

Abstract
Epidemiological studies estimate that greater than 60% of the adult US population may be categorized as either overweight or obese and there is a growing appreciation that the complications of obesity extend to the central nervous system (CNS). While the vast majority of these studies have focused upon the hypothalamus, more recent studies suggest that the complications of obesity may also affect the structural and functional integrity of the hippocampus. A potential contributor to obesity-related CNS abnormalities is the adipocyte-derived hormone leptin. In this regard, decreases in CNS leptin activity may contribute to deficits in hippocampal synaptic plasticity and suggest that leptin resistance, a well described phenomenon in the hypothalamus, may also be observed in the hippocampus. Unfortunately, the myriad of metabolic and endocrine abnormalities in diabetes/obesity phenotypes makes it challenging to assess the role of leptin in hippocampal neuroplasticity deficits associated with obesity models. To address this question, we examined hippocampal morphological and behavioral plasticity following lentivirus-mediated downregulation of hypothalamic insulin receptors (hypo-IRAS). Hypo-IRAS rats exhibit increases in body weight, adiposity, plasma leptin and triglyceride levels. As such, hypo-IRAS rats develop a phenotype that is consistent with features of the metabolic syndrome. In addition, hippocampal morphological plasticity and performance of hippocampal-dependent tasks are adversely affected in hypo-IRAS rats. Leptin-mediated signaling is also decreased in hypo-IRAS rats. We will discuss these findings in the context of how hyperleptinemia and hypertriglyceridemia may represent mechanistic mediators of the neurological consequences of impaired hippocampal synaptic plasticity in obesity.
Keywords: triglycerides, leptin, adiposity, fear conditioning, STAT3, morphology, lentivirus
1. Introduction
The escalating obesity epidemic is an important health care issue that has critical socio-economic consequences. There is a growing appreciation that the complications of metabolic diseases extend to the central nervous system (CNS) and, as a result, the neurological consequences of obesity have been receiving greater attention [1;2]. In this regard, increases in obesity and/or body mass index (BMI) are associated with decreased cognitive function in humans [3-6]. In rodents, hippocampal-dependent behaviors are impaired in experimental models of obesity [7-19], as well as in genetic mutations that result in disrupted leptin signaling, such as in the db/db mouse and the Zucker fa/fa rat [8;17]. Some of these studies have investigated hippocampal insulin resistance as a mechanistic mediator of these behavioral deficits, including our previous demonstration that insulin receptor (IR) signaling is reduced in obese Zucker rats [17]. However, the potential impact of hyperleptinemia has often been overlooked in these studies. In obesity phenotypes, hypothalamic leptin signaling is impaired, which leads to and/or contributes to leptin resistance (For review, see [20]). Additionally, leptin transport across the blood-brain barrier (BBB) is impaired in obesity phenotypes [21-24]. This has lead to the suggestion that decreases in hippocampal leptin signaling and/or BBB leptin transport contribute to the decreases in hippocampal synaptic plasticity observed in obesity.
We have developed a lentiviral vector that contains an antisense sequence selective for the IR (LV-IRAS) [25]. When injected into the third ventricle to target IRs expressed in the hypothalamus (hypo-IRAS), IRAS-treated rats exhibit significant decreases in IR expression and signaling in the hypothalamus when compared to rats treated with the control virus (hypo-Con); these parameters are unaffected in the hippocampus. Downregulation of hypothalamic IRs produces the expected increases in body weight, body adiposity, plasma leptin levels and plasma triglyceride levels [25;26], features that are consistent with aspects of the metabolic syndrome. As such, hypo-IRAS rats exhibit an obesity phenotype that provides an experimental model to examine the impact of elevated plasma leptin and triglyceride levels upon hippocampal synaptic plasticity. In this regard, we recently reported that high-frequency stimulation of the Schaffer collaterals fails to elicit long term potentiation (LTP) in the CA1 region of hypo-IRAS rats, a deficit in synaptic transmission that may be related to decreases in Ser845 phosphorylation of hippocampal GluA1 receptor subunits [26]. These results indicate that some measures of synaptic plasticity are impaired in the hippocampus of hypo-IRAS rats. In view of these observations, the aim of the current study was to determine whether the hyperleptinemic/obesity phenotype observed in hypo-IRAS rats adversely affects other aspects of hippocampal synaptic plasticity, including morphological and behavioral measures of neuroplasticity. Additionally, since leptin resistance may involve impairments in leptin signaling and/or BBB leptin transport, we also examined leptin signaling in hypothalamus following peripheral administration of leptin in hypo-IRAS rats.
2. Materials and methods
2.1. Animal protocols
Adult male Sprague Dawley rats (CD strain, Charles River) weighing 200-250 g were housed in groups of three with ad libitum access to food and water, in accordance with all guidelines and regulations of The University of South Carolina Animal Care and Use Committee. Lentivirus production and hypothalamic administration were performed as described in our previous studies [25;26]. Briefly, rats were anesthetized, placed in the stereotaxic apparatus and lentivirus was injected into the third ventricle using the following coordinates: AP: −2.6 mm; L: 0.0 mm; DV: −10.0 mm. The viral stock (6 ul of 5×106 tu/ul) was injected at a speed of 1 ul/min with a 10 ul Hamilton syringe driven by a motorized stereotaxic injector (Stoelting 53310). Rats were injected with either the LV-IRAS construct (hypo-IRAS) or the LV-control construct (hypo-Con). After surgery, hypo-IRAS and hypo-Con rats were housed individually in a BSL2 facility for at least three weeks prior to subsequent analyses. At the end of these experiments, blood was collected by tail bleed for plasma endocrine analysis.
2.2. Plasma endocrine analysis
Enzyme-linked immunosorbent assay (ELISA) analysis was performed for leptin (Linco Research, Billerica, MA) in plasma isolated from hypo-Con and hypo-IRAS rats [25;26]. ELISA plates were analyzed according to the manufacturer’s instructions using a Tecan SPECTRAFluor plate reader (Tecan U.S., Inc., Durham, NC). Plasma triglycerides were determined using an enzymatic kit (modified Trinder) according to the manufacturer’s instructions (Pointe Scientific, Inc., Canton, MI, USA). Statistical analysis was performed using an unpaired t-test with P < 0.05 as the criterion for statistical significance.
