracis is complex, involving many events, not limited simply to the acquisition of the pathogenicity island and cap genes on pXO1 and pXO2. The evolution of B. anthracis or its predecessors appears to be characterized by reductions in the number of genes involved in metabolism of arginine and proline, biosynthesis of glutamate-, aspartate- and pyruvate derivatives involved in amino acid biosynthesis, degradation of proteins, peptides, and glycopeptides, degradation of amino acids and amines, biosynthesis of thiamine, metabolism of butanoate, starch and sucrose, sugar transport, biosynthesis of secondary metabolites, metabolism in diverse environments, adaptation to atypical conditions as well as DNA recombination and repair. In addition, all these alterations appear to be associated with the acquisition of mobile elements and CDSs of unknown functions.
The finding that Clade 4 has a significantly reduced number of features involved in DNA recombination and repair is interesting and may provide support to the hypothesis of Didelot et al [77] that suggests that the pattern of gene acquisition may be indicative of a shift in recombination boundaries. A similar pattern has been previously observed between Campylobacter jejuni and C. coli resulting in changes in their ecology [78]. According to this hypothesis, different from other bacteria, the mismatch repair system in Bacillus may play a moderate role in the prevention of recombination resulting in higher promiscuity of gene acquisition. The CGH data we present in this report are congruent with those from the MLST study by Didelot et al. [77] indicating that there is a major gene acquisition shift that appears to have taken place within Clade 4 that strongly correlates with the emergence of a lineage—B. anthracis—that is highly invasive for mammals.
Comparative genome analyses enabled the identification of several mechanisms employed by pathogens to survive within the host. Observed differences between Clade 1 and Clade 3 with respect to urea/amide transporters are interesting, as some pathogens, especially those associated with enteric infections, are known to use urea metabolism as a means to neutralize acidic conditions [59,79,80]. The finding that B. anthracis possesses more transporters involved in iron acquisition is also significant, further underscoring the importance of iron in the virulence process [60,66]. Another mechanism pertained to myo-inositol degradation. Myo-Inositol is highly abundant within the host serving as a structural component of the eukaryotic cell membrane, which also acts as a signaling molecule. It appears that Clade 4 members including B. anthracis, employ scavenging mechanism for energy resources. This pattern is congruent with the sialic acid degradation pathway employed by pathogenic vibrios [81].
The acquisition of specific genes, e.g. VAGs, in strains with elevated pathogenicity, appears to have occurred in concert with an overall genome size reduction in B. anthracis, indicating niche speciation [82]. Gene acquisition events may lead to a shift in host environment or tissue tropism. This shift may have a fundamental impact on the genome since previously useful genes may no longer serve as such. In such cases, the rate of fixed mutations increases dramatically and involves pseudogene formation, IS element activity and movement, genome rearrangements and gene loss [74,83]. Adaptation to the host environment may be driving the loss of genes that are no longer essential in the host environment. The gene loss profile observed among the seven genomes comprising Clade 4, and especially B. anthracis, suggests that genome evolution is driving the microbial cell toward deeper dependency on the host for energy and metabolites. At the same time, this process has been accompanied by the acquisition of VAGs by early Clade 3 and especially Clade 4 members that may represent the initial steps towards niche speciation i.e. in a mammalian host [29,84]. Then, the acquisition of the pathogenicity islands coding for the three toxins and cap gene locus may have further enhanced the invasive capabilities of a clone, resulting in a genomic variant that is able to consistently cause anthrax. Therefore, acquisition of these two loci must have been a critical point in the evolution of the B. anthracis. The combined evidence from comparative clade analysis and plasmid profile suggests that members of this lineage have been undergoing rapid evolution towards a higher level of niche adaptation.
Analysis of strains by the marker set representing presumed virulence factors suggests that the evolution of highly virulent B. anthracis from its predecessors is associated with the fixation of a specific set of VAGs. In addition to being functionally characterized as virulence determinants, some of these proteins have been shown to have immunogenic properties and are possible therapeutic targets [85-87]. The identification of genes encoding proteins containing or involved in the acquisition of trace elements, besides those involved in cytadherence or invasion, is significant. Enzymes that bind trace elements are mainly involved in redox processes which are essential for microbial growth and survival, especially under stress conditions e.g. inflammation or phagocytosis [70,88-93]. Therefore it is no surprise that many pathogens, including B. anthracis, appear to possess a large repertoire of genes involved in trace element metabolism [59-71,94]. These genes may play an important role in establishing and maintaining infection, particularly within the phagolysosomes of macrophages or polymorpho-nuclear leukocytes [70,88-93].
Extensive research on B. cereus pathogenesis has resulted in the identification of many virulence factors and our knowledge about their mechanisms and interactions is still expanding. As an opportunistic pathogen, B. cereus is often associated with diarrheal and emetic food poisoning. Additionally, in rare cases involving individuals with impaired immune defenses or trauma B. cereus has been shown to cause severe systemic or local infection [17,31,34]. On the other hand, there is mounting evidence originating from genome research and eco-microbiological studies indicating that members of the B. cereus group may be normal inhabitants of arthropod intestines [6,95,96]. From the analysis of sequences obtained from gene discovery we found that almost all of the annotated VAGs were found among non-clinical isolates. CGH profiling also demonstrated that non-clinical strains, in general, had larger sets of putative virulence factors. It is known that the pleotropic regulatory gene plcR, is fully functional among the B. cereus but inactive in B. anthracis due to a frame-shift mutation [6,95,96]. In the case of Clade 4 genomes and B. anthracis in particular, evolution seem to entail reduction of VAGs for which regulatory factors such as PlcR are no longer necessary. It is worth noting here that almost all the chromosomal VAGs found to be characteristic for either B. anthracis or its near neighbors within Clade 4 were found only occasionally in genomes belonging to non-clinical B. cereus strains from distant lineages.
