

Abstract
Motivation: Discovering variation among high-throughput sequenced genomes relies on efficient and effective mapping of sequence reads. The speed, sensitivity and accuracy of read mapping are crucial to determining the full spectrum of single nucleotide variants (SNVs) as well as structural variants (SVs) in the donor genomes analyzed.
Results: We present drFAST, a read mapper designed for di-base encoded ‘color-space’ sequences generated with the AB SOLiD platform. drFAST is specially designed for better delineation of structural variants, including segmental duplications, and is able to return all possible map locations and underlying sequence variation of short reads within a user-specified distance threshold. We show that drFAST is more sensitive in comparison to all commonly used aligners such as Bowtie, BFAST and SHRiMP. drFAST is also faster than both BFAST and SHRiMP and achieves a mapping speed comparable to Bowtie.
Availability: The source code for drFAST is available at http://drfast.sourceforge.net
Contact: calkan@u.washington.edu
1 INTRODUCTION
Genomic variation between individuals or across species ranges from single nucleotide polymorphisms (SNPs) and structural variation to larger chromosomal rearrangements (Alkan et al., 2011). Thanks to the improvements in sequencing technologies, large-scale genome variation studies such as the 1000 Genomes Project (1000 Genomes Project Consortium, 2010; Mills et al., 2011) have made it possible to better characterize normal human genomic variation and disease (Lupski et al., 2010; Ng et al., 2010; Vissers et al., 2010).
The development of high-throughput sequencing (HTS) technologies has changed the landscape of genome research. The first commercially available HTS technology was from Roche/454 Life Sciences (Margulies et al., 2005) and was used to sequence the genome of James Watson (Wheeler et al., 2008). It was followed by other ‘second generation’ sequencing platforms that generate orders of magnitude more data for a fraction of the cost, such as Illumina Genome Analyzer (Bentley et al., 2008) and AB SOLiD (McKernan et al., 2009). Third-generation sequencing platforms are now under development, and HeliScope (Pushkarev et al., 2009) and PacBio RS (Eid et al., 2009) were recently made available; however, for the time being, they produce reads with higher error rates.
Analysis of genomic variation using sequencing starts with mapping the randomly sheared and ideally uniformly sampled DNA fragments from the genome. Different properties and error models of sequence reads generated by these technologies require the development of specialized read mapping algorithms for each platform for accurate read alignment and characterization of genomic variants. This becomes more complicated for short reads: due to repeats and duplications in genomes, they can map to multiple locations with equal sequence identity. Leveraging the high sequence coverage and randomly selecting one ‘best’ location when a read cannot be unambiguously placed has proven to be effective in discovering SNPs and small indels in relatively non-complex areas of the genome (Li et al., 2008a; 2009). However, structural variation detection sensitivity is shown to benefit from tracking all map locations of the reads including suboptimal alignments (Hormozdiari et al., 2009; Lee et al., 2009; Mills et al., 2011), and characterization of segmental duplications is extremely resistant against mapping the reads uniquely (Alkan et al., 2009; Sudmant et al., 2010).
Read mappers can be broadly classified into two categories according to the method used to index the reference genome using either hash tables or suffix arrays [compressed through the Ferragina-Manzini index (Ferragina et al., 2000) with the use of the Burrows-Wheeler Transform (Burrows et al., 1994)]. Hash-based aligners such as MAQ (Li et al., 2008a), SHRiMP (Rumble et al., 2009), mrFAST (Alkan et al., 2009), mrsFAST (Hach et al., 2010) and BFAST (Homer et al., 2009) have poorer performance in comparison to suffix array-based aligners [e.g. BWA (Li et al., 2009), Bowtie (Langmead et al., 2009)] when dealing with short reads; however, their relative performance increases considerably and surpasses the suffix array-based aligners when the read length and thus the number of errors (mismatches or indels) that need to be tolerated increase.
In this article, we describe a hash-based read mapping algorithm named ‘di-base read fast alignment search tool’ (drFAST) designed for the di-base encoded color-space reads generated with the SOLiD platform. The main advantage of di-base encoding is increased base call accuracy due to each base being represented by two ‘colors’. This helps in differentiating base calling errors (color-space errors) from real sequence variance, thereby increasing the reliability of detected genomic variants. We show that mapping speed of drFAST is higher than other SOLiD-enabled hash-based read mappers, BFAST (Homer et al., 2009) and SHRiMP (Rumble et al., 2009), and comparable to suffix array-based aligner Bowtie (Langmead et al., 2009). In addition, drFAST was able to map more reads than all the tools we benchmarked. Furthermore, drFAST achieves 100% sensitivity if the maximum allowed edit distance is less than L/k , where L is the sequence length and k is the length of the k-mers stored in the hash table (k is set to 12 by default). Coupled with its ability to store all map locations within a user-specified distance threshold and its paired-end (PE) mapping capabilities, drFAST can be used to characterize segmental duplications (Alkan et al., 2009; Sudmant et al., 2010) and increase sensitivity of structural variation discovery using VariationHunter (Hormozdiari et al., 2009; Hormozdiari et al., 2010).