2.3. Contextually-Conditioned Freezing
Conditioned freezing was assessed as described previously [27]. Rats were placed in a 46 × 24 × 22 cm shock box inside an acoustic isolation chamber and allowed to explore for 3 minutes to assess unconditioned freezing (Unconditioned freezing). Rats were conditioned with 3 context-footshock pairings (1 mA, 1 sec shocks) delivered with a 60 sec intershock interval (ISI; acquisition freezing). Forty-eight hours later rats were returned to the context for 8 minutes for analysis of contextually-conditioned freezing (retention test). Freezing behavior, defined as the absence of movement except for respiration, was assessed from video tapes by an observer blind to the treatment status of the animal. Freezing was scored for the 3 minutes before shocks, for 1 min after each shock during acquisition, and for 8 min during the retention test. Freezing behavior was analyzed using a one-way (virus treatment) repeated measures ANOVA with time as the within-subjects factor. Significance level was set at P<0.05, and the Student-Newman-Keuls test was used for post-hoc analysis. Two hours after the completion of the retention test, rats were anesthetized and transcardially perfused with phosphate-buffered saline (PBS) and 4% paraformaldehyde. All brains were then processed for c-Fos immunoreactivity.
2.4. Immunohistochemical approaches
Immunohistochemistry was performed as described in our previous studies [27-30]. For immunofluorescence studies, rat brain sections were washed with PBS and then incubated with primary antisera raised against the presynaptic protein synaptophysin (1:1000 dilution; Santa Cruz Biotechnology, Inc.) and the postsynaptic protein PSD-95 (1:1000 dilution; Upstate Biotechnology). Following an overnight incubation at 4°C, sections were washed in PBS and then incubated with secondary antisera conjugated with Alexa 488-labeled anti-rabbit IgGs (1:200, Molecular Probes) and Alexa 568-labeled anti-mouse IgGs (1:200, Molecular Probes) for 3-4 hours at room temperature. Following incubation with secondary antisera, sections were rinsed in PBS, mounted on glass slides and coverslipped with Aquamount (Polysciences, Inc.). Sections were examined with a Zeiss LSM510 confocal laser scanning microscope.
Immunohistochemistry for c-Fos was performed as described in our previous studies [27;30]. Briefly, sections were washed with tris-buffered saline (TBS) and incubated with c-Fos primary antisera at a dilution of 1:15000 for 24 hours at 4°C (Calbiochem, La Jolla, CA). Rat brain sections were washed with TBS and then incubated in biotinylated donkey anti-rabbit secondary (1:1000) for 90 minutes at room temperature. Following washes with TBS, sections were incubated horseradish peroxidase-conjugated streptavidin (1:600) at room temperature for 1 hour. Fos-like immunoreactivity was developed using diaminobenzidine as a substrate. Counting of c-Fos-positive neurons was performed on six rats per group using two representative sections from similar rostral-caudal levels. Statistical analysis was performed using an unpaired t-test with P < 0.05 as the criterion for statistical significance.
2.5. Immunoblot analysis
Immunoblot analysis was performed as described in our previous studies [25;26;31]. Briefly, 20 μg of total membrane fractions were separated by SDS/PAGE (10%), transferred to nitrocellulose (NC) membranes and blocked in TBS plus 10% non fat dry milk. NC membranes were then incubated with primary antisera (PSD-95 or synaptophysin) in TBS/5% non fat dry milk. After overnight incubation at 4°C, blots were washed with TBS plus 0.05% Tween 20 (TBST) and incubated with peroxidase-labeled species-specific secondary antibodies. NC membranes were then washed with TBST and developed using enhanced chemiluminescence reagents (GE Healthcare) as described by the manufacturer. Normalization for protein loading was performed using a mouse monoclonal primary antibody selective for actin (Sigma Chemical Company).
2.6. Immunohistochemical analysis of leptin signaling
Hypo-Con and hypo-IRAS rats were given an intraperitoneal (i.p.) injection of leptin (5 mg/kg) and 45 minutes later were transcardially perfused with saline followed by 4% paraformaldehyde in PBS. Rat brains were then processed for pSTAT3 immunohistochemistry as described by Levin and coworkers [32] based upon previously established protocols [33]. Briefly, free-floating sections were washed in potassium phosphate-buffered saline (KPBS) at room temperature, followed by an incubation in 1% NaOH, 1% H2O2 in KPBS. Sections were washed in KPBS and then incubated in 0.3% glycine/KPBS. Sections were then incubated in SDS/KPBS solution for 10 min, washed in KPBS and then blocked in 4% normal horse serum (NHS)/0.4% Triton X/KPBS. Sections were then incubated in this buffer with pSTAT3 primary antisera (Cell Signaling Technology) overnight at 4°C, followed by a 1 hour incubation at room temperature. Sections were washed in 1% NHS/0.02% Triton/KPBS and then incubated with biotinylated anti-rabbit IgGs for 1 hour at room temperature. Sections were washed, incubated with ABC reagents for 1 hour at room temperature, washed in KPBS and developed using diaminobenzidine as a substrate.
3. Results
3.1. Development of obesity/hyperleptinemic phenotype
Our previous studies determined that an IR antisense lentivirus downregulated hypothalamic IR expression, increased body weight and plasma leptin levels [25;26]. These parameters were used as biomarkers to evaluate the efficacy of LV-IRAS administration in the current studies. Approximately three weeks after hypothalamic LV administration, hypo-IRAS rats exhibited significant increases in plasma leptin levels when compared to hypo-Con rats. Hypo-IRAS rats also exhibited significant increases in body weight gain and in plasma triglycerides (Table 1). Additionally, western blot analysis revealed that IR expression was significantly decreased in the hypothalamus of hypo-IRAS rats when compared with hypo-Con rats (Table 1). Collectively, these data confirm the efficacy of the hypo-IRAS construct, and the development of the obesity phenotype.