The finding of a PagA variant in the genome of an environmental isolate (B. weihenstephanensis AH1143) may provide some additional insights about the natural reservoir and extent of diversity of one of the B. anthracis toxin genes. Our finding, together with previously published evidence [13,97], further supports the hypothesis that variants of B. anthracis toxin genes such as pag and lef may exist among other members of B. cereus sensu lato group, although their role outside the mammalian host has yet to be determined.
Taken together, results of this study allowed us to better recognize the sources of genome diversity, understand the relationships among members of B. cereus group, and further elucidate aspects of their genome evolution, especially events associated with the evolution of B. anthracis lineage and its close relatives within this taxon. Furthermore, the type of analytical approach we presented here will help improve diagnostics and open the avenues for developing predictive cladistic models to improve our understanding of genome evolution associated with clone emergence. Such efforts will enable the identifications genomic markers that can be used for diagnostic purposes and finally assist both drug- and vaccine development.
Supplementary Material
Gene Discovery (GD). Random genomic libraries are generated for each B. cereus strain. Cloned genomic fragments are common (uncolored boxes) or unique (colored boxes). Plasmid DNAs are printed onto glass slide microarrays and subsequently probed with Cy-labeled query B. cereus and B. anthracis in a competitive hybridization. Those plasmids that do not hybridize with B. anthracis were considered to contain unique genomic fragments (novel genes) and were sequence characterized. Other plasmids that hybridized weakly were considered to contain divergent sequences were also sequenced.
Summary of the bioinformatic approach used to characterize the unique genomic fragments from B. cereus strains subjected gene discovery.
Relative frequencies of novel genes discovered among seven query B. cereus genomes based on predicted functional role categories.
Total gene content estimates for Bacillus cereus group members.
Comparative cluster analysis of strains based on CGH data. Left, clustering based on the global gene hybridization patterns (~29977 70-mer oligonucleotides) using the trinary designations “0”, “0.5” and “1” corresponding to “absent”, “divergent” and “present” CDSs respectively. Right, clustering of the 950 markers representing known plasmid sequences.
Acknowledgments
We thank Dr. Jacques Ravel for kindly providing us with genomic DNA from B. anthracis Vollum, Australia 94, Tsiankovskii-I, as well as Dr. Martin Blaser for providing us with genomic DNA B. anthracis stains Sterne 34F2 and Pasteur. We also thank Mr. Dritan Papazisi for his suggestions regarding linear modeling predictions. This work was supported by the NIAID contract No. N01-AI-15447 to Pathogen Functional Genomics Resource Center at the JCVI.
Footnotes
Sequence Accession Numbers. The sequence data from this study have been submitted to the NCBI trace archive under the following accession numbers: B. cereus AH819: 2260380778-2260383516; B. cereus AH607: 2260383517-2260385933; B. cereus AH535: 2260385934-2260389759; B. cereus AH1123: 2260389760-2260391507; B. cereus AH812: 2260391508-2260391717; B. cereus AH259: 2260396559-2260397178; B. weihenstephanensis AH1143: 2260391718-2260396558. Sequences can also be accessed at the web site of the Pathogen Functional Genomics Resource Center at the JCVI http://pfgrc.jcvi.org/index.php/white_papers/project_description/2008/2008_b_anthracis_characterization.html#project1.