2 METHODS
For each read sequenced from a donor genome, a mapping algorithm aims to find locations in a ‘reference genome’ where the read can be aligned exactly or within a small number of errors in the form of substitutions or insertions/deletions (indels). drFAST is a read mapper designed for color-space reads generated with the SOLiD platform, and finds ‘all’ possible map locations for each read of length r in the reference genome within a user-specified e mismatches.
drFAST is a seed-and-extend type algorithm and it builds an index of the reference genome by creating a collision-free hash table for all subsequences of length k (k-mers) of the reference genome. To map the reads, it first partitions each read to (e+1) k-mers and searches for each of these k-mers in the hash table. For each hit in the hash table, it then tests if the remainder of the read can be ‘extended’ by aligning to the reference genome starting at the determined hit location.
How exactly this is done is described below.
2.1 Genome transformation
The sequence data produced with the SOLiD platform are in color-space format (S={0, 1, 2, 3}*), where the reference genome sequence is in letter-space (i.e. R={A, C, G, T}*). Each color encodes two adjacent base pairs in the read, and each base pair is represented by two colors. Transformation of reads from color-space to letter-space before mapping may result in generating incorrect reads where base call errors exist, as depicted in Figure 1. To avoid such incorrect decoding of reads, we translate the reference genome to color-space and use this transformed genome to create the index.
Fig. 1.

Translating the read from color-space to letter-space may result in a new sequence different from the original read if there exists a color-space error.
2.2 Indexing the reference genome
drFAST creates a collision-free hash table for all k-mers in the reference genome. Each entry of this index is a 2-tuple τ=(s, L), where s is a k-mer from the genome (k=12 by default) and L is a list of all positions of the genome starting with this subsequence. The index is maintained in lexicographically sorted order with respect to their subsequences. For a reference genome of length n, the upper bound for the size of its index is O(n); but due to the repetitive nature of genome sequences, the index size is smaller in practice.
2.3 Indexing the reads
drFAST partitions each read of length r into e+1 non-overlapping blocks of length k where e is the user-specified maximum Hamming distance allowed for mapping. In the case where k≤⌊r/(e+1)⌋, the pigeon hole principle guarantees that at least one of these blocks maps to the reference genome with no errors. Similar to the indexing described in Section 2.2, drFAST creates an index of blocks computed from all reads in 2-tuples τr=(s, Lr), where Lr denotes the list of reads that include the k-mer s.
2.4 Searching
drFAST compares the reference genome index keys with read index keys to find the locations in the reference genome where a read can be mapped with at most e errors. For each partition of the read, drFAST first finds the locations of the reference genome with the identical subsequence (same keys). It then tries to extend the location through sequence alignment of the reads to the genome, and reports those locations where the Hamming distance of the alignment is at most e. A simple loop scans both indices (both are lexicographically sorted); if the keys of the indices are the same (same subsequence) for entries τ=(s, L) in the reference and τr=(s, Lr) in the read index. Then all entries in L are candidate map locations for each read entry in Lr, and thus the entire list L should be compared with Lr (extending step).
Similar to mrsFAST (Hach et al., 2010), drFAST performs ‘all-to-all’ list comparison using a recursive divide-and-conquer strategy that guarantees cache obliviousness; i.e. asymptotically minimizing the number of costly cache misses (Frigo et al., 1999).
2.5 Extending
The final step is to verify if each read can be aligned to candidate map locations within the user-specified error threshold e. drFAST aims to align the color-space read (Sc) to the letter-space sequence (Sl). The aligning process can be considered as finding a letter-space read S′l that aligns to Sl, and highly similar to Sc if transformed to color-space:
| (1) |
where CCG is the function that transforms the letter-space to color-space as defined by the SOLiD technology, and Sim is the similarity function.
Maximizing the similarity between two sequences is equivalent to minimizing their distance. We use Hamming distance (i.e. the number of mismatches) as the distance measure between two sequences.
| (2) |
To address the problem, drFAST introduces two efficient methods.