Table 1.
Physiological and endocrine parameters in hypo-IRAS and hypo-Con rats
| Hypo-Con | Hypo-IRAS | |
|---|---|---|
| Plasma leptin, (ng/ml) | 8.8 ± 0.8 | 13.3 ± 0.7* |
| Plasma triglycerides (mg/dl) | 166.8 ± 9.0 | 237.2 ± 17.0* |
| Body weight gain (g) | 56.3 ± 5.4 | 85.2 ± 9.4* |
| Hypothalamic IR levels (% of hypo-Con) | 100.0 ± 8.6 | 55.2 ± 6.1# |
= P< 0.05
= P< 0.001
3.2. Contextually-conditioned freezing is impaired in hypo-IRAS rats
To determine whether the obesity phenotype elicited by downregulation of hypothalamic IRs produced deficits in hippocampal-dependent behaviors, contextually-conditioned freezing was performed in hypo-IRAS and hypo-Con rats. As shown in Figure 1, unconditioned freezing (minutes 1-3 before shocks) and acquisition of freezing behavior (minutes 4-6) were similar in hypo-Con and hypo-IRAS rats during the conditioning session on day 1. However, similar to observations in mice provided a high-fat diet to induce obesity [14], hypo-IRAS rats exhibited significant decreases in freezing behavior in the retention test 48 hours later when compared to the hypo-Con rats. The absence of behavioral changes in unconditioned freezing period and in the acquisition period suggests that differences in pain sensitivity are not responsible for the differences in freezing behavior observed during the retention test.
Figure 1.
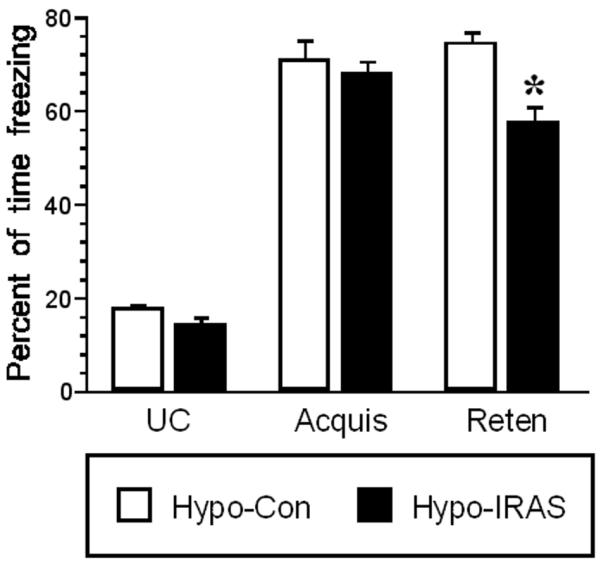
Downregulation of hypothalamic IRs impairs contextually-conditioned fear responses. Contextually-conditioned freezing responses were examined in hypo-IRAS and hypo-Con rats. Freezing behavior on the day of acquisition was similar in both groups of rats. [F(2,46) = −0.453, P = 0.65] However, performance in the retention test 48 hours later revealed that hypo-IRAS rats exhibited significant decreases in freezing behavior when compared to hypo-Con rats. This decrease in freezing behavior is particularly evident during the last 3 minutes of the retention test (Reten bars; [F(5, 210)=76, P<0.0001]. These results demonstrate that hypo-IRAS rats exhibit deficits in a learning task that is dependent at least in part upon hippocampal function. [Data are expressed as percentage of time spent freezing and are based upon at least 14 rats per group. * = significantly different from hypo-Con rats. UC = unconditioned freezing; Acquis = freezing during acquisition; Reten = retention freezing behavior]
3.3. Fear conditioning-induced c-Fos expression is reduced in the CA1 region of hypo-IRAS rats
As a measure of neuronal activation, freezing-induced c-Fos expression was examined in the hippocampus of hypo-IRAS and hypo-Con rats. Two hours after completion of the retention test, hypo-IRAS and hypo-Con rats were perfused with 4% paraformaldehyde and processed for c-Fos immunohistochemistry. As shown in Figure 2, Fos-like immunoreactivity (FLI) was observed in neuronal nuclei in the CA1 region of the hippocampus of hypo-Con rats two hours following fear conditioning (Panel A); however, hypo-IRAS rats did not exhibit appreciable FLI in the CA1 region following fear conditioning (Panel B). In the dentate gyrus, FLI induced by fear conditioning was similar in hypo-Con and hypo-IRAS rats (Panels C and D, respectively). Cell counting confirmed these observations. While FLI was unchanged in the dentate gyrus, c-Fos expression was significantly reduced in the CA1 region of hypo-IRAS rats two hours following contextual fear conditioning when compared to hypo-Con rats (Figure 3).
Figure 2.
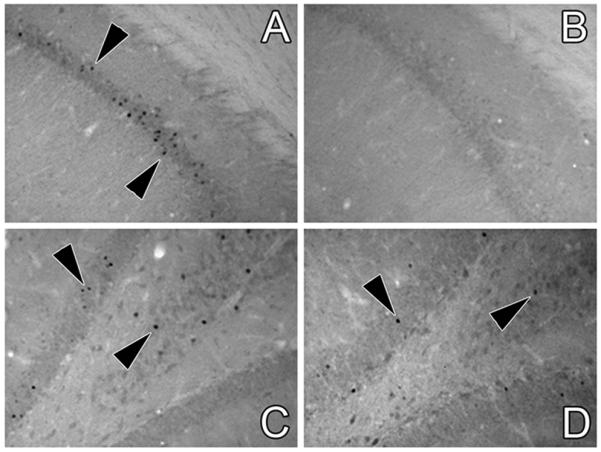
Hypo-IRAS rats exhibit decreased neuronal activation in the CA1 region following contextual fear conditioning. Fos-like immunoreactivity (FLI) was examined in the hippocampus of hypo-IRAS and hypo-Con rats two hours following the conditioned freezing retention test. Representative light photomicrographs of FLI in the CA1 region of the hippocampus of hypo-Con rats and hypo-IRAS rats are depicted in Panels A and B, respectively. Contextual fear conditioning-induced FLI in the dentate gyrus of hypo-Con rats and hypo-IRAS rats are depicted in Panels C and D, respectively.
