

Abstract
Combining microtubule-targeting anti-mitotic drugs with targeted apoptosis potentiators is a promising new chemotherapeutic strategy to treat cancer. In this study we investigate the cellular mechanism by which Navitoclax (previously called ABT-263), a Bcl-2 family inhibitor, potentiates apoptosis triggered by paclitaxel and an inhibitor of Kinesin-5 (KSP), across a panel of epithelial cancer lines. Using time-lapse microscopy, we show that Navitoclax has little effect on cell death during interphase, but strongly accelerates apoptosis during mitotic arrest, and greatly increases the fraction of apoptosis-resistant cells that die. By systematically knocking down individual Bcl-2 proteins we determined that Mcl-1 and Bcl-xL are the primary negative regulators of apoptosis during prolonged mitotic arrest. Mcl-1 levels decrease during mitotic arrest due to an imbalance between synthesis and turnover, and turnover depends in part on the MULE/HUWE1 E3 ligase. The combination of Mcl-1 loss with inhibition of Bcl-xL by Navitoclax causes rapid apoptosis in all lines tested. Variation in expression levels of Mcl-1 and Bcl-xL largely determine variation in response to anti-mitotics alone, and anti-mitotics combined with Navitoclax, across our panel. We conclude that Bcl-xL is a critical target of Bcl-2 family inhibitors for enhancing the lethality of anti-mitotic drugs in epithelial cancers, and combination treatment with Navitoclax and a spindle specific anti-mitotic, such as a Kinesin-5 inhibitor, might be more effective than paclitaxel alone.
Keywords: Navitoclax, paclitaxel, apoptosis response
Introduction
Anti-mitotic drugs, such as paclitaxel, are important for treatment of a variety of solid tumors. They induce prolonged mitotic arrest through activation of the spindle assembly checkpoint [1]. Drug treated cells then either die in mitotic arrest, or slip out of mitotic arrest into an abnormal G1 state, in which they may die, arrest in a tetraploid G1 state, or continue to proliferate [2-6]. While anti-mitotics at sufficiently high concentration can cause mitotic arrest in all proliferating cells, sensitivity to induction of cell death during or after this arrest is highly variable across different cancer cell lines in syngeneic mouse tumors [7] and cultured human cell lines [8]. This poorly-understood variation in cell death sensitivity may contribute to large variation in patient responses.
Although non-apoptotic death has been reported [9, 10], most of the available data, including our own, show that anti-mitotics induce cell death during mitotic arrest, or after mitotic slippage, by the intrinsic apoptosis pathway, where Mitochondrial Outer Membrane Permeabilization (MOMP) is the defining event [11-13]. Thus, one way to potentially enhance anti-mitotics induced cell death, and reduce variation in death sensitivity across cancers, is to combine anti-mitotics with drugs that directly stimulate MOMP. ABT-737 and its orally active analog, Navitoclax (formerly ABT-263), are among the most promising candidates for such drug combinations [14-16]. ABT-737 is a potent inhibitor (Ki <10 nM) of several Bcl-2 homologs that inhibit MOMP, including Bcl-2, Bcl-xL and Bcl-w [14]. These drugs have already been shown to potentiate the cytotoxic effect of paclitaxel in cell culture, animal models and early stage clinical trials [17, 18]. However, the cellular mechanism by which ABT-737 and Navitoclax enhance paclitaxel-mediated apoptosis, and how this mechanism varies across different cell types, are still poorly understood. For example, it is unclear whether the taxane + Navitoclax combination kills cells in interphase, during mitotic arrest, or after slippage from arrest. It is also unclear which of the various targets of Navitoclax are relevant for promoting cancer cell killing by taxanes, which precludes development of more selective drugs. Answering these questions might also provide clues to the still-unsolved problem of how anti-mitotics trigger MOMP at the molecular level.
Here, we investigate the cellular mechanism by which Navitoclax enhances cancer cell killing by two anti-mitotic drugs, the microtubule-stabilizing drug, paclitaxel, and an allosteric inhibitor of motor protein, Kinesin-5 (K5I, also called a KSP inhibitor), both of which promote prolonged mitotic arrest, but by different mechanisms. We test which Bcl-2 family members are important for inhibiting and enhancing cell death during mitotic arrest caused by these drugs in epithelial cancer cells, and thus identify relevant targets of Navitoclax when it is combined with an anti-mitotic. Our primary technique for measuring apoptosis was time-lapse microscopy, which allows conceptual synchronization of cell cycle without the need for additional drugs, and naturally accounts for the very large variation between individual cells in death sensitivity and kinetics [4, 6]. Our findings have important implications for how the Navitoclax + taxane combination may work or fail in the clinic. Moreover, as neither Kinesin-5 inhibitors nor Navitoclax are neurotoxic, our results also suggest that Navitoclax, or a comparable Bcl-xL inhibitor, would strongly potentiate the clinical response to K5Is, without causing neurotoxicity that is characteristic of microtubule-targeting anti-mitotics.
Materials and Methods
Cell culture
All cell lines were purchased from American Type Culture Collection (ATCC, USA) and cultured under 37°C and 5% CO2 in appropriate medium supplemented with 10% Fetal Calf Serum (FCS), 100U/ml penicillin and 100μg/ml streptomycin. HeLa was maintained in DMEM; U2OS was maintained in McCoy’s; OVCAR-5 was maintained in RPMI; and A549 was maintained in F-12K.