2.5.1 Method I: dynamic programming
Let ∑ ={A, C, G, T}, and σ, σ′∈∑, and let Score(i, σ) indicate the optimal alignment of two subsequences Sl[1..i] and Sc[1..i] (from the first to the i-th character) while σ is the last character of S′l. We then define
| (3) |
where d(a,b)=1 if a≠b, and d(a,b)=0 otherwise.
The detailed version of Equation (3) is as follows:
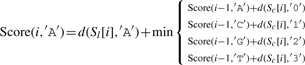 |
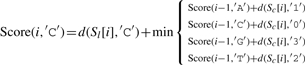 |
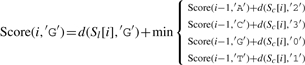 |
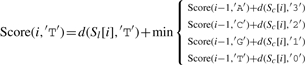 |
The minimum value of Score(|Sl|, ′A′), Score(|Sl|, ′C′), Score(|Sl|, ′G′) and Score(|Sl|, ′T′) is the Score of the best translation of Sc to Sl′.
Figure 2 shows an example of aligning a letter-space and a color-space sequence using the dynamic programming described in Equation (3). The minimum value in the last column represents the score of the best alignment. Using the backtracking pointer, we can then recover the best alignment sequence.
Fig. 2.

The dynamic programming table generated to align ATTGAATCA and 30121321 (0 = blue, 1 = green, 2 = yellow, 3 = red). The arrows represent the best alignment between the two sequences.
Remark 1. —
The dynamic programming formulation in Equation (3) will find the optimal solution to the objective function in Equation (2) if the costs of mismatches and read errors are equal to one.
Remark 2. —
Dynamic programming described in (3) can be modified to handle any cost function for mismatches and read errors.
Note that the Equation (3) uses Hamming distance but it can be easily generalized for edit distance to allow indels.
2.5.2 Method II: transformation-based detection
The second method is based on the theoretical design aspect of color-space reads (McKernan et al., 2009). A string of colors c1c2c3…ck can also be treated as transformations. For example, C102 can be written as f2(f0(f1(C))) where the transformation of the colors is applied one after the other. This specific transformation converts C to G, acting as color 3 (C102=C3=G). For any other base pair, color string 102 will behave exactly as color 3.
The set of color operations is isomorphic to the ‘Klein Four Group’ (Armstrong, 1988; McKernan et al., 2009). The Klein Four Group is the symmetry group of a rectangle, which has four elements: identity, vertical reflection, horizontal reflection and a 180° rotation. In other words, given the four bases in the corners of a rectangle, each color operation has a one-to-one correspondence with one of the Klein Group elements (Table 1). The Klein Four Group is closed under its elements meaning that if a b are two elements of this group a⊕b and b⊕a (a⊕b means a followed by b) is also an element of the this group. It also has associative, identity, reverse and commutative properties. This means that any sequence of color operations can be considered as one color operation.
Table 1.
Applying color transformation ‘3’ (left) is the same as applying 180° rotation (right)
 |
We use this property of the color-space reads to detect mismatches. Let two sets of color operations of the same length exist (c1…ck and r1…rk) with different starting color (c1≠r2). For both sets, if any two consecutive colors are replaced with their equivalent (closure property) starting from left-hand side, you will end up with one at the end. If the last color matches with no intermediate matching colors then these two operations show a mismatch of length k−1. To illustrate this, consider two color operations 313 and 100. For simplicity, we also consider a leading base C. After applying the color operations, strings GTA and AAA will be generated, respectively. It can be seen that the last base pair generated using both operations is A and intermediary base pairs are not matching. These two sets of operations have the same transformation. Thus although they generate different sets of base pairs in middle, the final ‘product’ is the same character.
Theorem 1. —
Let c=c1c2c3ck be a k-color substring of a read aligned with the corresponding color-space reference r=r1r2r3rk. Then c encodes an isolated (k−1)-base change if and only if the base position preceding c is not a variant, and the following two equations hold under the Color Addition (Table 2):
For all i from 1 to k−1:
Table 2.
Addition table code for strings of colors
| ⊕ | 0 | 1 | 2 | 3 |
|---|---|---|---|---|
| 0 | 0 | 1 | 2 | 3 |
| 1 | 1 | 0 | 3 | 2 |
| 2 | 2 | 3 | 0 | 1 |
| 3 | 3 | 2 | 1 | 0 |
We use Theorem 1 as the basis of our validation function (i.e. extending step). If there is a color mismatch between the read and the reference genome, we consider the next e+1 colors to test if there exists any same color transformation of size at most e+1 between the read and the genome. Considering a window of limited length, this sometimes may cause incorrect classification of a long stretch of mismatches as two independent read error calls. We refine such calls at the final step.
