

To the Editor
Atopic dermatitis (AD) is the most common inflammatory skin disease, affecting up to 20% of children in the US and is characterized by an increased susceptibility to cutaneous infections1, 2. One in 10 AD subjects has difficulty clearing cutaneous infections with a host of viruses including herpes simplex, vaccinia, human papilloma and/or molluscum contagiosum1. This typically manifests as more extensive cutaneous and sometimes systemic disease and/or resistance to standard therapies. One of the rarer but more severe viral complications is eczema herpeticum (EH), caused by herpes simplex virus (HSV)-1 that results in a widespread skin infection1. EH can be complicated by viremia and multi-organ involvement including keratoconjunctivitis, meningitis, and encephalitis1. Although we have shown that AD subjects with more severe disease, greater allergen sensitization and enhanced Th2 polarity are at greatest risk for this viral complication, the molecular mechanisms responsible for this remain unclear1.
Disruption of the epidermal barrier is now recognized as a cardinal feature in AD pathogenesis. The skin has two barrier structures: the stratum corneum (SC) and tight junctions (TJ). It is widely accepted that AD subjects' SC is dysfunctional. The loss-of-function mutations in filaggrin, a structural protein important for its hygroscopic properties, have been reproducibly and robustly associated with AD3 and more recently with EH4. TJ are found on opposing membranes of stratum granulosum keratinocytes, directly below the SC. TJ consist of a complex of adhesive and scaffolding proteins that control the passage of water, ions, and solutes through the paracellular pathway5. We have recently demonstrated that the nonlesional epithelium of AD subjects have remarkable bioelectric abnormalities indicative of a TJ defect which may be the consequence of reduced levels of claudin-1 (CLDN1), a key TJ adhesive protein6. This is in line with the landmark work of Furuse and Tsukita whose CLDN1 knockout mouse established the importance of epidermal TJ and claudin-1. These mice died within 24 hr of birth with wrinkled skin, severe dehydration, and increased epidermal permeability as measured by dye studies and transepidermal water loss7. In our AD studies, we also observed an inverse correlation between CLDN1 expression and markers of Th2 polarity (total eosinophil counts and serum total IgE)6. This has led us to speculate that TJ defects may promote greater Th2 polarity, which is a characteristic of EH subjects1. We hypothesized that TJ defects such as we observed in AD subjects, may promote HSV-1 infections on that basis.
Additionally, viruses often commandeer intercellular junction proteins as a means of viral entry5. For example, wild-type HSV-1 infects keratinocytes through a complex interaction involving the viral envelope glycoprotein gD with either nectin-1 (PVRL1) or herpes virus entry mediator (HVEM)8, 9. Nectin-1, a Ca2-independent cell adhesion protein of the immunoglobulin superfamily, co-localizes with E-cadherin and beta-catenin to form another intercellular junctional complex called adherens junctions. Previous studies have shown that the susceptibility of human keratinocytes to HSV-1 infection is inversely related to the degree of cell-cell contact and confluency9, 10. This suggests that healthy junctional complexes may be a key deterrent to the spread of HSV-1 from one keratinocyte to another.