Chemicals
Paclitaxel was purchased from Sigma. The potent kinesin-5 inhibitor (EMD534085) was provided by Merck-Serono. Responses to EMD534085 have been shown to be similar to s-trityl-cysteine, a commercially available Kinesin-5 inhibitor [4]. Navitoclax (ABT-263) was purchased from Selleck.
Time-lapse microscopy
Cells were plated in 35 mm imaging dish (μ-dish, ibidi, Germany) and cultured in phenol red-free CO2-independent medium (Invitrogen) supplemented with 10% FCS, 100 U/ml penicillin and 100 μl streptomycin. Cell images were acquired with the Nikon TE2000-PFS inverted microscope enclosed in a humidified chamber maintained at 37°C. Cells were imaged every 10 minutes using a motorized stage and a 20X objective (NA=0.95). Images were viewed and analyzed using the MetaMorph software (Molecular Dynamics).
Gene knockdown by RNA interference (RNAi)
siRNAs for knocking down individual Bcl-2 family proteins were obtained from the following sources: Bcl-2 (J-003307-16), Bcl-w (J-004384-07), Bcl-xL (J-003458-11), Bak (J-003305-07) and Bax (J-003308-12) from Dharmacon; Mcl-1 (s120644) and Cdc20 (4392420) from Ambion; Bim (#6461) from Cell Signaling. The above siRNAs were used at final concentration of 20 nM for RNAi in all four cell lines. siRNA against MULE/HUWE1 (5′-GAGUUUGGAGUUUGUGAAGtt-3′) was synthesized by Dharmacon and used at final concentration of 50 nM. Dharmacon On-Target plus siControl (D-001810-01) was used as non-targeting siRNA control. All siRNA transfections were performed using either DharmaFect 1 (Dharmacon) or HiPerFect (Qiagen) according to manufacturers’ instructions. Experiments were conducted after 48 hrs of gene silencing.
Gene expression by transient transfection
To express Mcl-1-GFP or Bak-EGFP, U2OS cells were seeded in 6-well plate and transiently transfected with the desired plasmids using Fugene6 (Roche) according to manufacturer’s instruction. Drug treatment experiments of U2OS cells with Mcl-GFP over-expression as well as time-lapse imaging experiment of U2OS cells expressing Bak-EGFP were conducted 48hrs after transient transfection. Both the Mcl-1-GFP and Bak-EGFP constructs were generous gift from Dr. Peter Sorger’s lab at the Department of Systems Biology, Harvard Medical School.
Western Blot Analysis
Cell lysates were obtained using LDS sample buffer (NuPAGE, Invitrogen). Proteins were resolved on 10% or 12% Tris-glycine gels and transferred onto PVDF membranes. Blots were probed with commercial primary antibodies and chemiluminescent detection using ECL-plus (Amersham). Antibodies: PARP1 (#9542), Bcl-w (#2724), Bcl-xL (#2762), Bim (#2933), Bak (#3814) and Cdc20 (#4823) were purchased from Cell Signaling; Bax (#sc-493) and Mcl-1 (#sc-819) from Santa Cruz; Bcl-2 (#M088901) from Dako. Anti-actin (#A5316) from Sigma was used as a loading control.
For western blot analysis of synchronized mitotic cells, we grew large volume of cells in 25cm dishes to 90% confluency, then treated the cells with 150nM paclitaxel or 1μM K5I to induce mitotic arrest. After 3 hrs of drug treatment, the mitotic fraction of cells was collected by gently shaking and washing the dish to detach the mitotic cells from the bottom.
Results
Bcl-2 family expression does not obviously predict apoptosis sensitivity
We previously profiled the sensitivity of a panel of cancer-derived, and one non-cancer telomerase-immortalized (RPE), cell lines for sensitivity to apoptosis in response to anti-mitotic drugs [8]. At drug concentrations that were saturating for mitotic arrest, the extent and speed of apoptosis varied greatly across the cell line panel, but correlated strongly for three different anti-mitotic drugs, suggesting they trigger apoptosis by similar mechanisms. In every case subsequently studied, cell death occurred during or after slippage from mitotic arrest, by MOMP followed by caspase activation [6 and Supplementary Figure 1]. To probe the mechanistic basis of differential MOMP sensitivity across the panel, we measured levels of known MOMP regulators by immunoblotting (Figure 1). Basal expression of Bax, Bak, Mcl-1 and Bcl-w were relatively similar across cell lines, while expression of Bcl-2 and Bcl-xL were quite variable. By visual inspection, there was no obvious correlation between overall expression levels of pro- and anti-apoptotic proteins and MOMP sensitivity in response to anti-mitotics.
Figure 1.