Figure 3.
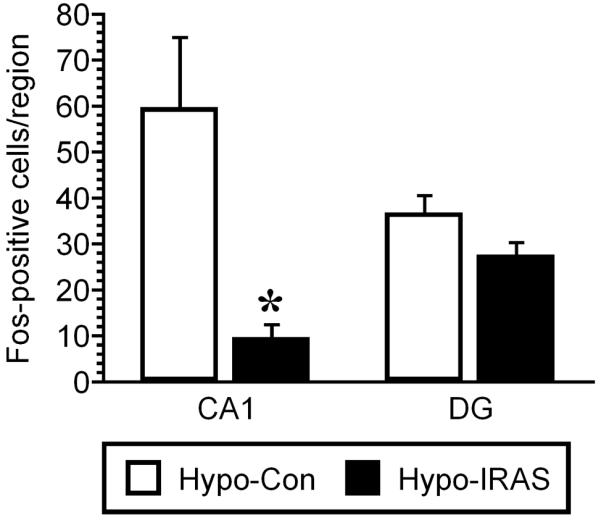
Quantitative analysis of c-Fos-positive nuclei in hypo-Con and hypo-IRAS rats. Fos-like immunoreactivity (FLI) was significantly reduced in the CA1 region of hypo-IRAS rats compared to hypo-Con-treated rats in response to the conditioned context. No changes in c-Fos expression were observed in the dentate gyrus of hypo-IRAS rats compared to hypo-Con rats. These data demonstrate that neuronal activation, as least as determined by FLI, is significantly decreased in response to contextual fear conditioning in the CA1 region of the hippocampus of hypo-IRAS rats. [Data based upon 6 rats/group. * = P ≤ 0.05 vs. control]
3.4. Downregulation of hypothalamic IRs alters hippocampal synaptic morphology
We have previously observed changes in the distribution of the presynaptic protein synaptophysin (SYN) and the postsynaptic protein PSD-95 in experimental paradigms that are associated with behavioral deficits in hippocampal plasticity, such as in type 1 diabetic rats and in rats exposed to stress levels of glucocorticoids [28]. In view of these observations, the distribution and expression of SYN and PSD-95 was determined in the hippocampus of hypo-IRAS rats and hypo-Con rats by confocal fluorescence immunohistochemistry. As shown in Figure 4, PSD-95 (red) and SYN (green) exhibited the expected localization profiles in the CA3 region of the hippocampus of hypo-Con rats (Panels A and C, respectively). Conversely, hypo-IRAS rats exhibited significant clustering and redistribution of both PSD-95 and SYN. Specifically, PSD-95 immunofluorescence was clustered and less organized in the CA3 region of hypo-IRAS rats (Panel B) when compared to hypo-Con rats (Panel A). Similarly, SYN immunofluorescence in hypo-IRAS rats exhibited a dramatic increase in clustering in the oriens layer of CA3 (Panel D), presumably representing mossy fibers terminals that form synaptic contacts with CA3 pyramidal neurons. Higher power magnification further identified SYN clustering in the CA3 region of hypo-IRAS rats (Panel F) compared to the expected profile of SYN expression observed in hypo-Con rats (Panel E). In order to determine whether these observations reflect changes in total protein expression, western blot analysis was performed in total membrane extracts isolated from the hippocampus of hypo-IRAS and hypo-Con rats. Immunoblot analysis revealed that the changes in the distribution of PSD-95 immunoreactivity identified by fluorescence immunohistochemistry were associated with increases in total hippocampal PSD-95 protein expression in hypo-IRAS rats. Increases in hippocampal SYN levels observed in hypo-IRAS rats did not achieve statistical significance (Figure 5).
Figure 4.

Representative confocal microscopic images for synaptophysin (SYN) and PSD-95 in the CA3 region of hypo-Con and hypo-IRAS rats. Panel A: Hypo-Con rats exhibit the expected distribution of the post-synaptic protein PSD-95 (red, Panel A), while PSD-95 immunoreactivity is clustered and less organized in the CA3 region of hypo-IRAS rats (Panel B). Immunofluorescence for the presynaptic protein SYN (green) in hypo-Con rats and hypo-IRAS rats are depicted in Panels C and D, respectively. Higher power magnification of SYN immunofluorescence in hypo-Con and hypo-IRAS rats is shown in Panels E and F. Unlike the expected distribution observed in hypo-Con rats, SYN immunofluorescence is clustered adjacent to the pyramidal cell layer in hypo-IRAS rats. These results suggest that downregulation of hypothalamic insulin receptors elicits synaptic re-organization in the CA3 region of the hippocampus.
Figure 5.

Immunoblot analysis of synaptic proteins in the hippocampus of hypo-IRAS and hypo-Con rats. Panel A: Representative immunoblot of SYN immunoreactivity in hippocampal whole membrane preparations isolated from hypo-IRAS (IRAS) or hypo-Con (Con) rats. Panel B. Representative immunoblot of PSD-95 immunoreactivity in hippocampal whole membrane preparations isolated from hypo-IRAS (IRAS) or hypo-Con (Con) rats. Autoradiographic analysis revealed that PSD-95 protein levels were significantly increased in the hippocampus of hypo-IRAS rats when compared to hypo-Con rats. [t(2,13) = −3.087, P=0.008]. Hypo-IRAS rats exhibited non-significant increases in (SYN) compared to hypo-Con rats. [t(2,13) = −1.939, P=0.07]. [Data based upon immunoblots performed on individual whole membrane preparations isolated from hippocampi of hypo-Con (n=7) and hypo-IRAS rats (n=7). * = significantly different from hypo-Con rats. Molecular weight standards are shown on right]
3.5. Downregulation of hypothalamic insulin receptors impairs leptin signaling
Leptin activation of the long form of the leptin receptor (LRb) stimulates the JAK/STAT pathway, resulting in phosphorylation of JAK2 and STAT3. Phospho-STAT3 (pSTAT3) serves as a transcriptional regulator to increase the expression of (among other genes) suppressor of cytokine signaling 3 (SOCS3), which mediates feedback inhibition of LRb. (For review of these pathways, see [34]). To determine whether downregulation of hypothalamic IRs affects leptin-mediated signaling, hypo-Con and hypo-IRAS rats received an i.p. injection of leptin (5 mg/kg) and 45 minutes later were perfused for immunohistochemical analysis for pSTAT3. As shown in Figure 6, peripheral leptin administration elicited the expected increases in pSTAT3 levels in the hypothalamus of hypo-Con rats (Panel A). Conversely, peripheral leptin administration failed to elicit phosphorylation of STAT3 in the hypothalamus of hypo-IRAS rats (Panel B). These results demonstrate that hypo-IRAS rats develop leptin resistance, which may result from decreases in BBB transport of leptin and/or decreases in hypothalamic LRb-mediated signaling.