In these studies, we tested the hypothesis that the silencing of epidermal CLDN1 will enhance HSV-1 infectivity of human keratinocytes. We had previously demonstrated that reductions in CLDN1 adversely affected measures of TJ integrity6. Primary human keratinocytes (PHK) were isolated from foreskin as previously described6 and transiently transfected with scrambled (control) or CLDN1 (100 nM) siRNA (Santa Cruz Biotechnology, Inc; CA) using Lipofectamine™ 2000 transfection reagent (Invitrogen, Carlsbad, CA; see complete method in the Online Repository). We have previously demonstrated that with this knockdown approach lead to a >50% reduction in claudin-1 transcripts, and adversely affected TJ function as demonstrated by a 50% reduction in TEER and a 50% increase in permeability6. Importantly, this level of claudin-1 reduction mirrored what we had observed in AD samples. Twenty-four to 48h post-transfection, PHK were differentiated in DMEM with 10% heat-inactivated fetal bovine serum (HI-FBS; Invitrogen/Gibco) for 24h and subsequently infected with the highly virulent HSV-1 strain F (provided by Dr. D.C. Johnson) at a multiplicity of infection (MOI) of 0.1 (see complete method in the Online Repository). The MOI was chosen based on findings from viral titration (0.01 to 10 MOI) studies on differentiated and undifferentiated PHK, respectively, used as negative and positive controls (data not shown). After 2h, the viral inoculum was removed and PHK were cultured for 24h in DMEM containing 5% HI-FBS and 0.4% human-γ-globulin (0.5 mg/ml; Sigma, St. Louis, MO) to neutralize extracellular virus. HSV-infected PHK were fixed in 4% formaldehyde and stained with a polyclonal rabbit anti-HSV-1 (1:500; Dako, Carpinteria, CA) antibody followed by Alexa Fluor 488 donkey-anti-rabbit IgG H+L (1:1000, Invitrogen/Molecular probe) and 4′,6-diamidino-2-phenyl-indole, dihydrochoride (DAPI; 1:10,000, Molecular Probes). For each sample, six random fields were captured at identical acquisition settings and analyzed computationally to objectively quantify differences in focal forming units (FFU). A FFU was defined as a cluster of 3 or more adjacent HSV-1 positive cells. Claudin-1 depletion significantly increased the number and size of HSV-1 FFU (Fig 1A) as compared to control siRNA transfected PHK (Fig 1B, C). An average of 4.8 ± 0.7 FFU/field were counted in CLDN1 knockdown PHK, while only 2.5 ± 0.5 FFU/field were detected in the control cells (P = 0.05; n= 6; Fig 1C). Adapted Fluorospot software was used to semi-quantify differences in dimensions of each FFU (see complete method in the Online Repository). The diameters of FFU were significantly greater in keratinocytes whose CLDN1 expression was reduced (207 ± 22 μm) compared to controls (152 ± 40 μm; P = 0.03; n= 6; Fig 1D). Similar findings were observed when evaluating FFU area (P = 0.04; n= 6; Fig 1E) in PHK after CLDN1 knockdown (15649 ± 4367 pixels) as compared to control transfection (9902 ± 4943 pixels). The increased frequency of HSV-1-infected cells in CLDN-1 knockdown PHK was confirmed by an infectious center assay (see complete method in the Online Repository). More CLDN1 siRNA transfected PHK were infected with HSV-1 (38 ± 6%; n=3) than control PHK (28 ± 7%; n= 3; P = 0.003) (see Fig E1 in the Online Repository). Importantly, the observed increase in HSV-1 infectiveness in CLDN1 knockdown PHK could not be explained by reduced expression of nectin-1, the HSV-1 receptor. Indeed, nectin-1 mRNA expression was not affected by CLDN1 silencing (n=5; see Fig E2 in the Online Repository). However, we cannot rule out the possibility that reductions in CLDN1 and the affects on TJ function might reduce nectin-1 membrane localization. It is also important to highlight that our in vitro model is sufficient to induce functional TJ as demonstrated by enhanced trans epithelial resistance and reduced permeability6. However, it is not enough to induce terminal differentiation (e.g. filaggrin expression and cornified envelope). Although, this might be a limitation of the methodology we believe this condition well reflect the in vivo situation. In ADEH often the virus spreading is due to reactivation of a silent infection (inside-out); thus suggesting that the first barrier structure the virus will encounter is indeed TJ (located in the stratum granulosum, just below stratum corneum).
Figure 1. Knockdown of claudin-1 in human keratinocytes increases HSV-1 infectivity.