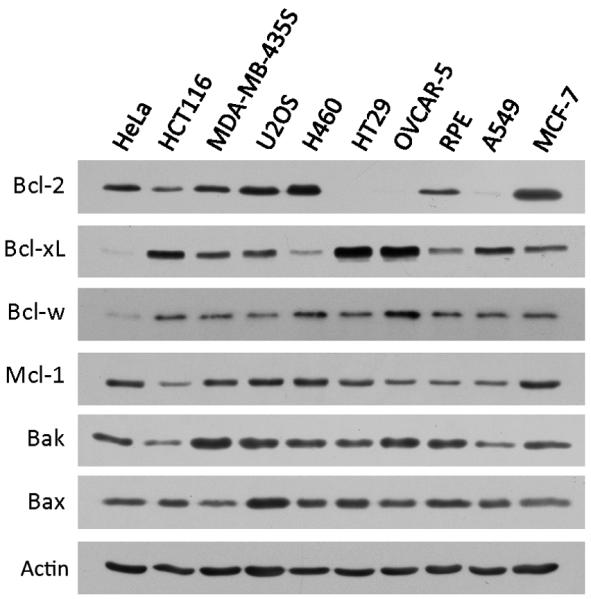
Basal expression of selected Bcl-2 family proteins in a panel of human cell lines we profiled before [8]. Cell lines were ordered from left to right in descending sensitivity to anti-mitotics induced cell death.
Navitoclax accelerates apoptosis during mitotic arrest
All the cell lines expressed anti-apoptotic Bcl-2 family members. As a first test of their roles in cell death during or after mitotic arrest, we measured the effect of Navitoclax, a potent small molecule inhibitor of Bcl-2, Bcl-w and Bcl-xL, but not Mcl-1, using time-lapse microscopy. We chose HeLa, U2OS, OVCAR-5 and A549 to cover the spectrum from most to least death sensitive, and also variation in expression levels of Bcl-2 and Bcl-xL. Navitoclax was used at 1μM, a clinically-relevant concentration that is probably saturating for Bcl-2 family binding [14]. In Navitoclax alone proliferation continued normally in HeLa, OVCAR-5 and A549 cells, and we observed < 5% cell death in these three cell lines after 60 hrs of drug treatment (gray lines in the upper panel of Fig. 2). In U2OS cells, Navitoclax alone was cytotoxic, leading to ~35% cell death after 60 hrs of drug treatment. When Navitoclax was combined with an anti-mitotic drug (paclitaxel or K5I, used at concentrations that are saturating for mitotic arrest in all lines), we observed strong enhancement of cell death for all four cell lines. MOMP was observed to precede death in > 95% of single cell death events, confirming that anti-mitotics + Navitoclax trigger cell death mainly through the mitochondria-dependent apoptosis pathway. Moreover, in anti-mitotic alone, or anti-mitotic + Navitoclax, no apoptosis occurred before cells entered mitotic arrest. Evidently Navitoclax enhances the pro-apoptotic signaling that initiates when cells enter mitotic arrest instead of at time of drug addition [6, 8]. We therefore scored death kinetics by measuring the time elapsed from the time of entry into mitotic arrest to the time of morphological death, i.e. cell blebbing and lysing, and plotted cumulative survival curves that provide a metric for the rate of apoptosis induction during and after mitotic arrest (Fig. 2).
Figure 2.

Cumulative survival curves for indicated treatments in four cancer cell lines: treatment with 1μM Navitoclax (ABT-263) alone (denoted in gray), 150 nM paclitaxel alone (red), 150nM paclitaxel + 1μM Navitoclax (green), 1μM K5I alone (black) and 1μM K5I + 1μM Navitoclax (blue). Total number of cells analyzed for each curve ranges from 76 to 120, varied between conditions and cell lines. Individual cells were monitored by phase-contrast and fluorescence time-lapse microscopy, and time from mitotic entry to morphological death was measured and plotted as cumulative survival curves. The upper panel quantified kinetics of all cell death and the lower panel quantified only death during mitotic arrest.
The upper panel of figure 2 shows kinetics of all cell death, pooling death during mitotic arrest with death after mitotic slippage. The lower panel shows only death during mitotic arrest. The black and red lines show death in anti-mitotic alone. HeLa is the most sensitive, with most cells dying during mitotic arrest, and A549 the least, with few cells dying. As reported previously [8], paclitaxel (red lines) is significantly more cytotoxic than K5I (black lines) in less death-sensitive lines, even though both drugs cause the same kinetics of mitotic arrest. Addition of Navitoclax strongly accelerated death during mitotic arrest in U2OS, OVCAR-5 and A549, and moderately accelerated it in HeLa. This effect was particularly strong for cell line/drug combinations where relatively little death occurred during mitosis for anti-mitotic alone, and in some cases caused > 5-fold enhancement of death (e.g. U2OS/K5I). Even in the most apoptosis-resistant A549 cells, Navitoclax strongly enhanced death during mitotic arrest for both anti-mitotics.
Note that we chose to investigate the kinetics of cell response at saturating drug concentrations of Navitoclax and anti-mitotics, instead of a dose-titration study at fixed time point. Dose-dependent effects of anti-mitotics are in general difficult to interpret, as they require consideration of drug efflux pump activity, and single cell behaviors at subsaturating concentrations become very complex. Therefore, we chose to use only one dose and investigate the cellular kinetics in detail so as to seek mechanistic understanding at the level of pathways that mediate mitotic arrest and apoptosis. We picked a saturating concentration to maximize the mechanistic strength of our results and conclusions.