Microarray data deposition. The sequence data from this study have been submitted to the NCBI Gene Expression omnibus (GEO) under accession number GSE1906.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ash C, Farrow JA, Dorsch M, Stackebrandt E, Collins MD. Comparative analysis of Bacillus anthracis, Bacillus cereus, and related species on the basis of reverse transcriptase sequencing of 16S rRNA. Int J Syst Bacteriol. 1991;41:343–6. doi: 10.1099/00207713-41-3-343. [DOI] [PubMed] [Google Scholar]
- 2.Chen ML, Tsen HY. Discrimination of Bacillus cereus and Bacillus thuringiensis with 16S rRNA and gyrB gene based PCR primers and sequencing of their annealing sites. J Appl Microbiol. 2002;92:912–9. doi: 10.1046/j.1365-2672.2002.01606.x. [DOI] [PubMed] [Google Scholar]
- 3.Daffonchio D, Cherif A, Borin S. Homoduplex and heteroduplex polymorphisms of the amplified ribosomal 16S-23S internal transcribed spacers describe genetic relationships in the “Bacillus cereus group”. Appl Environ Microbiol. 2000;66:5460–8. doi: 10.1128/aem.66.12.5460-5468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Helgason E, Okstad OA, Caugant DA, Johansen HA, Fouet A, Mock M, Hegna I, Kolsto AB. Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis--one species on the basis of genetic evidence. Appl Environ Microbiol. 2000;66:2627–30. doi: 10.1128/aem.66.6.2627-2630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turnbull PC, Hutson RA, Ward MJ, Jones MN, Quinn CP, Finnie NJ, Duggleby CJ, Kramer JM, Melling J. Bacillus anthracis but not always anthrax. J Appl Bacteriol. 1992;72:21–8. doi: 10.1111/j.1365-2672.1992.tb04876.x. [DOI] [PubMed] [Google Scholar]
- 6.Kolsto AB, Tourasse NJ, Okstad OA. What sets Bacillus anthracis apart from other Bacillus species? Annu Rev Microbiol. 2009;63:451–76. doi: 10.1146/annurev.micro.091208.073255. [DOI] [PubMed] [Google Scholar]
- 7.Gonzalez JM, Jr, Dulmage HT, Carlton BC. Correlation between specific plasmids and delta-endotoxin production in Bacillus thuringiensis. Plasmid. 1981;5:352–65. doi: 10.1016/0147-619x(81)90010-x. [DOI] [PubMed] [Google Scholar]
- 8.Han CS, Xie G, Challacombe JF, Altherr MR, Bhotika SS, Brown N, Bruce D, Campbell CS, Campbell ML, Chen J, Chertkov O, Cleland C, Dimitrijevic M, Doggett NA, Fawcett JJ, Glavina T, Goodwin LA, Green LD, Hill KK, Hitchcock P, Jackson PJ, Keim P, Kewalramani AR, Longmire J, Lucas S, Malfatti S, McMurry K, Meincke LJ, Misra M, Moseman BL, Mundt M, Munk AC, Okinaka RT, Parson-Quintana B, Reilly LP, Richardson P, Robinson DL, Rubin E, Saunders E, Tapia R, Tesmer JG, Thayer N, Thompson LS, Tice H, Ticknor LO, Wills PL, Brettin TS, Gilna P. Pathogenomic sequence analysis of Bacillus cereus and Bacillus thuringiensis isolates closely related to Bacillus anthracis. J Bacteriol. 2006;188:3382–90. doi: 10.1128/JB.188.9.3382-3390.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ivanova N, Sorokin A, Anderson I, Galleron N, Candelon B, Kapatral V, Bhattacharyya A, Reznik G, Mikhailova N, Lapidus A, Chu L, Mazur M, Goltsman E, Larsen N, D’Souza M, Walunas T, Grechkin Y, Pusch G, Haselkorn R, Fonstein M, Ehrlich SD, Overbeek R, Kyrpides N. Genome sequence of Bacillus cereus and comparative analysis with Bacillus anthracis. Nature. 2003;423:87–91. doi: 10.1038/nature01582. [DOI] [PubMed] [Google Scholar]
- 10.Rasko DA, Altherr MR, Han CS, Ravel J. Genomics of the Bacillus cereus group of organisms. FEMS Microbiol Rev. 2005;29:303–29. doi: 10.1016/j.femsre.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 11.Rasko DA, Ravel J, Okstad OA, Helgason E, Cer RZ, Jiang L, Shores KA, Fouts DE, Tourasse NJ, Angiuoli SV, Kolonay J, Nelson WC, Kolsto AB, Fraser CM, Read TD. The genome sequence of Bacillus cereus ATCC 10987 reveals metabolic adaptations and a large plasmid related to Bacillus anthracis pXO1. Nucleic Acids Res. 2004;32:977–88. doi: 10.1093/nar/gkh258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Read TD, Peterson SN, Tourasse N, Baillie LW, Paulsen IT, Nelson KE, Tettelin H, Fouts DE, Eisen JA, Gill SR, Holtzapple EK, Okstad OA, Helgason E, Rilstone J, Wu M, Kolonay JF, Beanan MJ, Dodson RJ, Brinkac LM, Gwinn M, DeBoy RT, Madpu R, Daugherty SC, Durkin AS, Haft DH, Nelson WC, Peterson JD, Pop M, Khouri HM, Radune D, Benton JL, Mahamoud Y, Jiang L, Hance IR, Weidman JF, Berry KJ, Plaut RD, Wolf AM, Watkins KL, Nierman WC, Hazen A, Cline R, Redmond C, Thwaite JE, White O, Salzberg SL, Thomason B, Friedlander AM, Koehler TM, Hanna PC, Kolsto AB, Fraser CM. The genome sequence of Bacillus anthracis Ames and comparison to closely related bacteria. Nature. 2003;423:81–6. doi: 10.1038/nature01586. [DOI] [PubMed] [Google Scholar]