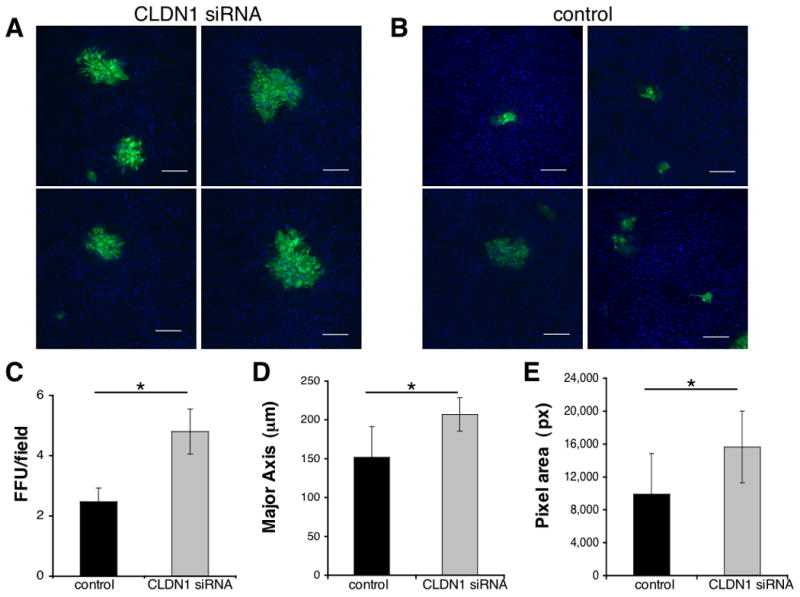
The green color indicates HSV-1 staining. The DAPI (blue) nuclear stain provides an assessment of cell density (Bars=200 μm). Larger and more numerous FFU were detected in PHK treated with CLDN1 siRNA (A) as compared to the control siRNA (B; Representative images of n=6 experiments). Cells stained with a rabbit IgG isotype control were all negative (data not shown). (C) An average of 4.8 ± 0.7 FFU/field were counted in CLDN1 knockdown PHK and 2.5 ± 0.5 FFU/field in the control samples (*P=0.05; n=6). (D) The diameter of the major axis of FFU was significantly greater in PHK whose CLDN1 expression was reduced (207 ± 21 μm) compared to controls (152 ± 40 μm; *P=0.03; n=6). (E) Similar observations were noted in FFU area (*P=0.04; n=6) in CLDN1 knockdown (15649 ± 4367 pixels) as compared to control transfection (9902 ± 4943 pixels).
As part of the NIAID-funded Atopic Dermatitis and Vaccinia Network (ADVN) (reviewed in a previous publication by Beck et al1), we performed a preliminary study to evaluate whether CLDN1 polymorphisms were associated with EH. To do this, we utilized a haplotype-tagging SNP approach using genetic markers available in the public arena (n = 132 in dbSNP) in 414 European Americans (EA) and 328 African Americans (AA; see complete methods and Table E1 in Online repository). We previously reported modest associations (P = 0.003 -0.05) between SNP variants throughout the CLDN1 gene and AD that were more significant among African Americans6. In this study, we addressed whether genetic variations in CLDN1 contribute to risk of EH in both North American AD populations. The EH subphenotype was defined as AD subjects that had at least one EH episode documented either by an ADVN investigator (or a physician affiliated with the same academic center) or diagnosed clinically by an outside physician where the HSV infection was confirmed by PCR, tissue immunofluorescence, Tzanck smear and/or culture. The study was approved by the institutional review boards at National Jewish Health, Johns Hopkins University, Oregon Health & Science University, University of California San Diego, Children's Hospital of Boston and University of Rochester Medical Center. All subjects gave written informed consent prior to participation.