Navitoclax accelerates apoptosis mainly by inhibiting Bcl-xL
To pinpoint the specific Bcl-2 family target(s) of Navitoclax during mitotic arrest, we used RNAi to knock down individual anti-apoptotic Bcl-2 proteins, including Bcl-2, Bcl-w, Bcl-xL and Mcl-1, and compared death kinetics to drug treatment. The upper panel of Figure 3 and Table 1 compare kinetics of overall cell death following entry into K5I-induced mitotic arrest, pooling both death in mitosis and death after slippage, under seven conditions: control (no siRNA), control siRNA (control for the pro-apoptotic effect of transfection), Bcl-2 siRNA, Bcl-w siRNA, Bcl-xL siRNA, Mcl-1 siRNA and Navitoclax. The lower panel of Figure 3 and Table 2 compare kinetics of cell death during mitotic arrest only. Knockdown efficiencies were generally > 80%, and are documented in supplementary figure 2. Control siRNA transfection (red lines) slightly sensitized cells to K5I induced apoptosis. Bcl-2 knockdown (blue lines) was similar to control siRNA for all four cell lines. Bcl-w knockdown (green lines) also had little effect, except in OVCAR-5 cells, where it increased the percentage of cell death by ~2 fold, but the median time to death, Td, did not change substantially (Table 1). Mcl-1 knockdown (magenta lines) sensitized all lines to apoptosis, especially HeLa, where it had the strongest effect. This line has high expression of Mcl-1 but little Bcl-xL (Fig. 1). Bcl-xL knockdown (yellow lines) had overall the strongest effect, dramatically accelerating apoptosis in the three less apoptosis sensitive lines. In U2OS and OVCAR-5 the effect of Bcl-xL knockdown was comparable to Navitoclax treatment. In A549 it was less effective, but knockdown was noticeably incomplete in this line (supp. Fig. 2). As we saw previously with Navitoclax treatment, Bcl-xL knockdown, and to a lesser extent Mcl-1 knockdown, had the effect of increasing the fraction of cells that died in the three less apoptosis sensitive lines, as well as shifting death from post-slippage to inside mitotic arrest. These data suggest that Bcl-xL is the major target of Navitoclax for accelerating MOMP during mitotic arrest, and thus for potentiating cell death caused by anti-mitotic drugs.
Figure 3.

Cumulative survival curves for indicated siRNA treatments or Navitoclax in combination with 1μM K5I in four cancer cell lines. The upper panel quantified kinetics of all cell death and the lower panel quantified only death during mitotic arrest. Total number of cells analyzed for each curve ranges from 76 to 144, varied between conditions and cell lines.
Table 1.
Percentage of overall cell death and the median death time (Td) in response to Kinesin-5 Inhibitor under transfection of different siRNA oligos.
| K5I+ | HeLa | U2OS | OVCAR-5 | A549 | ||||
|---|---|---|---|---|---|---|---|---|
| Death (%) |
Td (hour) |
Death (%) |
Td (hour) |
Death (%) |
Td (hour) |
Death (%) |
Td (hour) |
|
| no siRNA | 95 | 19.5 | 45 | 24.6 | 16 | 17.5 | 14 | 12.0 |
| scramble siRNA | 97 | 17.8 | 63 | 21.5 | 23 | 16.8 | 25 | 13.5 |
| Bcl-2 siRNA | 96 | 19.7 | 64 | 24.7 | 18 | 17.2 | 20 | 18.0 |
| Bcl-w siRNA | 99 | 16.8 | 62 | 18.7 | 37 | 23.5 | 16 | 18.0 |
| Bcl-xL siRNA | 100 | 12.0 | 94 | 3.8 | 82 | 13.6 | 47 | 10.3 |
| Mcl-1 siRNA | 99 | 8.3 | 85 | 15.0 | 54 | 11.3 | 26 | 12.3 |
| Navitoclax | 100 | 11.3 | 99 | 5.7 | 89 | 10.8 | 91 | 11.3 |
total number of cells analyzed from the time-lapse movies ranges from 76 to 144, varied between conditions and cell lines. Death denotes the percentage of cell death, and Td is the median time to death since mitotic entry.
Table 2.
Percentage of cell death in mitosis and the median time to mitotic death (Tmd) in response to Kinesin-5 Inhibitor under transfection of different siRNA oligos.
| K5I+ | HeLa | U2OS | OVCAR-5 | A549 | ||||
|---|---|---|---|---|---|---|---|---|
| D in M (%) |
Tmd (hour) |
D in M (%) |
Tmd (hour) |
D in M (%) |
Tmd (hour) |
D in M (%) |
Tmd (hour) |
|
| no siRNA | 84 | 19.3 | 6 | 13.7 | 16 | 17.5 | 8 | 8.8 |
| scramble siRNA | 86 | 16.2 | 20 | 12.8 | 20 | 13.8 | 14 | 10.5 |
| Bcl-2 siRNA | 84 | 19.0 | 13 | 8.5 | 17 | 17.2 | 3 | 12.3 |
| Bcl-w siRNA | 86 | 16.0 | 32 | 12.8 | 31 | 22.0 | 3 | 7.0 |
| Bcl-xL siRNA | 99 | 11.8 | 79 | 3.0 | 73 | 13.2 | 38 | 9.2 |
| Mcl-1 siRNA | 95 | 8.2 | 33 | 4.7 | 40 | 9.5 | 11 | 6.0 |
| Navitoclax | 100 | 11.3 | 96 | 5.5 | 82 | 10.8 | 62 | 10.3 |
D in M denotes the percentage of cell death occurring in mitotic arrest, and Tmd is the median time to mitotic death since mitotic entry.