- 13.Hoffmaster AR, Ravel J, Rasko DA, Chapman GD, Chute MD, Marston CK, De BK, Sacchi CT, Fitzgerald C, Mayer LW, Maiden MC, Priest FG, Barker M, Jiang L, Cer RZ, Rilstone J, Peterson SN, Weyant RS, Galloway DR, Read TD, Popovic T, Fraser CM. Identification of anthrax toxin genes in a Bacillus cereus associated with an illness resembling inhalation anthrax. Proc Natl Acad Sci U S A. 2004;101:8449–54. doi: 10.1073/pnas.0402414101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu X, Van der Auwera G, Timmery S, Zhu L, Mahillon J. Distribution, diversity, and potential mobility of extrachromosomal elements related to the Bacillus anthracis pXO1 and pXO2 virulence plasmids. Appl Environ Microbiol. 2009;75:3016–28. doi: 10.1128/AEM.02709-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klee SR, Ozel M, Appel B, Boesch C, Ellerbrok H, Jacob D, Holland G, Leendertz FH, Pauli G, Grunow R, Nattermann H. Characterization of Bacillus anthracis-like bacteria isolated from wild great apes from Cote d’Ivoire and Cameroon. J Bacteriol. 2006;188:5333–44. doi: 10.1128/JB.00303-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rasko DA, Rosovitz MJ, Okstad OA, Fouts DE, Jiang L, Cer RZ, Kolsto AB, Gill SR, Ravel J. Complete sequence analysis of novel plasmids from emetic and periodontal Bacillus cereus isolates reveals a common evolutionary history among the B. cereus-group plasmids, including Bacillus anthracis pXO1. J Bacteriol. 2007;189:52–64. doi: 10.1128/JB.01313-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoffmaster AR, Hill KK, Gee JE, Marston CK, De BK, Popovic T, Sue D, Wilkins PP, Avashia SB, Drumgoole R, Helma CH, Ticknor LO, Okinaka RT, Jackson PJ. Characterization of Bacillus cereus isolates associated with fatal pneumonias: strains are closely related to Bacillus anthracis and harbor B. anthracis virulence genes. J Clin Microbiol. 2006;44:3352–60. doi: 10.1128/JCM.00561-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Modrie P, Beuls E, Mahillon J. Differential transfer dynamics of pAW63 plasmid among members of the Bacillus cereus group in food microcosms. J Appl Microbiol. 2010;108:888–97. doi: 10.1111/j.1365-2672.2009.04488.x. [DOI] [PubMed] [Google Scholar]
- 19.Timmery S, Modrie P, Minet O, Mahillon J. Plasmid capture by the Bacillus thuringiensis conjugative plasmid pXO16. J Bacteriol. 2009;191:2197–205. doi: 10.1128/JB.01700-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van der Auwera GA, Timmery S, Hoton F, Mahillon J. Plasmid exchanges among members of the Bacillus cereus group in foodstuffs. Int J Food Microbiol. 2007;113:164–72. doi: 10.1016/j.ijfoodmicro.2006.06.030. [DOI] [PubMed] [Google Scholar]
- 21.Hoflack L, Wilcks A, Andrup L, Mahillon J. Functional insights into pGI2, a cryptic rolling-circle replicating plasmid from Bacillus thuringiensis. Microbiology. 1999;145(Pt 7):1519–30. doi: 10.1099/13500872-145-7-1519. [DOI] [PubMed] [Google Scholar]
- 22.Helgason E, Caugant DA, Olsen I, Kolsto AB. Genetic structure of population of Bacillus cereus and B. thuringiensis isolates associated with periodontitis and other human infections. J Clin Microbiol. 2000;38:1615–22. doi: 10.1128/jcm.38.4.1615-1622.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu X, Swiecicka I, Timmery S, Mahillon J. Sympatric soil communities of Bacillus cereus sensu lato: population structure and potential plasmid dynamics of pXO1- and pXO2-like elements. FEMS Microbiol Ecol. 2009;70:344–55. doi: 10.1111/j.1574-6941.2009.00771.x. [DOI] [PubMed] [Google Scholar]
- 24.Koehler TM. Bacillus anthracis. In: Fischetti V, editor. Gram-Positive Pathogens. 2. Washington DC: ASM Press; 2006. pp. 659–671. [Google Scholar]
- 25.Pannucci J, Okinaka RT, Sabin R, Kuske CR. Bacillus anthracis pXO1 plasmid sequence conservation among closely related bacterial species. J Bacteriol. 2002;184:134–41. doi: 10.1128/JB.184.1.134-141.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pannucci J, Okinaka RT, Williams E, Sabin R, Ticknor LO, Kuske CR. DNA sequence conservation between the Bacillus anthracis pXO2 plasmid and genomic sequence from closely related bacteria. BMC Genomics. 2002;3:34. doi: 10.1186/1471-2164-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okinaka RT, Cloud K, Hampton O, Hoffmaster AR, Hill KK, Keim P, Koehler TM, Lamke G, Kumano S, Mahillon J, Manter D, Martinez Y, Ricke D, Svensson R, Jackson PJ. Sequence and organization of pXO1, the large Bacillus anthracis plasmid harboring the anthrax toxin genes. J Bacteriol. 1999;181:6509–15. doi: 10.1128/jb.181.20.6509-6515.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okinaka R, Cloud K, Hampton O, Hoffmaster A, Hill K, Keim P, Koehler T, Lamke G, Kumano S, Manter D, Martinez Y, Ricke D, Svensson R, Jackson P. Sequence, assembly and analysis of pX01 and pX02. J Appl Microbiol. 1999;87:261–2. doi: 10.1046/j.1365-2672.1999.00883.x. [DOI] [PubMed] [Google Scholar]