Figure 6.

Peripheral leptin administration stimulates phosphorylation of STAT3 (pSTAT3) in the hypothalamus of hypo-Con, but not hypo-IRAS rats. Rats were given an i.p. injection of leptin (5 mg/kg) and were perfused 45 minutes later; brains were then processed for pSTAT3 immunohistochemistry as described above. Panel A: Representative bright-field photomicrograph of pSTAT3 immunoreactivity in the hypothalamus of leptin-treated hypo-Con rats. Panel B: Representative bright-field photomicrograph of leptin-treated hypo-IRAS rats, indicating that peripheral leptin treatment failed to elicit a significant increase in hypothalamic pSTAT3 levels. [f: fornix; VMN: ventromedial nucleus of the hypothalamus; Arc: arcuate nucleus]
4. Discussion
The results of the current study confirm and extend our previous findings [26] that the development of an obesity/hyperleptinemic phenotype elicited by the region-specific downregulation in hypothalamic IR expression adversely affects the structural integrity and functional plasticity of the hippocampus. Specifically, hypo-IRAS rats exhibit decreases in contextual fear conditioning that are accompanied by decreases in c-Fos expression in the CA1 region of the hippocampus. Interestingly, our previous studies determined that hypo-IRAS rats exhibit electrophysiological deficits in the CA1 region, thereby providing another example of decreased neuronal activation in the hippocampus of hypo-IRAS rats. Beyond these functional measures, hypo-IRAS rats exhibit significant clustering and redistribution of SYN and PSD-95 in the CA3 region. Changes in the expression and distribution of PSD-95 observed in this study may contribute to the decreases in Ser845 phosphorylation of GluA1, as well as decreases in hippocampal synaptic transmission observed in hypo-IRAS rats [26]. The inability of peripherally administered leptin to elicit phosphorylation of STAT3 suggests that decreases in BBB transport of leptin and/or deficits in LRb signaling may also contribute to these deficits in hippocampal plasticity. Indeed, since leptin transport is impaired in obesity phenotypes [21-23], these results support the hypothesis that cognitive deficits in obesity may result in part from deficits in BBB leptin transport and decreased central LRb signaling.
4.1. Leptin signaling and neuroplasticity
Leptin is synthesized and secreted by adipocytes and is transported across the BBB via a saturable transport system [21]. The actions of leptin in the hypothalamus are well characterized and include the regulation of food intake, metabolism, body weight and body composition (For review see, [35]). Beyond the hypothalamus, leptin receptor expression and activity also regulates hippocampal synaptic plasticity. For example, intrahippocampal administration of leptin [36], as well as peripheral administration of leptin [37] enhances hippocampal-dependent behavioral performance in a dose-dependent manner. Mechanistically, LRb activation of JAK/STAT and/or Akt signaling may contribute to leptin’s ability to enhance hippocampal synaptic plasticity [38]. Farther downstream of these signaling events, leptin may enhance cognitive function through modulation of glutamatergic neurotransmission [39-41] and glutamate receptor trafficking [42;43], as well as through modification of the structural properties of hippocampal neurons [44]. If increases in leptin activity contribute to neuroplasticity, then reductions in CNS leptin activity may impair hippocampal synaptic plasticity; clinical and pre-clinical data support this hypothesis. As described above, cognitive function is impaired in obese humans [3-6] and performance of hippocampal-dependent behaviors is impaired in rodent models of obesity, including genetic mutations that result in disrupted leptin signaling [7-19]. The complexity of metabolic and endocrine disturbances in obesity makes it challenging to specifically determine the mechanisms that are responsible for, or participate in, the cognitive deficits observed in obesity phenotypes. In hypo-IRAS rats, hypothalamic-pituitary-adrenal axis activity and metabolic responses to an oral glucose tolerance test are unaffected, at least under the experimental conditions described in our current and previous studies. Therefore, hypo-IRAS rats provide an obesity model to examine the potential role of hyperleptinemia and hypertriglyceridemia in the hippocampal synaptic plasticity deficits observed in obesity.