3 ADDITIONAL FEATURES
Parallelization: an embarrassingly parallel wrapper for drFAST can be easily written to split the reads into smaller ‘chunks’ (~1–5 million reads per file) and align on cluster nodes. This approach is the best practice because:
drFAST requires <700 MB to load the genome and its index and only a total of ~1.3 GB of memory to map 1 million reads to the genome.
Mapping of each read is independent from mapping the others (except in the case of PE sequences where both ends need to be processed in the same chunk).
PE mapping: SOLiD, like most other HTS technologies, can generate PE sequences. A pair of PE sequences are generated from the prefix and suffix of the same sheared DNA fragment, thus they can be used to increase mapping accuracy and discover structural variation (Alkan et al., 2011; Mills et al., 2011). Current implementation of drFAST supports tracking the PE information, enabling direct use of VariationHunter for structural variation (Hormozdiari et al., 2009) and transposon insertion (Hormozdiari et al., 2010) discovery, as well as NovelSeq (Hajirasouliha et al., 2010) for characterization of novel sequence insertions.
4 RESULTS
To measure the performance of drFAST, we compared its two variants to popular color-space read mappers currently available.
Benchmarked software:
drFAST-DP (Dynamic programming variant) (version 0.0.0.2);
drFAST-CT (Color transformation variant) (version 0.0.0.0);
BFAST (Homer et al., 2009) (version 0.6.4);
Bowtie (Langmead et al., 2009) (version 0.12.0);
SHRiMP (Rumble et al., 2009) (version 2.0.1);
SOCS (Ondov et al., 2008) (version 2.0.3);
Mapreads (McKernan et al., 2009) (version 2.4.1); and
PerM (Chen et al., 2009) (version 0.3.3);
Parameters: we used the following parameter settings for these mappers:
drFAST: e=2,3 (error threshold for different runs).
BFAST: parameters recommended in the BFAST manual.
Bowtie: n,v = 2,3 (error threshold for different runs); -a (for reporting all); -S (output in SAM format); -C (color-space mapping).
SHRiMP: -m 1 (score 1 for match); -i -1 (score -1 for mismatch) -x -1 (score -1 for color-space error); -U (ungapped alignment) -o 10 000 (maximum number of alignments for a read); -N 1 (number of threads); -h 96% (≥96% alignment identity).
SOCS: -x 0 (number of bases to trim); -s 2 (mismatch sensitivity); -t 4 (mismatch tolerance); -m 0 (maximum number of alignments for a read, 0 indicates to report all); -T 1 (number of threads); -l yes (consider the lower case bases in genome).
Mapreads: S = 0 (color-space mapping); M = 2 (number of mismatches allowed); A = 2 (count adjacent mismatches as one mismatch); Z = 10 000 (maximum number of alignment for a read).
PerM: –seed S20 (full sensitivity for two SNPs); -v 4 (number of mismatches).
We used the same parameters (for reporting ‘all’ mapping locations) when available to ensure a fair comparison.
Note that BWA and MAQ are not considered here since they ignore the first two characters of SOLiD reads.
Data, reference genome and computing power: we used both simulated and real datasets for comparisons. We simulated three sets, each with 4 million reads of length 50 bp sampled randomly from chromosome 1 of human reference genome (NCBI build 35) as follows:
Set 1: we transformed the reads to color-space with no color errors and no mismatches.
Set 2: reads are transformed with two color errors. To achieve this, we transformed the reads to color-space and then changed the color of two arbitrarily selected non-consecutive positions. Note that if two color errors are consecutive, this might make it impossible to distinguish a read error from a SNP.
Set 3: generated with no color errors but one SNP.
In addition, we randomly selected 1 million (50 bp long) reads from publicly available color-space reads generated from the genomes of NA18507 (McKernan et al., 2009) (SRX004555), NA10847 (SRX008164) and NA12156. We used the human reference genome (NCBI build 35, unmasked) as the reference genome in all our experiments. The benchmarking results we report are performed on a server with 64 bit Intel Xeon processor and 8 GB of RAM.
Time, accuracy and sensitivity results: we give the comparison results for all the mappers above with respect to the proportion of the reads that have at least one map location on the reference genome (sensitivity), total number of map locations found (comprehensiveness) and time needed to map the reads.
Table 3 shows the results on simulated datasets with error threshold of 2 (color errors and mismatches), except in the case of PerM where we allowed up to four mismatches due to recommendations of its developers. drFAST maps all the reads from simulated datasets back to the reference genome very efficiently. The closest competitor to drFAST appears to be Bowtie, which is, in general, slower than drFAST-CT and is not 100% sensitive. Although Bowtie with a parameter setting of v = 2 seems to map each read to more locations than drFAST, when no substitutions are present (Set 1), or a single color error is added (Set 2), this is simply due to Bowtie not being stringent on the number of errors it permits disregarding the parameter setting; we noticed that there are mapping locations with more than five color errors.