- 29.Mock M, Fouet A. Anthrax. Annu Rev Microbiol. 2001;55:647–71. doi: 10.1146/annurev.micro.55.1.647. [DOI] [PubMed] [Google Scholar]
- 30.Jensen GB, Hansen BM, Eilenberg J, Mahillon J. The hidden lifestyles of Bacillus cereus and relatives. Environ Microbiol. 2003;5:631–40. doi: 10.1046/j.1462-2920.2003.00461.x. [DOI] [PubMed] [Google Scholar]
- 31.Drobniewski FA. Bacillus cereus and related species. Clin Microbiol Rev. 1993;6:324–38. doi: 10.1128/cmr.6.4.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schuch R, Fischetti VA. The secret life of the anthrax agent Bacillus anthracis: bacteriophage-mediated ecological adaptations. PLoS One. 2009;4:e6532. doi: 10.1371/journal.pone.0006532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schuch R, Pelzek AJ, Kan S, Fischetti VA. Prevalence of Bacillus anthracis-like organisms and bacteriophages in the intestinal tract of the earthworm Eisenia fetida. Appl Environ Microbiol. 2010;76:2286–94. doi: 10.1128/AEM.02518-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beecher DJ. The Bacillus cereus group. In: Sussman M, editor. Molecular medical microabiology. Academic press; 2002. pp. 1162–1190. [Google Scholar]
- 35.Agaisse H, Gominet M, Okstad OA, Kolsto AB, Lereclus D. PlcR is a pleiotropic regulator of extracellular virulence factor gene expression in Bacillus thuringiensis. Mol Microbiol. 1999;32:1043–53. doi: 10.1046/j.1365-2958.1999.01419.x. [DOI] [PubMed] [Google Scholar]
- 36.Cohan FM. Bacterial species and speciation. Syst Biol. 2001;50:513–24. doi: 10.1080/10635150118398. [DOI] [PubMed] [Google Scholar]
- 37.Pearson T, Busch JD, Ravel J, Read TD, Rhoton SD, U’Ren JM, Simonson TS, Kachur SM, Leadem RR, Cardon ML, Van Ert MN, Huynh LY, Fraser CM, Keim P. Phylogenetic discovery bias in Bacillus anthracis using single-nucleotide polymorphisms from whole-genome sequencing. Proc Natl Acad Sci U S A. 2004;101:13536–41. doi: 10.1073/pnas.0403844101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Priest FG, Barker M, Baillie LW, Holmes EC, Maiden MC. Population structure and evolution of the Bacillus cereus group. J Bacteriol. 2004;186:7959–70. doi: 10.1128/JB.186.23.7959-7970.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Helgason E, Tourasse NJ, Meisal R, Caugant DA, Kolsto AB. Multilocus sequence typing scheme for bacteria of the Bacillus cereus group. Appl Environ Microbiol. 2004;70:191–201. doi: 10.1128/AEM.70.1.191-201.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Papazisi L, Ratnayake S, Remortel BG, Bock GR, Liang W, Saeed AI, Fleischmann RD, Kilian M, Peterson SN. Tracing phylogenomic events leading to diversity of Haemophilus influenzae and the emergence of Brazilian Purpuric Fever (BPF)-associated clones. Genomics. 2010;96:290–302. doi: 10.1016/j.ygeno.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985;33:103–19. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- 42.Moore DD. Overview of recombinant DNA libraries. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, editors. Current protocols in molecular biology. New York: John Wiley & Sons; 1993. pp. 5.1.1–5.1.3. [Google Scholar]
- 43.Peterson SN, Sung CK, Cline R, Desai BV, Snesrud EC, Luo P, Walling J, Li H, Mintz M, Tsegaye G, Burr PC, Do Y, Ahn S, Gilbert J, Fleischmann RD, Morrison DA. Identification of competence pheromone responsive genes in Streptococcus pneumoniae by use of DNA microarrays. Mol Microbiol. 2004;51:1051–70. doi: 10.1046/j.1365-2958.2003.03907.x. [DOI] [PubMed] [Google Scholar]
- 44.Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, Kravitz SA, Mobarry CM, Reinert KH, Remington KA, Anson EL, Bolanos RA, Chou HH, Jordan CM, Halpern AL, Lonardi S, Beasley EM, Brandon RC, Chen L, Dunn PJ, Lai Z, Liang Y, Nusskern DR, Zhan M, Zhang Q, Zheng X, Rubin GM, Adams MD, Venter JC. A whole-genome assembly of Drosophila. Science. 2000;287:2196–204. doi: 10.1126/science.287.5461.2196. [DOI] [PubMed] [Google Scholar]
- 45.Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, Deboy RT, Davidsen TM, Mora M, Scarselli M, Margarit y Ros I, Peterson JD, Hauser CR, Sundaram JP, Nelson WC, Madupu R, Brinkac LM, Dodson RJ, Rosovitz MJ, Sullivan SA, Daugherty SC, Haft DH, Selengut J, Gwinn ML, Zhou L, Zafar N, Khouri N, Radune D, Dimitrov G, Watkins K, O’Connor KJ, Smith S, Utterback TR, White O, Rubens CE, Grandi G, Madoff LC, Kasper DL, Telford JL, Wessels MR, Rappuoli R, Fraser CM. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci U S A. 2005;102:13950–5. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27:4636–41. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 48.Bozdech Z, Zhu J, Joachimiak MP, Cohen FE, Pulliam B, DeRisi JL. Expression profiling of the schizont and trophozoite stages of Plasmodium falciparum with a long-oligonucleotide microarray. Genome Biol. 2003;4:R9. doi: 10.1186/gb-2003-4-2-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–8. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 50.Champion OL, Gaunt MW, Gundogdu O, Elmi A, Witney AA, Hinds J, Dorrell N, Wren BW. Comparative phylogenomics of the food-borne pathogen Campylobacter jejuni reveals genetic markers predictive of infection source. Proc Natl Acad Sci U S A. 2005;102:16043–8. doi: 10.1073/pnas.0503252102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leavis HL, Willems RJ, van Wamel WJ, Schuren FH, Caspers MP, Bonten MJ. Insertion sequence-driven diversification creates a globally dispersed emerging multiresistant subspecies of E. faecium. PLoS Pathog. 2007;3:e7. doi: 10.1371/journal.ppat.0030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–4. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 53.Stabler RA, Gerding DN, Songer JG, Drudy D, Brazier JS, Trinh HT, Witney AA, Hinds J, Wren BW. Comparative Phylogenomics of Clostridium difficile Reveals Clade Specificity and Microevolution of Hypervirulent Strains. J Bacteriol. 2006;188:7297–7305. doi: 10.1128/JB.00664-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zar JH. Biostatistical analysis. 4. Upper Saddle River NJ, USA: Prentice Hall; 1999. [Google Scholar]
- 55.Brenner DJ, Mayer LW, Carlone GM, Harrison LH, Bibb WF, Brandileone MC, Sottnek FO, Irino K, Reeves MW, Swenson JM, et al. Biochemical, genetic, and epidemiologic characterization of Haemophilus influenzae biogroup aegyptius (Haemophilus aegyptius) strains associated with Brazilian purpuric fever. J Clin Microbiol. 1988;26:1524–34. doi: 10.1128/jcm.26.8.1524-1534.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brenner DJ, Staley JT, Krieg NR. Classification of prokaryotic organisms and the concept of bacteria species. In: Garrity GM, editor. Bergey’s manual of systematic bacteriology. 2. New York: Springer-Verlag; 2001. pp. 27–31. [Google Scholar]
- 57.Jackson PJ, Hill KK, Laker MT, Ticknor LO, Keim P. Genetic comparison of Bacillus anthracis and its close relatives using amplified fragment length polymorphism and polymerase chain reaction analysis. J Appl Microbiol. 1999;87:263–9. doi: 10.1046/j.1365-2672.1999.00884.x. [DOI] [PubMed] [Google Scholar]
- 58.Hill KK, Ticknor LO, Okinaka RT, Asay M, Blair H, Bliss KA, Laker M, Pardington PE, Richardson AP, Tonks M, Beecher DJ, Kemp JD, Kolsto AB, Wong AC, Keim P, Jackson PJ. Fluorescent amplified fragment length polymorphism analysis of Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis isolates. Appl Environ Microbiol. 2004;70:1068–80. doi: 10.1128/AEM.70.2.1068-1080.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bosse JT, Gilmour HD, MacInnes JI. Novel genes affecting urease acivity in Actinobacillus pleuropneumoniae. J Bacteriol. 2001;183:1242–7. doi: 10.1128/JB.183.4.1242-1247.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cendrowski S, MacArthur W, Hanna P. Bacillus anthracis requires siderophore biosynthesis for growth in macrophages and mouse virulence. Mol Microbiol. 2004;51:407–17. doi: 10.1046/j.1365-2958.2003.03861.x. [DOI] [PubMed] [Google Scholar]
- 61.Kim BE, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol. 2008;4:176–85. doi: 10.1038/nchembio.72. [DOI] [PubMed] [Google Scholar]
- 62.Krishnakumar R, Craig M, Imlay JA, Slauch JM. Differences in enzymatic properties allow SodCI but not SodCII to contribute to virulence in Salmonella enterica serovar Typhimurium strain 14028. J Bacteriol. 2004;186:5230–8. doi: 10.1128/JB.186.16.5230-5238.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee H, Chang YC, Varma A, Kwon-Chung KJ. Regulatory diversity of TUP1 in Cryptococcus neoformans. Eukaryot Cell. 2009;8:1901–8. doi: 10.1128/EC.00256-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Passalacqua KD, Bergman NH, Lee JY, Sherman DH, Hanna PC. The global transcriptional responses of Bacillus anthracis Sterne (34F2) and a Delta sodA1 mutant to paraquat reveal metal ion homeostasis imbalances during endogenous superoxide stress. J Bacteriol. 2007;189:3996–4013. doi: 10.1128/JB.00185-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pfleger BF, Lee JY, Somu RV, Aldrich CC, Hanna PC, Sherman DH. Characterization and analysis of early enzymes for petrobactin biosynthesis in Bacillus anthracis. Biochemistry. 2007;46:4147–57. doi: 10.1021/bi6023995. [DOI] [PubMed] [Google Scholar]
- 66.Taylor CM, Osman D, Cavet JS. Differential expression from two iron-responsive promoters in Salmonella enterica serovar Typhimurium reveals the presence of iron in macrophage-phagosomes. Microb Pathog. 2009;46:114–8. doi: 10.1016/j.micpath.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 67.Suksrichavalit T, Prachayasittikul S, Nantasenamat C, Isarankura-Na-Ayudhya C, Prachayasittikul V. Copper complexes of pyridine derivatives with superoxide scavenging and antimicrobial activities. Eur J Med Chem. 2009;44:3259–65. doi: 10.1016/j.ejmech.2009.03.033. [DOI] [PubMed] [Google Scholar]