Mcl-1 is lost by turnover during mitotic arrest
Mcl-1 expression has been shown to negatively correlate with ABT-737 sensitivity in some studies [15]. All our cell lines expressed Mcl-1 at significant levels (Fig. 1), so we next asked why this important anti-apoptotitc Bcl-2 family member did not protect against MOMP during mitotic arrest when Bcl-xL was inhibited with Navitoclax or knockdown by RNAi.
Uniquely among characterized Bcl-2 family proteins, Mcl-1 turns over rapidly in interphase cells, with a typical half-life of < 1 hr [19, 20]. It is constitutively ubiquitinated by E3 ligases and then degraded by proteasomes [21]. Transcription is silenced during mitotic arrest, and translation is inhibited [22-24], so we suspected Mcl-1 levels might decay during arrest by a continuation of the interphase turnover mechanism. We first measured time courses of Mcl-1 levels in cells treated with anti-mitotic drugs, and found that its levels decrease at the time point where mitotic arrest peaks (~15-30hrs) (Fig. 4a). In HeLa, the level subsequently decreases to zero as all cells die, but in the more resistant line it recovers as cells slip out of mitotic arrest. To more accurately measure Mcl-1 levels during mitotic arrest, we synchronized cells by mitotic shake-off in drug, and measured Mcl-1 levels by immunoblotting. Mcl-1 levels were found to decay with a half life of ~ 4 hrs in HeLa and ~ 1.6 hrs in A549 during mitotic arrest (Fig. 4b). In A549 Mcl-1 plateaus at ~20% of the initial level. In HeLa it may continue to drop, and we suspect complete loss of Mcl-1 may trigger MOMP.
Figure 4.

a) Mcl-1 levels at 12 selected time points (unit: hour) in the four selected cell lines treated with either 150nM paclitaxel or 1μM K5I. Actin, which served as loading control, was not shown, as loading were similar in all samples. b) Comparison of Mcl-1 kinetics during mitotic arrest in HeLa (death sensitive) and A549 cells (death resistant). Pure mitotic HeLa and A549 cells were obtained by synchronizing cells in mitosis with 3hr treatment of 1μM K5I and then gently shaking off the mitotic faction. Mcl-1 level was quantified from western blots, normalized to initial level at time 0 and fit to an exponential decay to derive the half life τ. Values of Mcl-1 level were averaged from three separate experiments. c) Attenuation of Mcl-1 depletion by RNAi of MULE/HUWE1 (50nM) and the resulting reduction in apoptosis (indicated by Parp1 cleavage) compared to no knockdown of MULE/HUWE1.
To test if loss of Mcl-1 during mitotic arrest contributes to apoptosis induction, we attenuated Mcl-1 depletion by RNAi knockdown of MULE/HUWE1. This is the first E3 ligase that was shown to play a role in Mcl-1 turnover [21], subsequently others have been implicated (see below). We used a custom synthesized siRNA used in previous study [25], and measured the resulting apoptosis response to anti-mitotics in HeLa cells by Parp1 cleavage (Fig. 4c). MULE/HUWE1 knockdown decreased the extent of Mcl-1 loss, and also decreased Parp1 cleavage. These data confirm that depletion of Mcl-1 is a pro-apoptotic signal that contributes to apoptosis during mitotic arrest. Because Mcl-1 levels were only partially protected by RNAi of MULE/HUWE1, we suspect other turnover mechanisms are important; Anaphase Promoting Complex (APC/C) [26], NOXA [27] and another E3 ligase, FBW7 [28, 29] have all been implicated in recent studies. In supplementary figure 3, we compared kinetics of Mcl-1 depletion in mitotic shake-off HeLa cells under three conditions: control, MULE/HUWE1 siRNA and Cdc20 siRNA (Cdc20 RNAi was used to inhibit APC/C activity). It is evident that RNAi of both MULE/HUWE1 and Cdc20 delayed and reduced Mcl-1 depletion during mitotic arrest. However, neither completely rescued Mcl-1 depletion, pointing to the existence of multiple degradation factors for Mcl-1 during mitotic arrest.
Bim does not trigger MOMP during mitotic arrest
The activator BH-3 protein Bim was previously implicated as the major trigger of cell death in BMK cells treated with paclitaxel, but where in the cell cycle Bim-dependent apoptosis occurred was not determined [30]. To determine whether Bim is required to activate apoptosis during mitotic arrest, we knocked it down using siRNA, and measured kinetics of cell death in HeLa, which die mainly during mitotic arrest (Fig. 5). Knockdown was largely complete by immunoblotting analysis (supp. Fig. 4). Both the extent and kinetics of death in mitosis were very similar under treatments of K5I alone (black line in Fig. 5), K5I plus control siRNA (red line) and K5I plus Bim siRNA (blue line), with 80~90% cell death occurring during mitotic arrest and a median time to death of 19hrs. Together with western blot analysis that showed no attenuation in Parp1 cleavage under Bim knockdown (supp. Fig. 4), we conclude that Bim is not required to activate cell death during mitotic arrest in HeLa. Note that Bim is a fast turnover protein with a half life of ~3 hrs [31]. Therefore, on the western blot levels of Bim exhibited significant reduction during mitotic arrest due to transcriptional silence (24hr time point in supp. Fig. 4). In contrast, knockdown of Bax (green line) and Bak (yellow line) caused substantial protection as expected for a MOMP-dependent death pathway. Immunoblotting analysis showed no up-regulation of any known sensitizer BH-3 only proteins (supp. Fig. 5), and we previously found no cleavage of Bid in anti-mitotic treated cells [8]. These data suggest that activator BH3 proteins or up-regulation of sensitizer BH-3 only proteins are not required for cell death during mitotic arrest.