Table 3.
Performance results of all tested color-space read aligners on simulated data with error threshold of two mismatches
| Dataset | Mapper | Time (min) | Map locations | Reads mapped (%) |
|---|---|---|---|---|
| Set 1 | drFAST-DP | 65 | 138 715 908 | 100 |
| drFAST-CT | 40 | 137 483 484 | 100 | |
| BFAST | 88 | 8 803 840 | 96.1 | |
| Bowtie v = 2 | 17 | 25 581 176 | 99.4 | |
| Bowtie n = 2 | 67 | 168 307 651 | 99.4 | |
| SHRiMP | 414 | 13 961 155 | 99.8 | |
| SOCS | 45 | 13 357 519 | 100 | |
| Mapreads | 50 | 55 569 848 | 100 | |
| PerM | 17 | 14 441 796 | 96.2 | |
| Set 2 | drFAST-DP | 42 | 37 652 313 | 100 |
| drFAST-CT | 26 | 36 458 468 | 100 | |
| BFAST | 101 | 8 098 581 | 98.0 | |
| Bowtie v = 2 | 13 | 9 738 234 | 60.8 | |
| Bowtie n = 2 | 31 | 57 550 920 | 61.9 | |
| SHRiMP | 519 | 11 977 512 | 99.8 | |
| SOCS | 90 | 12 909 860 | 100 | |
| Mapreads | 31 | 21 749 155 | 100 | |
| PerM | 15 | 12 679 070 | 98 | |
| Set 3 | drFAST-DP | 47 | 76 588 622 | 100 |
| drFAST-CT | 32 | 75 970 911 | 100 | |
| BFAST | 105 | 8 982 132 | 97.4 | |
| Bowtie v = 2 | 16 | 11 030 554 | 49.4 | |
| Bowtie n = 2 | 43 | 70 508 835 | 51.66 | |
| SHRiMP | 472 | 11 859 215 | 99.8 | |
| SOCS | 96 | 9 780 960 | 100 | |
| Mapreads | 37 | 29 799 473 | 100 | |
| PerM | 15 | 13 140 561 | 97.5 | |
In the case of PerM, we allowed for mapping with up to four mismatches as recommended by its developers, yet its sensitivity failed to reach 100%. Reads are simulated from human reference genome build 35 (chromosome 1). Set 1: no errors; Set 2: color errors; Set 3: substitutions. Values in bold show alignments with 100% sensitivity, higher value implies more sensitivity in the reported alignment.
When the reads involve a nucleotide substitution (Set 3), the number of mapping locations are lower than that of drFAST. What is more interesting is the number of reads that can be mapped to the reference genome. It seems like Bowtie can map at most 61.9% of the reads even when they include a single color error (Set 2), in contrast, drFAST (both variants) map 100% of the reads. When the errors are in the form of nucleotide substitutions, the proportion of reads mapped by Bowtie drops to 51.66%.
Since Bowtie was the closest competitor to drFAST, we performed another experiment on the same datasets by increasing the error threshold to 3 (Table 4). Interestingly for this setting, the proportion of reads mapped by Bowtie is 99.4%, almost matching the 100% mapping sensitivity of drFAST. However, both in terms of time and the number of map locations, drFAST (both variants) perform better than Bowtie, especially when errors (Set 2 for color errors and Set 3 for nucleotide errors) are present.
Table 4.
Performance comparison on simulated datasets between drFAST-DP, drFAST-CT and Bowtie where error threshold is set to three mismatches
| Dataset | Mapper | Time (min) | Map locations | Reads mapped (%) |
|---|---|---|---|---|
| Set 1 | drFAST-DP | 88 | 364 601 231 | 100 |
| drFAST-CT | 75 | 363 472 241 | 100 | |
| Bowtie v = 3 | 60 | 56 407 732 | 99.4 | |
| Bowtie n = 3 | 101 | 252 735 117 | 99.4 | |
| Set 2 | drFAST-DP | 42 | 118 053 818 | 100 |
| drFAST-CT | 45 | 113 349 741 | 100 | |
| Bowtie v = 3 | 45 | 23 365 015 | 99.4 | |
| Bowtie n = 3 | 50 | 111 931 387 | 99.4 | |
| Set 3 | drFAST-DP | 47 | 215 274 860 | 100 |
| drFAST-CT | 50 | 215 261 940 | 100 | |
| Bowtie v = 3 | 48 | 28 321 746 | 99.4 | |
| Bowtie n = 3 | 75 | 137 015 425 | 99.4 | |
Values in bold show alignments with 100% sensitivity, higher value implies more sensitivity in the reported alignment.