4.2. Hyperleptinemia and impaired neuroplasticity: putative mechanisms
While our current and previous studies demonstrate that the obesity phenotype elicited by downregulation of hypothalamic IRs impairs hippocampal plasticity, the mechanism(s) through which these deficits occurs remains to be elucidated. For example, do hippocampal plasticity deficits reflect decreases in hippocampal IR levels or do they result from the development of the obesity phenotype: our previous studies support the latter hypothesis. In this regard, we have previously shown that lentivirus expression is limited to the hypothalamus when the LV constructs are injected into the third ventricle [25]. In addition, we have also demonstrated that hippocampal IR levels and activity are unaffected in hypo-IRAS rats [25;26]. Therefore, while insulin is recognized as an important mediator of hippocampal synaptic plasticity [45], our data illustrate that the deficits in synaptic plasticity observed in hypo-IRAS rats is not the result of decreased IR expression or activity. However, previous observations involving leptin and triglycerides provide a conceptual framework that allow for the development of a potential hypothesis regarding obesity-induced hippocampal plasticity deficits. As described above, leptin transport across the BBB is reduced in obesity phenotypes [21-24]. Multiple mechanisms may regulate leptin transport across the BBB, but increases in plasma levels of triglycerides may be most relevant to the current studies since hypo-IRAS rats exhibit increases in plasma triglycerides ([26]; also see Table 1). In addition to decreasing BBB transport of leptin, triglycerides directly impair hippocampal synaptic transmission and behavioral performance [7]. Such results suggest that increases in plasma triglyceride levels observed in obesity may impair synaptic plasticity by acting directly in the hippocampus, as well as by decreasing BBB transport of leptin. Collectively, these findings provide the framework for the mechanisms through which decreasing hypothalamic IRs adversely affects hippocampal synaptic plasticity (Figure 7). As observed in knockout mice and in antisense oligonucleotide approaches that decreased hypothalamic IR expression [46;47], lentivirus-mediated downregulation of hypothalamic IRs increases body weight and adiposity. Increases in adiposity are accompanied by increases in plasma levels of leptin, as well as by increases in plasma triglyceride levels. Plasma triglycerides impair BBB transport of leptin [48], thereby decreasing the ability of leptin to enhance hippocampal synaptic plasticity. Additionally, triglycerides may directly impair hippocampal plasticity [7]. Several lines of evidence provide additional support for this hypothesis. To begin, impairments in BBB transport of leptin in rodents can be reversed by lowering plasma triglyceride levels with gemfibrozil [48], which could account for the ability of gemfibrozil to improve behavioral performance in obese mice [7] and enhance cognitive function in elderly populations [49]. In addition, reductions in BBB leptin transport can be reversed by fasting [50]. Our recent studies demonstrated that mild food restriction of hypo-IRAS rats reduces adiposity, returns plasma leptin and triglyceride levels to control values and also restores hippocampal synaptic transmission [26]. Accordingly, some important future experiments could include examination of the ability of food restriction or dyslipidemia treatments to reverse the structural and functional deficits observed in the hippocampus of hypo-IRAS rats. Such studies may begin to clarify the role of hyperleptinemia and hypertriglyceridemia in obesity-induced cognitive deficits.
Figure 7.
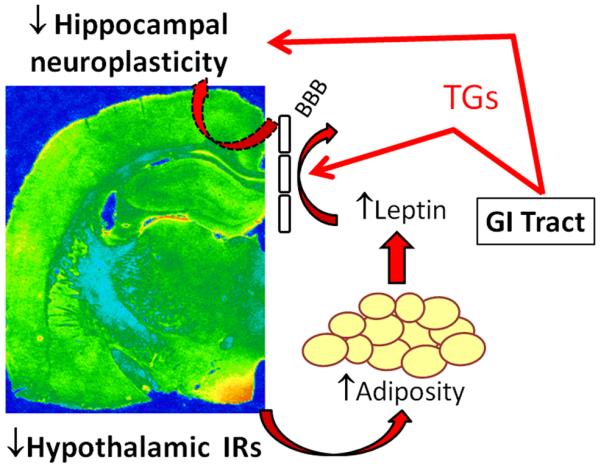
Development of obesity/hyperleptinemic phenotype following downregulation of hypothalamic IRs impairs hippocampal synaptic plasticity. Decreases in BBB transport of leptin (dashed arrow) and/or direct actions of triglycerides in the CNS (solid red arrow) may contribute to hippocampal plasticity deficits in obesity phenotypes, such as the hypo-IRAS rats. See text for details.
4.3. Conclusions
In summary, the results of the current study demonstrate that downregulation of hypothalamic IRs elicits a phenotype that is consistent with features of the metabolic syndrome and this phenotype adversely affects structural and functional plasticity in the rat hippocampus. These deficits in hippocampal synaptic plasticity may provide a mechanistic basis for cognitive deficits associated with obesity phenotypes. Additionally, these data support the hypothesis that leptin resistance, a concept more often associated with the hypothalamus, may also be observed in the hippocampus in obesity phenotypes.
Acknowledgements
Supported by NIH grant numbers NS047728 (LPR), DK017844 (LPR), MH086067 (LPR and DDM), MH063344 (MAW) and the University of South Carolina Research Foundation. The authors would like to thank Dr. Barry Levin for advice and consultation related to pSTAT3 immunohistochemistry.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Benoit SC, Davis JF, Davidson TL. Learned and cognitive controls of food intake. Brain Res. 2010;1350:71–76. doi: 10.1016/j.brainres.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morley JE, Banks WA. Lipids and cognition. J. Alzheimers. Dis. 2010;20:737–747. doi: 10.3233/JAD-2010-091576. [DOI] [PubMed] [Google Scholar]
- 3.Naderali EK, Ratcliffe SH, Dale MC. Obesity and Alzheimer’s disease: a link between body weight and cognitive function in old age. Am. J. Alzheimers. Dis. Other Demen. 2009;24:445–449. doi: 10.1177/1533317509348208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cournot M, Marquie JC, Ansiau D, Martinaud C, Fonds H, Ferrieres J, Ruidavets JB. Relation between Body Mass Index and cognitive function in healthy middle-aged men and women. European Heart Journal. 2006;27:937. doi: 10.1212/01.wnl.0000238082.13860.50. [DOI] [PubMed] [Google Scholar]