- 68.Suksrichavalit T, Prachayasittikul S, Piacham T, Isarankura-Na-Ayudhya C, Nantasenamat C, Prachayasittikul V. Copper complexes of nicotinic-aromatic carboxylic acids as superoxide dismutase mimetics. Molecules. 2008;13:3040–56. doi: 10.3390/molecules13123040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cybulski RJ, Jr, Sanz P, Alem F, Stibitz S, Bull RL, O’Brien AD. Four superoxide dismutases contribute to Bacillus anthracis virulence and provide spores with redundant protection from oxidative stress. Infect Immun. 2009;77:274–85. doi: 10.1128/IAI.00515-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fang FC. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol. 2004;2:820–32. doi: 10.1038/nrmicro1004. [DOI] [PubMed] [Google Scholar]
- 71.Fang FC, DeGroote MA, Foster JW, Bäumler AJ, Ochsner U, Testerman T, Bearson S, Giárd J-C, Xu Y, Campbell G, Laessig T. Virulent Salmonella typhimurium has two periplasmic Cu, Zn-superoxide dismutases. Proc Natl Acad Sci U S A. 1999;96:7502–7507. doi: 10.1073/pnas.96.13.7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Frost LS, Leplae R, Summers AO, Toussaint A. Mobile genetic elements: the agents of open source evolution. Nat Rev Microbiol. 2005;3:722–32. doi: 10.1038/nrmicro1235. [DOI] [PubMed] [Google Scholar]
- 73.Woese CR. On the evolution of cells. Proc Natl Acad Sci U S A. 2002;99:8742–7. doi: 10.1073/pnas.132266999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wren BW. The yersiniae--a model genus to study the rapid evolution of bacterial pathogens. Nat Rev Microbiol. 2003;1:55–64. doi: 10.1038/nrmicro730. [DOI] [PubMed] [Google Scholar]
- 75.Lapidus A, Goltsman E, Auger S, Galleron N, Segurens B, Dossat C, Land ML, Broussolle V, Brillard J, Guinebretiere MH, Sanchis V, Nguen-The C, Lereclus D, Richardson P, Wincker P, Weissenbach J, Ehrlich SD, Sorokin A. Extending the Bacillus cereus group genomics to putative food-borne pathogens of different toxicity. Chem Biol Interact. 2008;171:236–49. doi: 10.1016/j.cbi.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 76.Klee SR, Brzuszkiewicz EB, Nattermann H, Bruggemann H, Dupke S, Wollherr A, Franz T, Pauli G, Appel B, Liebl W, Couacy-Hymann E, Boesch C, Meyer FD, Leendertz FH, Ellerbrok H, Gottschalk G, Grunow R, Liesegang H. The genome of a Bacillus isolate causing anthrax in chimpanzees combines chromosomal properties of B. cereus with B. anthracis virulence plasmids. PLoS One. 2010;5:e10986. doi: 10.1371/journal.pone.0010986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Didelot X, Barker M, Falush D, Priest FG. Evolution of pathogenicity in the Bacillus cereus group. Syst Appl Microbiol. 2009;32:81–90. doi: 10.1016/j.syapm.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 78.Sheppard SK, McCarthy ND, Falush D, Maiden MC. Convergence of Campylobacter species: implications for bacterial evolution. Science. 2008;320:237–9. doi: 10.1126/science.1155532. [DOI] [PubMed] [Google Scholar]
- 79.Stahler FN, Odenbreit S, Haas R, Wilrich J, Van Vliet AH, Kusters JG, Kist M, Bereswill S. The novel Helicobacter pylori CznABC metal efflux pump is required for cadmium, zinc, and nickel resistance, urease modulation, and gastric colonization. Infect Immun. 2006;74:3845–52. doi: 10.1128/IAI.02025-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tsuda M, Karita M, Morshed MG, Okita K, Nakazawa T. A urease-negative mutant of Helicobacter pylori constructed by allelic exchange mutagenesis lacks the ability to colonize the nude mouse stomach. Infect Immun. 1994;62:3586–9. doi: 10.1128/iai.62.8.3586-3589.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Almagro-Moreno S, Boyd EF. Sialic acid catabolism confers a competitive advantage to pathogenic vibrio cholerae in the mouse intestine. Infect Immun. 2009;77:3807–16. doi: 10.1128/IAI.00279-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dagan T, Blekhman R, Graur D. The “domino theory” of gene death: gradual and mass gene extinction events in three lineages of obligate symbiotic bacterial pathogens. Mol Biol Evol. 2006;23:310–6. doi: 10.1093/molbev/msj036. [DOI] [PubMed] [Google Scholar]
- 83.Lawrence JG, Roth JR. Genomic Flux: Genome Evolution by Gene Loss and Acquisition. In: Charlebois LR, editor. Organization of Prokaryotic Genome. Washington DC: American Society for Microbiology; 1999. pp. 263–289. [Google Scholar]