As all three sets are generated from chromosome 1 with at most two errors added, a sensitive mapper should be able to map all reads to chromosome 1 when the error threshold is set to 2. In order to experimentally check the accuracy of all locations found by drFAST, we simulated the corresponding Illumina reads (letter-space) and aligned to chromosome 1 using mrsFAST. As seen in Table 5 for Sets 1 and 3, drFAST finds slightly more mapping locations than mrsFAST, where the sensitivities of both aligners are 100%. The reason drFAST can find more mapping locations for SOLiD reads compared with the corresponding Illumina reads is because drFAST could map a read like T0000 to two different positions with base pair contents TTTT, and also CCCC when one color error is ‘corrected’. This is not the case with letter-space reads generated by a platform like Illumina Genome Analyzer. Although it would not be correct to arbitrarily select one ‘version’ above the other, or returning both alignments as possibilities, we propose to correct such artifacts by incorporating the base pair quality values. This problem will arise only in polyN regions, and thus, we propose to disable error correction of polyN reads where the base quality value of the first base is sufficiently high (i.e. q>30).
Table 5.
Number of mapping locations reported by mrsFAST for the same set of simulated reads in letter-space
| Dataset | Mapper | Time (min) | Map locations | Reads mapped (%) |
|---|---|---|---|---|
| Set 1 | mrsFAST | 20 | 135 450 193 | 100 |
| Set 3 | mrsFAST | 20 | 75 115 629 | 100 |
BFAST and SHRiMP results are not presented for the three real datasets (Table 6) due to: (i) in our experiments, BFAST terminated with error in indexing step; (ii) SHRiMP requires 16 GB of main memory for alignment. Furthermore, both programs are slower than drFAST or Bowtie. As a result, we compared drFAST with Bowtie, SOCS, Mapreads and PerM with an error threshold of 2 (Table 6) and we compared drFAST with Bowtie (closest competitor) with an error threshold of 3 (Table 7). In five out of the six cases, drFAST maps significantly more reads, and to substantially more locations, in comparable time. The performance of the two programs are comparable only for NA18507 for n = 2, in terms of mapped reads; however, drFAST-CT is slightly faster on this dataset.
Table 6.
Performance comparison on real datasets between drFAST-DP, drFAST-CT, Bowtie, SOCS and PerM on 1 million randomly selected reads from three different sequencing experiments
| Dataset | Mapper | Time (min) | Map locations | Reads mapped (%) |
|---|---|---|---|---|
| NA18507 | drFAST-DP | 114 | 189 276 027 | 36.8 |
| drFAST-CT | 54 | 149 362 540 | 35.6 | |
| Bowtie v = 2 | 21 | 64 092 233 | 27.8 | |
| Bowtie n = 2 | 63 | 202 948 323 | 35.2 | |
| SOCS | 320 | 21 081 941 | 35.3 | |
| Mapreads | 80 | 17 032 680 | 37.3 | |
| PerM | 76 | 10 068 062 | 35.3 | |
| NA10847 | drFAST-DP | 200 | 667 928 813 | 47.1 |
| drFAST-CT | 100 | 512 599 230 | 45.9 | |
| Bowtie v = 2 | 84 | 280 928 112 | 38.0 | |
| Bowtie n = 2 | 91 | 270 996 634 | 36.0 | |
| SOCS | 420 | 53 668 622 | 44.8 | |
| Mapreads | 140 | 39 589 079 | 48.5 | |
| PerM | 100 | 20 699 652 | 44.8 | |
| NA12156 | drFAST-DP | 136 | 491 158 791 | 33.5 |
| drFAST-CT | 91 | 440 317 111 | 32.5 | |
| Bowtie v = 2 | 99 | 329 916 108 | 25.0 | |
| Bowtie n = 2 | 99 | 318 621 596 | 23.7 | |
| SOCS | 400 | 38 246 530 | 31.2 | |
| Mapreads | 110 | 22 182 469 | 35.1 | |
| PerM | 140 | 10 798 496 | 31.2 | |
We set the error threshold to 2 bp for all aligners, except PerM, where we set the threshold to four as per the PerM developers suggested. We then removed the alignments with more than 2 bp mismatches for comparison purposes.
Table 7.