- 5.Elias MF, Elias PK, Sullivan LM, Wolf PA, D’Agostino RB. Obesity, diabetes and cognitive deficit: The Framingham Heart Study. Neurobiol. Aging. 2005;26:S11–S16. doi: 10.1016/j.neurobiolaging.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 6.Bruehl H, Wolf OT, Sweat V, Tirsi A, Richardson S, Convit A. Modifiers of cognitive function and brain structure in middle-aged and elderly individuals with type 2 diabetes mellitus. Brain Res. 2009;1280:186–194. doi: 10.1016/j.brainres.2009.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farr SA, Yamada KA, Butterfield DA, Abdul HM, Xu L, Miller NE, Banks WA, Morley JE. Obesity and hypertriglyceridemia produce cognitive impairment. Endocrinology. 2008;149:2628–2636. doi: 10.1210/en.2007-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li XL, Aou S, Oomura Y, Hori N, Fukunaga K, Hori T. Impairment of long-term potentiation and spatial memory in leptin receptor-deficient rodents. Neuroscience. 2002;113:607–615. doi: 10.1016/s0306-4522(02)00162-8. [DOI] [PubMed] [Google Scholar]
- 9.Buchenauer T, Behrendt P, Bode FJ, Horn R, Brabant G, Stephan M, Nave H. Diet-induced obesity alters behavior as well as serum levels of corticosterone in F344 rats. Physiol Behav. 2009;98:563–569. doi: 10.1016/j.physbeh.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 10.Jurdak N, Kanarek RB. Sucrose-induced obesity impairs novel object recognition learning in young rats. Physiol Behav. 2009;96:1–5. doi: 10.1016/j.physbeh.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 11.Molteni R, Barnard RJ, Ying Z, Roberts CK, Gomez-Pinilla F. A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience. 2002;112:803–814. doi: 10.1016/s0306-4522(02)00123-9. [DOI] [PubMed] [Google Scholar]
- 12.McNay EC, Ong CT, McCrimmon RJ, Cresswell J, Bogan JS, Sherwin RS. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol. Learn. Mem. 2010;93:546–553. doi: 10.1016/j.nlm.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldbart AD, Row BW, Kheirandish-Gozal L, Cheng Y, Brittian KR, Gozal D. High fat/refined carbohydrate diet enhances the susceptibility to spatial learning deficits in rats exposed to intermittent hypoxia. Brain Res. 2006;1090:190–196. doi: 10.1016/j.brainres.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 14.Hwang LL, Wang CH, Li TL, Chang SD, Lin LC, Chen CP, Chen CT, Liang KC, Ho IK, Yang WS, Chiou LC. Sex differences in high-fat diet-induced obesity, metabolic alterations and learning, and synaptic plasticity deficits in mice. Obesity. (Silver. Spring) 2010;18:463–469. doi: 10.1038/oby.2009.273. [DOI] [PubMed] [Google Scholar]
- 15.Granholm AC, Bimonte-Nelson HA, Moore AB, Nelson ME, Freeman LR, Sambamurti K. Effects of a saturated fat and high cholesterol diet on memory and hippocampal morphology in the middle-aged rat. J. Alzheimers. Dis. 2008;14:133–145. doi: 10.3233/jad-2008-14202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winocur G, Greenwood CE. The effects of high fat diets and environmental influences on cognitive performance in rats. Behav. Brain Res. 1999;101:153–161. doi: 10.1016/s0166-4328(98)00147-8. [DOI] [PubMed] [Google Scholar]
- 17.Winocur G, Greenwood CE, Piroli GG, Grillo CA, Reznikov LR, Reagan LP, McEwen BS. Memory Impairment in Obese Zucker Rats: An Investigation of Cognitive Function in an Animal Model of Insulin Resistance and Obesity. Behav. Neurosci. 2005;119:1389–1395. doi: 10.1037/0735-7044.119.5.1389. [DOI] [PubMed] [Google Scholar]
- 18.Greenwood CE, Winocur G. Cognitive impairment in rats fed high-fat diets: a specific effect of saturated fatty-acid intake. Behav. Neurosci. 1996;110:451–459. doi: 10.1037//0735-7044.110.3.451. [DOI] [PubMed] [Google Scholar]
- 19.Greenwood CE, Winocur G. Learning and memory impairment in rats fed a high saturated fat diet. Behav. Neural Biol. 1990;53:74–87. doi: 10.1016/0163-1047(90)90831-p. [DOI] [PubMed] [Google Scholar]
- 20.Munzberg H, Myers MG., Jr. Molecular and anatomical determinants of central leptin resistance. Nat. Neurosci. 2005;8:566–570. doi: 10.1038/nn1454. [DOI] [PubMed] [Google Scholar]
- 21.Banks WA. The many lives of leptin. Peptides. 2004;25:331–338. doi: 10.1016/j.peptides.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 22.Burguera B, Couce ME, Curran GL, Jensen MD, Lloyd RV, Cleary MP, Poduslo JF. Obesity is associated with a decreased leptin transport across the blood-brain barrier in rats. Diabetes. 2000;49:1219–1223. doi: 10.2337/diabetes.49.7.1219. [DOI] [PubMed] [Google Scholar]
- 23.Banks WA, DiPalma CR, Farrell CL. Impaired transport of leptin across the blood-brain barrier in obesity. Peptides. 1999;20:1341–1345. doi: 10.1016/s0196-9781(99)00139-4. [DOI] [PubMed] [Google Scholar]
- 24.Kastin AJ, Pan W, Maness LM, Koletsky RJ, Ernsberger P. Decreased transport of leptin across the blood-brain barrier in rats lacking the short form of the leptin receptor. Peptides. 1999;20:1449–1453. doi: 10.1016/s0196-9781(99)00156-4. [DOI] [PubMed] [Google Scholar]