To determine whether SNPs in CLDN1 were associated with EH, the Cochran-Armitage trend test using the PLINK software (http://pngu.mgh.harford.edu/∼purcell/plink/to) was performed within each AD population and confirmed with the adaptive permutation test. We observed only modest associations between CLDN1 SNPs and EH in our North American AD populations. We wondered if the strong association we observed between both FLG null mutations R501X and 2282del4 and EH (OR = 10.1 [4.7-22.1]; P=1.99 × 1011) was masking a lesser effect from CLDN1 variants4. To address this, we excluded subjects who had one or both of these FLG mutations and reanalyzed the associations with all CLDN1 SNPs. In the European American group, an intronic SNP (rs3774032; OR = 0.59 [0.35-0.98] P=0.037) was marginally associated with EH (Fig 2), but not significant after adaptive permutations adjustment (P=0.10). When excluding subjects with a FLG mutation (n=29 [36.3%]; Table 1), the association became slightly more significant (OR = 0.44 [0.22-0.85], P=0.0225) and still significant after adaptive permutations adjustment (P=0.035). Interestingly, one additional intronic SNP emerged (rs3732923, OR = 1.93 [1.21-3.07], P=0.0010; adaptive permutation P=0.0005; Fig 2). Although the EH sample size in the African American population was too small (n=21) for meaningful analyses, a SNP in the promoter region was associated with EH (rs3954259; OR = 2.16 [1.02-4.59], P=0.040; adaptive permutation P=0.033). Limitations of these studies are the small sample size and the superficial coverage of CLDN1 loci. Nevertheless, these findings implicate CLDN1 mutations in susceptibility to widespread HSV skin infections in subjects with AD, especially those who do not have a FLG mutation.
Figure 2. Excluding subjects with the two most common FLG mutations significantly enhanced CLDN1 associations with Eczema Herpeticum.
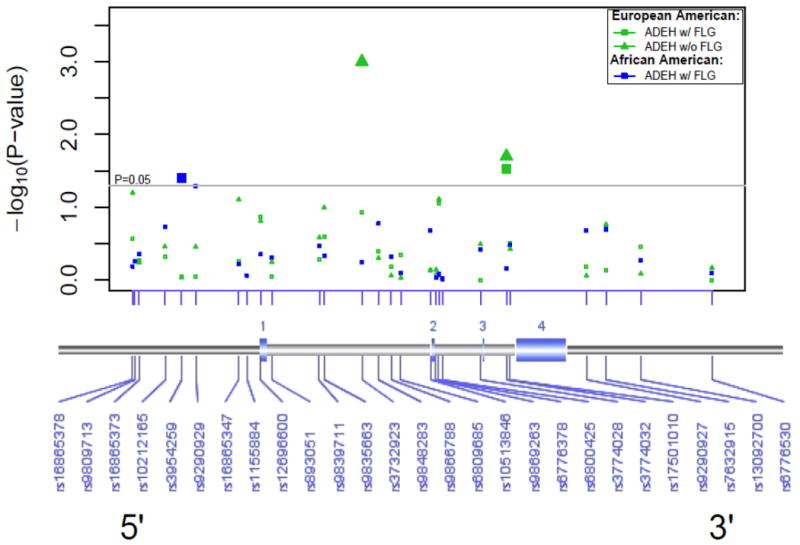
A SNP (rs3774032, P=0.03) was significantly associated with EH in the European American population. When subjects were stratified for FLG (e.g. w/o FLG mutations) the association was slightly more significant (P=0.02). In this group, we observed one additional intronic SNP that was significantly associated with EH (rs3732923, P=0.001). In the African American population, a SNP (rs3954259; P=0.040), in the promoter region was associated with EH.