Performance comparison on real datasets between drFAST-DP, drFAST-CT and Bowtie on 1 million randomly selected reads from three different sequencing experiments. We set the error threshold to 3 bp
| Dataset | Mapper | Time (min) | Map locations | Reads mapped (%) |
|---|---|---|---|---|
| NA18507 | drFAST-DP | 154 | 309 994 599 | 41.5 |
| drFAST-CT | 61 | 302 237 779 | 40.5 | |
| Bowtie v = 3 | 63 | 145 473 423 | 37.1 | |
| Bowtie n = 3 | 78 | 290 357 005 | 36.35 | |
| NA10847 | drFAST-DP | 300 | 1 121 281 408 | 52.1 |
| drFAST-CT | 141 | 1 092 259 727 | 51.6 | |
| Bowtie v = 3 | 182 | 565 114 739 | 47.8 | |
| Bowtie n = 3 | 76 | 270 885 799 | 35.8 | |
| NA12156 | drFAST-DP | 310 | 655 648 865 | 42.4 |
| drFAST-CT | 120 | 639 667 174 | 39.5 | |
| Bowtie v = 3 | 187 | 585 191 747 | 34.6 | |
| Bowtie n = 3 | 98 | 318 527 434 | 23.6 | |
We also compared the amount of memory used by each program when mapping 1 million reads to the human reference genome assembly (Table 8).
Table 8.
Memory required by each software to map 1 million 35 base reads to human reference genome. The memory requirement increases with the number of reads and/or the read length; this increase is typically linear with the increase in the number of base pairs in the dataset
| Mapper | Memory usage (GB) |
|---|---|
| drFAST-DP | 1.3 |
| drFAST-CT | 1.3 |
| BFAST | ≥10 |
| Bowtie | 4.5 |
| SHRiMP | 16 |
| SOCS | ≥5a |
| PerM | 2 |
| Mapread | ≥8b |
aWe set the memory usage of SOCS to 5 GB.
bWe set the memory usage of Mapreads to 8 GB.
One important issue to note is that the drFAST aligner is aimed at SV/CNV inference and it does not return mapping quality values, which are still essential for accurate SNP detection. However, drFAST also returns ‘best’ map locations for PE and mate pair reads in addition to all possible discordant configurations where ‘best’ is defined as the mapping with the lowest Hamming distance and with span size closest to the library average. Future releases of drFAST will have the capability of returning mapping quality for these best map locations, which will effectively increase the appeal of drFAST, and users will be able to use it for both structural variation discovery through multimapping PE and mate pair reads, and SNP discovery. Until this feature is available in drFAST, one may need to run other aligners in parallel to discover SNPs.
5 CONCLUSION
This is an exciting time for genomics research. The amount of available (1000 Genomes Project Consortium, 2010) and anticipated (Genome 10K Community of Scientists, 2009) sequence data now arms us to expand our understanding human variation, disease susceptibility and genome evolution. Although there are inherent accuracy and bias problems associated with different sequencing platforms (Smith et al., 2008), we can also leverage the different ‘strengths’ of these technologies to increase confidence and comprehensiveness of SNP (Nothnagel et al., 2011) and structural variation (Mills et al., 2011) discovery.
For species where a reference genome is available as in human, mapping sequence reads to this reference assembly is the first step in genome analysis. Sensitivity and accuracy, as well as the speed of read alignment, are crucial for precise characterization of genomic variants. To this end, many mapping algorithms were developed (Alkan et al., 2009; Hach et al., 2010; Homer et al., 2009; Li et al., 2008a; b; Li et al., 2009; Rumble et al., 2009) focusing mainly on the Illumina Genome Analyzer data, and very little effort was devoted to analyze color-space reads generated with the SOLiD platform (Homer et al., 2009; McKernan et al., 2009; Rumble et al., 2009). The main limitation of the SOLiD-aware read aligners is that they were not optimized for structural variation detection [except for SHRiMP (Rumble et al., 2009), which is more powerful in mapping to more complex areas of the genome], and they are unusable for segmental duplication analysis due to their unique mapping approach (Alkan et al., 2009). On the other hand, by tracking all possible map locations and underlying sequence variation, drFAST provides an opportunity to better access and increase ‘mappability’ in repeat and duplication-rich areas of the genome that are known to harbor much structural variation (Kidd et al., 2008). Although the sensitivity of drFAST is higher than the other aligners, we also demonstrate speed enhancements of both dynamic programming and color transformation versions. Through its readiness to be integrated to VariationHunter (Hormozdiari et al., 2009) for more sensitive SV discovery, to NovelSeq (Hajirasouliha et al., 2010) to characterize novel sequence insertions, and usability for segmental variation detection (Alkan et al., 2009). drFAST is an important step forward for recovering additional genetic variation from di-base encoded color-space sequencing.