Figure 5.
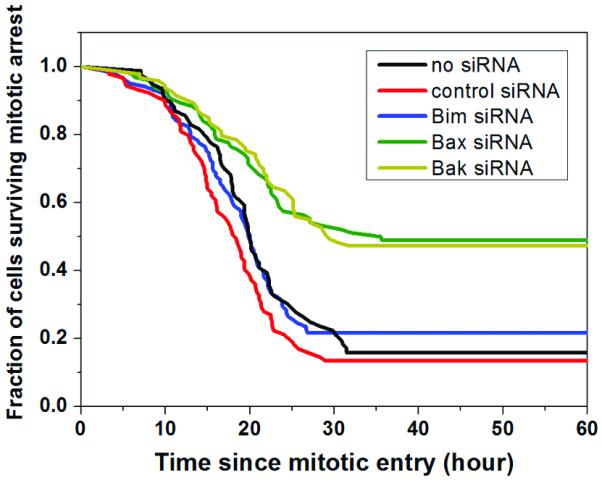
Cumulative survival curves for indicated siRNA treatments in combination with 1μM K5I in HeLa cells. The survival statistics quantified only death during mitotic arrest.
Discussion
The strong acceleration of apoptosis during mitotic arrest we observed when Navitoclax was combined with anti-mitotic drugs, as well as recent data from Xenograft cancer models [32], point to exciting potential of this combination for treatment of common epithelial cancers. Navitoclax has been known to potentiate paclitaxel-mediated cytotoxicity since the first description of ABT-737 [17], but ours is the first study to pinpoint where in the cell cycle this potentiation occurs, and to document its kinetics at the single-cell level. The major target of Navitoclax in the cells we tested was Bcl-xL, not Bcl-2 or Bcl-w, as evidenced by similar acceleration of apoptosis during mitotic arrest with drug treatment and RNAi knockdown of Bcl-xL alone. The potent lethality of inhibiting Bcl-xL during mitotic arrest is due to the fact that another important and widely-expressed anti-apoptotic protein, Mcl-1, is naturally depleted during mitotic arrest as a result of imbalance between synthesis and degradation. The pathway by which Mcl-1 is proteolyzed has been the subject of several recent studies. MULE/HUWE1 was implicated early as an E3 ligase [21]. Our data (Fig. 4c) suggest it indeed plays a role during mitosis, though it seems not to act alone. APC/C was recently implicated as an alternative E3 ligase [26]. Our data are consistent for a role of this factor (supp. Fig. 3), though caution is required in interpreting this observation, since APC/C plays a central role in mitotic regulation, and its effects on Mcl-1 might be indirect. We have not yet tested the role of other candidates, including NOXA [27] and FBW7 [28, 29]. Whatever the precise mechanism of Mcl-1 proteolysis during mitosis, the important conclusion from our perspective is that its loss is strongly synergistic with inhibition of Bcl-xL by Navitoclax, leading to rapid apoptosis during mitotic arrest, even in lines where little apoptosis is seen in anti-mitotic alone.
The mechanism by which mitotic arrest promotes apoptosis is clarified, but not completely solved, by the finding from us and others [25-29] that Mcl-1 is lost during mitotic arrest due to an imbalance between synthesis and proteolysis. Conceptually, this finding supports a kinetic model in which cells that enter mitotic arrest activate two independent and competing pathways, one that leads to MOMP, and the other to slippage by slow proteolysis of Cyclin-B [33, 34]. Combination of Mcl-1 loss with inhibition of Bcl-xL by Navitoclax or siRNA knockdown greatly accelerates the death pathway, so it wins over the slippage pathway in a much larger fraction of cells. Loss of Mcl-1 is clearly an important component of the MOMP-activation pathway, less clear is whether this is the only MOMP-activating process during mitotic arrest. It is also not clear to what extent acceleration of apoptosis activation matters clinically, i.e. if a cell is going to die eventually, does it matter if it dies within hours following drug addition, as opposed to days or weeks? We suspect it might, that a rapid bolus of apoptosis might alter the tumor environment in ways that a more delayed response did not. The issue of whether apoptosis kinetics really matters for clinical responses will have to be addressed in the tumor context.