- 25.Grillo CA, Tamashiro KL, Piroli GG, Melhorn S, Gass JT, Newsom RJ, Reznikov LR, Smith A, Wilson SP, Sakai RR, Reagan LP. Lentivirus-mediated downregulation of hypothalamic insulin receptor expression. Physiol Behav. 2007;92:691–701. doi: 10.1016/j.physbeh.2007.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grillo CA, Piroli GG, Evans AN, Macht VA, Wilson SP, Scott KA, Sakai RR, Mott DD, Reagan LP. Obesity/hyperleptinemic phenotype adversely affects hippocampal plasticity: Effects of dietary restriction. Physiol Behav. 2010 doi: 10.1016/j.physbeh.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burghardt PR, Pasumarthi RK, Wilson MA, Fadel J. Alterations in fear conditioning and amygdalar activation following chronic wheel running in rats. Pharmacol. Biochem. Behav. 2006;84:306–312. doi: 10.1016/j.pbb.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 28.Grillo CA, Piroli GG, Wood GE, Reznikov LR, McEwen BS, Reagan LP. Immunocytochemical analysis of synaptic proteins provides new insights into diabetes-mediated plasticity in the rat hippocampus. Neuroscience. 2005;136:477–486. doi: 10.1016/j.neuroscience.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 29.Reagan LP, Gorovits N, Hoskin EK, Alves SE, Katz EB, Grillo CA, Piroli GG, McEwen BS, Charron MJ. Localization and regulation of GLUTx1 glucose transporter in the hippocampus of streptozotocin diabetic rats. Proc. Natl. Acad. Sci. USA. 2001;98:2820–2825. doi: 10.1073/pnas.051629798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reznikov LR, Reagan LP, Fadel JR. Activation of phenotypically distinct neuronal subpopulations in the anterior subdivision of the rat basolateral amygdala following acute and repeated stress. J. Comp Neurol. 2008;508:458–472. doi: 10.1002/cne.21687. [DOI] [PubMed] [Google Scholar]
- 31.Piroli GG, Grillo CA, Reznikov LR, Adams S, McEwen BS, Charron MJ, Reagan LP. Corticosterone impairs insulin-stimulated translocation of GLUT4 in the rat hippocampus. Neuroendocrinology. 2007;85:71–80. doi: 10.1159/000101694. [DOI] [PubMed] [Google Scholar]
- 32.Levin BE, Dunn-Meynell AA, Banks WA. Obesity-prone rats have normal blood-brain barrier transport but defective central leptin signaling before obesity onset. Am. J. Physiol Regul. Integr. Comp Physiol. 2004;286:R143–R150. doi: 10.1152/ajpregu.00393.2003. [DOI] [PubMed] [Google Scholar]
- 33.Peterson WM, Wang Q, Tzekova R, Wiegand SJ. Ciliary neurotrophic factor and stress stimuli activate the Jak-STAT pathway in retinal neurons and glia. J. Neurosci. 2000;20:4081–4090. doi: 10.1523/JNEUROSCI.20-11-04081.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bates SH, Myers MG., Jr. The role of leptin receptor signaling in feeding and neuroendocrine function. Trends Endocrinol. Metab. 2003;14:447–452. doi: 10.1016/j.tem.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 35.Schwartz MW, Woods SC, Porte D, Jr., Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 36.Farr SA, Banks WA, Morley JE. Effects of leptin on memory processing. Peptides. 2006;27:1420–1425. doi: 10.1016/j.peptides.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 37.Oomura Y, Hori N, Shiraishi T, Fukunaga K, Takeda H, Tsuji M, Matsumiya T, Ishibashi M, Aou S, Li XL, Kohno D, Uramura K, Sougawa H, Yada T, Wayner MJ, Sasaki K. Leptin facilitates learning and memory performance and enhances hippocampal CA1 long-term potentiation and CaMK II phosphorylation in rats. Peptides. 2006;27:2738–2749. doi: 10.1016/j.peptides.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 38.Burgos-Ramos E, Chowen JA, Argente J, Barrios V. Regional and temporal differences in leptin signaling in rat brain. Gen. Comp Endocrinol. 2010;167:143–152. doi: 10.1016/j.ygcen.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 39.Shanley LJ, Irving AJ, Harvey J. Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. Journal of Neuroscience. 2001;21 doi: 10.1523/JNEUROSCI.21-24-j0001.2001. art-RC186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moult PR, Milojkovic B, Harvey J. Leptin reverses long-term potentiation at hippocampal CA1 synapses. J. Neurochem. 2009;108:685–696. doi: 10.1111/j.1471-4159.2008.05810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Durakoglugil M, Irving AJ, Harvey J. Leptin induces a novel form of NMDA receptor-dependent long-term depression. J. Neurochem. 2005;95:396–405. doi: 10.1111/j.1471-4159.2005.03375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moult PR, Cross A, Santos SD, Carvalho AL, Lindsay Y, Connolly CN, Irving AJ, Leslie NR, Harvey J. Leptin regulates AMPA receptor trafficking via PTEN inhibition. J. Neurosci. 2010;30:4088–4101. doi: 10.1523/JNEUROSCI.3614-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moult PR, Harvey J. Regulation of glutamate receptor trafficking by leptin. Biochem. Soc. Trans. 2009;37:1364–1368. doi: 10.1042/BST0371364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Malley D, MacDonald N, Mizielinska S, Connolly CN, Irving AJ, Harvey J. Leptin promotes rapid dynamic changes in hippocampal dendritic morphology. Molecular and Cellular Neuroscience. 2007;35:559–572. doi: 10.1016/j.mcn.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reagan LP. Insulin signaling effects on memory and mood. Curr. Opin. Pharmacol. 2007;7:633–637. doi: 10.1016/j.coph.2007.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bruning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, Klein R, Krone W, Muller-Wieland D, Kahn CR. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289:2122–2125. doi: 10.1126/science.289.5487.2122. [DOI] [PubMed] [Google Scholar]
- 47.Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat. Neurosci. 2002;5:566–572. doi: 10.1038/nn0602-861. [DOI] [PubMed] [Google Scholar]
- 48.Banks WA, Coon AB, Robinson SM, Moinuddin A, Shultz JM, Nakaoke R, Morley JE. Triglycerides induce leptin resistance at the blood-brain barrier. Diabetes. 2004;53:1253–1260. doi: 10.2337/diabetes.53.5.1253. [DOI] [PubMed] [Google Scholar]
- 49.Rogers RL, Meyer JS, McClintic K, Mortel KF. Reducing hypertriglyceridemia in elderly patients with cerebrovascular disease stabilizes or improves cognition and cerebral perfusion. Angiology. 1989;40:260–269. [PubMed] [Google Scholar]
- 50.Banks WA, Farrell CL. Impaired transport of leptin across the blood-brain barrier in obesity is acquired and reversible. Am. J. Physiol Endocrinol. Metab. 2003;285:E10–E15. doi: 10.1152/ajpendo.00468.2002. [DOI] [PubMed] [Google Scholar]