ACKNOWLEDGEMENTS
E.E.E. is a Howard Hughes Medical Institute Investigator.
Funding: Natural Sciences and Engineering Research Council of Canada (NSERC to S.C.S. in parts); Bioinformatics for Combating Infectious Diseases (BCID to S.C.S. in parts); Michael Smith Foundation for Health Research grants (to S.C.S. in parts); US National Institutes of Health (grants HG004120 and HG005209 to E.E.E.).
Conflict of Interest: E.E.E. is an SAB member of the Pacific Biosciences.
REFERENCES
- Alkan C., et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 2009;41:1061–1067. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan C., et al. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011;12:363–376. doi: 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong M. Groups and Symmetry. New York: Springer; 1988. p. 53. [Google Scholar]
- Bentley D.R., et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows M., et al. A block sorting lossless data compression algorithm. Digital Equipment Corporation Technical Report. 1994:124. [Google Scholar]
- Chen Y., et al. PerM: efficient mapping of short sequencing reads with periodic full sensitive spaced seeds. Bioinformatics. 2009;25:2514–2521. doi: 10.1093/bioinformatics/btp486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid J., et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- Ferragina P., et al. Proceedings of the 41st Annual Symposium on Foundations of Computer Science (FOCS 2000) IEEE Computer Society Press; 2000. Opportunistic data structures with applications; pp. 390–398. [Google Scholar]
- Frigo M., et al. 40th Annual Symposium on Foundations of Computer Science. New York, NY: IEEE Computer Society; 1999. Cache-oblivious algorithms; pp. 285–297. [Google Scholar]
- Genome 10K Community of Scientists. Genome 10K: a proposal to obtain whole-genome sequence for 10 000 vertebrate species. J. Hered. 2009;100:659–674. doi: 10.1093/jhered/esp086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hach F., et al. mrsFAST: a cache-oblivious algorithm for short-read mapping. Nat. Methods. 2010;7:576–577. doi: 10.1038/nmeth0810-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajirasouliha I., et al. Detection and characterization of novel sequence insertions using paired-end next-generation sequencing. Bioinformatics. 2010;26:1277–1283. doi: 10.1093/bioinformatics/btq152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homer N., et al. BFAST: an alignment tool for large scale genome resequencing. PLoS One. 2009;4:12. doi: 10.1371/journal.pone.0007767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F., et al. Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res. 2009;19:1270–1278. doi: 10.1101/gr.088633.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F., et al. Next-generation VariationHunter: combinatorial algorithms for transposon insertion discovery. Bioinformatics. 2010;26:350–357. doi: 10.1093/bioinformatics/btq216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd J.M., et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., et al. MoDIL: detecting small indels from clone-end sequencing with mixtures of distributions. Nat. Methods. 2009;6:473–474. doi: 10.1038/nmeth.f.256. [DOI] [PubMed] [Google Scholar]
- Li H., et al. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008a;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R., et al. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008b;24:713–714. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- Li H., et al. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski J., et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N. Engl. J. Med. 2010;362:1181–1191. doi: 10.1056/NEJMoa0908094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M., et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKernan K.J., et al. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009;19:1527–1541. doi: 10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills R.E., et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng S.B., et al. Exome sequencing identifies mll2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nothnagel M., et al. Technology-specific error signatures in the 1000 Genomes Project data. Hum. Genet. 2011 doi: 10.1007/s00439-011-0971-3. [Epub ahead of print, doi: 10.1007/s00439-011-0971-3, 23 February 2011] [DOI] [PubMed] [Google Scholar]
- Ondov B.D., et al. Efficient mapping of Applied Biosystems SOLiD sequence data to a reference genome for functional genomic applications. Bioinformatics. 2008;24:2776–2777. doi: 10.1093/bioinformatics/btn512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushkarev D., et al. Single-molecule sequencing of an individual human genome. Nat. Biotechnol. 2009;27:847–850. doi: 10.1038/nbt.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumble S.M., et al. SHRiMP: accurate mapping of short color-space reads. PLoS Comput. Biol. 2009;5:11. doi: 10.1371/journal.pcbi.1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D.R., et al. Rapid whole-genome mutational profiling using next-generation sequencing technologies. Genome Res. 2008;18:1638–1642. doi: 10.1101/gr.077776.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant P.H., et al. Diversity of human copy number variation and multicopy genes. Science. 2010;330:641–646. doi: 10.1126/science.1197005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers L.E., et al. A de novo paradigm for mental retardation. Nat. Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- Wheeler D.A., et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]