Our data have shed new light on cell-line variation in anti-mitotic drug responses. In cells with low levels of Bcl-xL, such as HeLa, loss of Mcl-1 alone is sufficient to release Bax/Bak from sequestration, which allows their spontaneous oligomerization, and triggers MOMP. In cells with substantial levels of Bcl-xL, such as U2OS, OVCAR-5 and A549, loss of Mcl-1 alone is insufficient, but combining it with Bcl-xL inhibition or knockdown is sufficient to trigger Bax/Bak oligomerization. It is known that Bcl-xL and Mcl-1, but not Bcl-2 or Bcl-w, can directly sequester Bax/Bak, in particular Bak on the mitochondria [35]. This might explain why Bcl-2 levels seem less important than Bcl-xL in determining the response to anti-mitoics, despite wide variation in Bcl-2 expression between cells in our panel. Consistent with a role for Bak, we observed oligomerization of Bak-GFP preceding cell death in response to anti-mitotics + Navitoclax by fluorescence microscopy (supp. Fig. 6). We also found that Navitoclax remains highly effective in enhancing and accelerating mitotic death in cells over-expressing Mcl-1 (supp. Fig. 7). Is loss of Mcl-1 and Bcl-xL activity sufficient to trigger MOMP, or is another signal required? Depletion of Mcl-1 does not greatly accelerate apoptosis in response to anti-mitotics in any line (Fig. 2), which may indicate the need for an additional signal. Although our data on Bim (Fig. 5) and tBid [8] suggest that neither of the known activator BH3 proteins is required for initiation of apoptosis during mitotic arrest, we cannot rule out that some other, yet to be identified, Bcl-2 family member acts as activator. Further study to determine the molecular model for mitotic death is important for progress on understanding the action of current anti-mitotics, and finding better targets for future drugs.
Anti-mitotic drugs that are specific to the mitotic spindle – inhibitors of Kinesin-5 (KSP), Polo kinase-1 and Aurora kinases – have so far shown somewhat disappointing clinical results [36]. K5Is are less pro-apoptotic than paclitaxel in cell culture (e.g. Fig. 2 and [8]), which may contribute to this clinical difference. The reasons are unclear, though the extra death seen with paclitaxel mostly occurs after slippage. One possible cause is greater micronucleation; cells that slip out of paclitaxel-induced arrest always have multiple micronuclei, while cells that slip out of K5I-induced arrest typically have one large nucleus. Combination with Navitoclax changes this equation, by shifting most death into mitosis, where the two drugs are almost equally pro-apoptotic. The combination of a K5I and Navitoclax is much more pro-apoptotic than K5I alone or paclitaxel alone, and essentially equal to paclitaxel + Navitoclax (Fig. 2). Thus the K5I + Navitoclax combination has the promise of efficacy greater than paclitaxel alone, comparable to paclitaxel + Navitoclax. Neither K5Is or Navitoclax are neurotoxic, so this combination might realize the promise of an anti-mitotic treatment that is efficacious but lacks neurotoxicity.
Supplementary Material
Acknowledgements
We thank Dr. Peter Sorger (Systems Biology, Harvard Medical School) for the IMS-RP retroviral vector, Mcl-1-GFP vector and Bak-EGFP vector, and the Nikon Imaging Center at Harvard Medical School for conducting part of our imaging experiments. This work was supported by the National Cancer Institute (CA139980) to TJ Mitchison, and Hong Kong Research Grant Council (#261310) to J Shi. EMD534085 was supplied by Merck Serono. The authors declare no financial interest.
References
- 1.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4(4):253–65. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 2.Rieder CL, Maiato H. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell. 2004;7(5):637–51. doi: 10.1016/j.devcel.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 3.Weaver BA, Cleveland DW. Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell. 2005;1:7–12. doi: 10.1016/j.ccr.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 4.Orth JD, Tang Y, Shi J, Loy CT, Amendt C, Wilm C, Zenke FT, et al. Quantitative live imaging of cancer and normal cells treated with Kinesin-5 inhibitors indicates significant differences in phenotypic responses and cell fate. Mol Cancer Ther. 2008;7(11):3480–9. doi: 10.1158/1535-7163.MCT-08-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14:111–22. doi: 10.1016/j.ccr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Huang HC, Shi J, Orth JD, Mitchison TJ. Evidence that mitotic exit is a better cancer therapeutic target than spindle assembly. Cancer Cell. 2009;16(4):347–58. doi: 10.1016/j.ccr.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Milross CG, Mason KA, Hunter NR, Chung WK, Peters LJ, Milas L. Relationship of mitotic arrest and apoptosis to antitumor effect of paclitaxel. Natl Cancer Inst. 1996;88(18):1308–14. doi: 10.1093/jnci/88.18.1308. [DOI] [PubMed] [Google Scholar]
- 8.Shi J, Orth JD, Mitchison T. Cell type variation in responses to antimitotic drugs that target microtubules and kinesin-5. Cancer Res. 2008;68:3269–76. doi: 10.1158/0008-5472.CAN-07-6699. [DOI] [PubMed] [Google Scholar]
- 9.Broker LE, Huisman C, Span SW, Rodriguez JA, Kruyt FA, Giaccone G. Cathepsin B mediates caspase-independent cell death induced by microtubule stabilizing agents in non-small cell lung cancer cells. Cancer Res. 2004;64(1):27–30. doi: 10.1158/0008-5472.can-03-3060. [DOI] [PubMed] [Google Scholar]