- 84.Keim PS, Wagner DM. Humans and evolutionary and ecological forces shaped the phylogeography of recently emerged diseases. Nat Rev Microbiol. 2009;7:813–21. doi: 10.1038/nrmicro2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ariel N, Zvi A, Makarova KS, Chitlaru T, Elhanany E, Velan B, Cohen S, Friedlander AM, Shafferman A. Genome-based bioinformatic selection of chromosomal Bacillus anthracis putative vaccine candidates coupled with proteomic identification of surface-associated antigens. Infect Immun. 2003;71:4563–79. doi: 10.1128/IAI.71.8.4563-4579.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gat O, Grosfeld H, Ariel N, Inbar I, Zaide G, Broder Y, Zvi A, Chitlaru T, Altboum Z, Stein D, Cohen S, Shafferman A. Search for Bacillus anthracis potential vaccine candidates by a functional genomic-serologic screen. Infect Immun. 2006;74:3987–4001. doi: 10.1128/IAI.00174-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Popov SG, Popova TG, Hopkins S, Weinstein RS, MacAfee R, Fryxell KJ, Chandhoke V, Bailey C, Alibek K. Effective antiprotease-antibiotic treatment of experimental anthrax. BMC Infect Dis. 2005;5:25. doi: 10.1186/1471-2334-5-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dahlgren C, Karlsson A. Respiratory burst in human neutrophils. J Immunol Methods. 1999;232:3–14. doi: 10.1016/s0022-1759(99)00146-5. [DOI] [PubMed] [Google Scholar]
- 89.Hampton MB, Kettle AJ, Winterbourn CC. Inside the Neutrophil Phagosome: Oxidants, Myeloperoxidase, and Bacterial Killing. Blood. 1998;92:3007–3017. [PubMed] [Google Scholar]
- 90.Newman SL. Macrophages in host defense against Histoplasma capsulatum. Trends Microbiol. 1999;7:67–71. doi: 10.1016/s0966-842x(98)01431-0. [DOI] [PubMed] [Google Scholar]
- 91.Gobert AP, Semballa S, Daulouede S, Lesthelle S, Taxile M, Veyret B, Vincendeau P. Murine Macrophages Use Oxygen- and Nitric Oxide-Dependent Mechanisms To Synthesize S-Nitroso-Albumin and To Kill Extracellular Trypanosomes. Infect Immun. 1998;66:4068–4072. doi: 10.1128/iai.66.9.4068-4072.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Katsuragi H, Ohtake M, Kurasawa I, Saito K. Intracellular production and extracellular release of oxygen radicals by PMNs and oxidative stress on PMNs during phagocytosis of periodontopathic bacteria. Odontology. 2003;91:13–8. doi: 10.1007/s10266-003-0022-1. [DOI] [PubMed] [Google Scholar]
- 93.Sureda A, Hebling U, Pons A, Mueller S. Extracellular H2O2 and not superoxide determines the compartment-specific activation of transferrin receptor by iron regulatory protein 1. Free Radic Res. 2005;39:817–24. doi: 10.1080/10715760500164045. [DOI] [PubMed] [Google Scholar]
- 94.Kirillina O, Bobrov AG, Fetherston JD, Perry RD. Hierarchy of iron uptake systems: Yfu and Yiu are functional in Yersinia pestis. Infect Immun. 2006;74:6171–8. doi: 10.1128/IAI.00874-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mignot T, Mock M, Robichon D, Landier A, Lereclus D, Fouet A. The incompatibility between the PlcR- and AtxA-controlled regulons may have selected a nonsense mutation in Bacillus anthracis. Mol Microbiol. 2001;42:1189–98. doi: 10.1046/j.1365-2958.2001.02692.x. [DOI] [PubMed] [Google Scholar]
- 96.Slamti L, Perchat S, Gominet M, Vilas-Boas G, Fouet A, Mock M, Sanchis V, Chaufaux J, Gohar M, Lereclus D. Distinct mutations in PlcR explain why some strains of the Bacillus cereus group are nonhemolytic. J Bacteriol. 2004;186:3531–8. doi: 10.1128/JB.186.11.3531-3538.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Petosa C, Collier RJ, Klimpel KR, Leppla SH, Liddington RC. Crystal structure of the anthrax toxin protective antigen. Nature. 1997;385:833–8. doi: 10.1038/385833a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gene Discovery (GD). Random genomic libraries are generated for each B. cereus strain. Cloned genomic fragments are common (uncolored boxes) or unique (colored boxes). Plasmid DNAs are printed onto glass slide microarrays and subsequently probed with Cy-labeled query B. cereus and B. anthracis in a competitive hybridization. Those plasmids that do not hybridize with B. anthracis were considered to contain unique genomic fragments (novel genes) and were sequence characterized. Other plasmids that hybridized weakly were considered to contain divergent sequences were also sequenced.
Summary of the bioinformatic approach used to characterize the unique genomic fragments from B. cereus strains subjected gene discovery.
Relative frequencies of novel genes discovered among seven query B. cereus genomes based on predicted functional role categories.
Total gene content estimates for Bacillus cereus group members.
Comparative cluster analysis of strains based on CGH data. Left, clustering based on the global gene hybridization patterns (~29977 70-mer oligonucleotides) using the trinary designations “0”, “0.5” and “1” corresponding to “absent”, “divergent” and “present” CDSs respectively. Right, clustering of the 950 markers representing known plasmid sequences.