Table 1. ADVN Registry Participant Characteristics.
| Characteristic | European American | African American | ||
|---|---|---|---|---|
| ADEH+ | ADEH- | ADEH+ | ADEH- | |
| Sample size | 93 | 165 | 21 | 155 |
| Males; N (%) |
49 (52.7%) |
47 (28.5%) |
8 (38.1%) |
35 (22.6%) |
| Age; mean (SD) |
22.9 (21.5) |
38.0 (14.5) |
22.8 (19.0) |
36.4 (11.0) |
| AD onset <5yrs; N (%) |
77 (97.5%) |
101 (78.3%) |
14 (93.3%) |
77 (68.1%) |
| Median IgE levels; (Interquartile Range)* | 2225 (611-9034) | 252 (85-1275) | 3505 (571-9429) | 425 (129-1194) |
| Median EASI†; (Interquartile Range)** | 10.0 (4.4-15.8) | 3.2 (1.3-8.4) | 10.9 (6.8-15.8) | 3.0 (1.5-7.4) |
| FLG Null Allele Carrier; N (%) |
29 (36.3%) |
36 (22.1%) |
2 (12.5%) |
9 (6.0%) |
The following abbreviations are used: AD, atopic dermatitis; ADEH+, atopic dermatitis with a history of eczema herpeticum; ADEH-, AD without a history of EH; EASI, Eczema area and severity index; and NA, not applicable.
EASI determined by the percentage of eczema area on a 7-point ordinal scale: 0= no eruption; 1=<10%; 2=10%-29%; 3=30%-49%; 4=50%-69%; 5=70%-89%; and 6=90%-100%.
Total serum IgE levels were significantly higher in ADEH+ patients compared to both ADEH- patients and non-atopic, healthy controls even after adjusting for age (P < 0.001).
EASI was significantly higher among ADEH+ patients compared to ADEH-patients in both racial groups even after adjusting for age (P < 0.001).
In conclusion, this study and previously reported work implicate both SC and TJ epidermal barrier defects as a mechanism to explain AD subjects susceptibility to widespread cutaneous infections with HSV and possibly other viral pathogens4. In support of this notion our EH patients more commonly experienced infections with molluscum contagiosum than AD subjects with no history of EH (29% vs 2%, respectively)1. More of our EH subjects (16%) suffered with recurrent HSV keratitis as compared to only 1% of AD subjects with no history of EH1. It is possible that the same mechanism may responsible for their ocular disease as corneal epithelium possess TJs and these structures express claudin111, 12. In this study, we show both mechanistic and genetic results that implicate tight junctions as a critical barrier structure important in containing the spread of epidermal HSV-1 infections. Defects in TJ as compared to the SC may be particularly relevant in viral infections where the virus enters from a basolateral direction as is the case with HSV reactivation. Collectively, this work highlights a previously underappreciated role for TJ proteins in cutaneous host defense and may lead to new therapeutic strategies.
Supplementary Material
Acknowledgments
We would like to acknowledge Melissa Dubois, Ph.D. and Todd W. Wisner at Oregon Health & Science University (Portland, OR) for helpful suggestions on HSV-1 experiments. Mr. Jonathan Rebhahn, M.S. at University of Rochester Medical Center (Rochester, NY) who developed the Exploraspot™ software to quantify FFU. Mary Bolognino, B.S. at University of Rochester Medical Center (Rochester, NY) for help with PHK studies.
We would also like to acknowledge several groups whose efforts made the clinical enrollment possible: ADVN Coordinators (Patricia Taylor, NP; Trista Berry, BS; Susan Tofte, FNP; Shahana Baig-Lewis, MPH; Peter Brown, BS; Lisa Heughan, BA, CCRC; Meggie Nguyen, BS; Doru Alexandrescu, MD; Lorianne Stubbs, RC; Reena Vaid MD; Diana Lee, MD), ADVN regulatory advisors (Judy Lairsmith, RN), biological sample tracking (JHU - Tracey Hand, MSc; Jessica Scarpola, and URMC - Mary Bolognino, MS), NIAID-DAIT oversight (Marshall Plaut, MD and Joy Laurienzo Panza, RN, BSN), Rho® Inc. statistical support (Daniel Zaccaro, MS and Brian Armstrong, MPH) Rho®, Inc. general study support and oversight (Jamie Reese, BS; Susi Lieff, PhD; and Gloria David, PhD, MHSc) and last but by no means least, all the patients who participated in this study.