- 10.Broker LE, Kruyt FA, Giaccone G. Cell death independent of caspases: a review. Clin Cancer Res. 2005;11(9):3155–62. doi: 10.1158/1078-0432.CCR-04-2223. [DOI] [PubMed] [Google Scholar]
- 11.Tao W, South VJ, Zhang Y, Davide JP, Farrell L, Kohl NE, et al. Induction of apoptosis by an inhibitor of the mitotic kinesin KSP requires both activation of the spindle assembly checkpoint and mitotic slippage. Cancer Cell. 2005;8(1):49–59. doi: 10.1016/j.ccr.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 12.Wang LG, Liu XM, Kreis W, Budman DR. The effect of antimicrotubule agents on signal transduction pathways of apoptosis: a review. Cancer Chemother Pharmacol. 1999;44(5):355–61. doi: 10.1007/s002800050989. [DOI] [PubMed] [Google Scholar]
- 13.Park SJ, Wu CH, Gordon JD, Zhong X, Emami A, Safa AR. Taxol induces caspase-10-dependent apoptosis. J Biol Chem. 2004;279(49):51057–67. doi: 10.1074/jbc.M406543200. [DOI] [PubMed] [Google Scholar]
- 14.Vogler M, Dinsdale D, Dyer MJ, Cohen GM. Bcl-2 inhibitors: small molecules with a big impact on cancer therapy. Cell Death Differ. 2009;16(3):360–7. doi: 10.1038/cdd.2008.137. [DOI] [PubMed] [Google Scholar]
- 15.van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10(5):389–99. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68(9):3421–8. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 17.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumors. Nature. 2005;435(7042):677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 18.Kutuk O, Letai A. Alteration of the mitochondrial apoptotic pathway is key to acquired paclitaxel resistance and can be reversed by ABT-737. Cancer Res. 2008;68(19):7985–94. doi: 10.1158/0008-5472.CAN-08-1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, et al. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003;17(12):1475–86. doi: 10.1101/gad.1093903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adams KW, Cooper GM. Rapid turnover of Mcl-1 couples translation to cell survival and apoptosis. J Biol Chem. 2007;282(9):6192–200. doi: 10.1074/jbc.M610643200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell. 2005;121(7):1085–95. doi: 10.1016/j.cell.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 22.Gottesfeld JM, Forbes DJ. Mitotic repression of the transcriptional machinery. Trends Biochem Sci. 1997;22(6):197–202. doi: 10.1016/s0968-0004(97)01045-1. [DOI] [PubMed] [Google Scholar]
- 23.Blagosklonny MV. Mitotic arrest and cell fate: why and how mitotic inhibition of transcription drives mutually exclusive events. Cell Cycle. 2007;6:70–4. doi: 10.4161/cc.6.1.3682. [DOI] [PubMed] [Google Scholar]
- 24.Sivan G, Elroy-Stein O. Regulation of mRNA Translation during cellular division. Cell Cycle. 2008;7:741–4. doi: 10.4161/cc.7.6.5596. [DOI] [PubMed] [Google Scholar]
- 25.Sánchez-Pérez T, Ortiz-Ferrón G, López-Rivas A. Mitotic arrest and JNK-induced proteasomal degradation of FLIP and Mcl-1 are key events in the sensitization of breast tumor cells to TRAIL by antimicrotubule agents. Cell Death Differ. 2010;17(5):883–94. doi: 10.1038/cdd.2009.176. [DOI] [PubMed] [Google Scholar]
- 26.Harley ME, Allan LA, Sanderson HS, Clarke PR. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 2010;29(14):2407–20. doi: 10.1038/emboj.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tunquist BJ, Woessner RD, Walker DH. Mcl-1 stability determines mitotic cell fate of human multiple myeloma tumor cells treated with the kinesin spindle protein inhibitor ARRY-520. Mol Cancer Ther. 2010;9(7):2046–56. doi: 10.1158/1535-7163.MCT-10-0033. [DOI] [PubMed] [Google Scholar]
- 28.Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471(7336):110–4. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- 29.Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, et al. SCFFBW7 regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471(7336):104–9. doi: 10.1038/nature09732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tan TT, Degenhardt K, Nelson DA, Beaudoin B, Nieves-Neira W, Bouillet P, et al. Key roles of BIM-driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell. 2005;7(3):227–38. doi: 10.1016/j.ccr.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 31.Meller R, Cameron JA, Torrey DJ, Clayton CE, Ordonez AN, Henshall DC, et al. Rapid degradation of Bim by the ubiquitin-proteasome pathway mediates short-term ischemic tolerance in cultured neurons. J Biol Chem. 2006;281(11):7429–36. doi: 10.1074/jbc.M512138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan N, Malek M, Zha J, Kassees R, Yue P, Berry L, et al. Navitoclax Enhances the Efficacy of Taxanes in Non-Small Cell Lung Cancer Models. Clin Cancer Res. 2011 Jan 10; doi: 10.1158/1078-0432.CCR-10-2353. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 33.Brito DA, Rieder CL. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr Biol. 2006;16:1194–1200. doi: 10.1016/j.cub.2006.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang HC, Mitchison TJ, Shi J. Stochastic competition between mechanistically independent slippage and death pathways determines cell fate during mitotic arrest. PLoS One. 2010;5(12):e15724. doi: 10.1371/journal.pone.0015724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19(11):1294–305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jackson JR, Patrick DR, Dar MM, Huang PS. Targeted anti-mitotic therapies: can we improve on tubulin agents? Nat Rev Cancer. 2007;7(2):107–17. doi: 10.1038/nrc2049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.