Funding: The Atopic Dermatitis and Vaccinia Network NIH/NIAID (contract N01 AI40029 and N01 AI40033) and Atopic Dermatitis Research Network (contract HHSN272201000020C and HHSN272201000017C), National Eczema Association (A.D.), the Oregon National Primate Research Center grant, RR000163 (M.K.S.) and Mary Beryl Patch Turnbull Scholar Program (K.C.B.).
Abbreviations
- AD
Atopic Dermatitis
- ADVN
Atopic Dermatitis and Vaccinia Network
- EH
Eczema Herpeticum
- HSV
Herpes Simplex Virus
- MOI
Multiplicity of Infection
- PHK
Primary Human Keratinocytes
- qPCR
Quantitative PCR (e.g. Real-time PCR)
- SNP
Single Nucleotide Polymorphism
- SC
Stratum Corneum
- TJ
Tight Junction
- TEER
Trans Epithelial Electrical Resistance
- ZO
Zonulae Occludens
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Beck LA, Boguniewicz M, Hata T, Schneider LC, Hanifin J, Gallo R, et al. Phenotype of atopic dermatitis subjects with a history of eczema herpeticum. J Allergy Clin Immunol. 2009;124:260–9. doi: 10.1016/j.jaci.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boguniewicz M, Leung DY. Recent insights into atopic dermatitis and implications for management of infectious complications. J Allergy Clin Immunol. 2010;125:4–13. doi: 10.1016/j.jaci.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O'Regan GM, Sandilands A, McLean WH, Irvine AD. Filaggrin in atopic dermatitis. J Allergy Clin Immunol. 2009;124:R2–6. doi: 10.1016/j.jaci.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 4.Gao PS, Rafaels NM, Hand T, Murray T, Boguniewicz M, Hata T, et al. Filaggrin mutations that confer risk of atopic dermatitis confer greater risk for eczema herpeticum. J Allergy Clin Immunol. 2009;124:507–13. doi: 10.1016/j.jaci.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Niessen CM. Tight junctions/adherens junctions: basic structure and function. J Invest Dermatol. 2007;127:2525–32. doi: 10.1038/sj.jid.5700865. [DOI] [PubMed] [Google Scholar]
- 6.De Benedetto A, Rafaels NM, McGirt LY, Ivanov AI, Georas SN, Cheadle C, et al. Tight Junctions Defects in Atopic Dermatitis. J Allergy Clin Immunol. 2010 doi: 10.1016/j.jaci.2010.10.018. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Furuse M, Hata M, Furuse K, Yoshida Y, Haratake A, Sugitani Y, et al. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J Cell Biol. 2002;156:1099–111. doi: 10.1083/jcb.200110122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gonzalez-Mariscal L, Garay E, Lechuga S. Virus interaction with the apical junctional complex. Front Biosci. 2009;14:731–68. doi: 10.2741/3276. [DOI] [PubMed] [Google Scholar]
- 9.Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science. 1998;280:1618–20. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- 10.Schelhaas M, Jansen M, Haase I, Knebel-Morsdorf D. Herpes simplex virus type 1 exhibits a tropism for basal entry in polarized epithelial cells. J Gen Virol. 2003;84:2473–84. doi: 10.1099/vir.0.19226-0. [DOI] [PubMed] [Google Scholar]
- 11.Shimazaki J, Higa K, Kato N, Satake Y. Barrier function of cultivated limbal and oral mucosal epithelial cell sheets. Invest Ophthalmol Vis Sci. 2009;50:5672–80. doi: 10.1167/iovs.09-3820. [DOI] [PubMed] [Google Scholar]
- 12.Yoshida Y, Ban Y, Kinoshita S. Tight junction transmembrane protein claudin subtype expression and distribution in human corneal and conjunctival epithelium. Invest Ophthalmol Vis Sci. 2009;50:2103–8. doi: 10.1167/iovs.08-3046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.