


Abstract
A typical G-rich telomeric DNA strand, which runs 5′→3′ toward the chromosome ends, protrudes by several nucleotides in lower eukaryotes. In human chromosomes long G-rich 3′-overhangs have been found. Apart from the standard G-rich tail, several non-canonical terminal structures have been proposed. However, the mechanism of long-tail formation, the presence and the role of these structures in telomere maintenance or shortening are not completely understood. In a search for a simple method to accurately measure the 3′-overhang we have established a protocol based on the ligation of telomeric oligonucleotide hybridized to non-denatured DNA under stringent conditions (oligonucleotide ligation assay with telomeric repeat oligonucleotide). This method enabled us to detect a large proportion of G-rich single-stranded telomeric DNA that was as short as 24 nt. Nevertheless, we showed G-tails longer than 400 nt. In all tested cells the lengths ranging from 108 to 270 nt represented only 37% of the whole molecule population, while 56–62% were <90 nt. Our protocol provides a simple and sensitive method for measuring the length of naturally occurring unpaired repeated DNA.
INTRODUCTION
The ends of eukaryotic chromosomes are protected from loss of DNA, end-to-end fusion and other potential errors by complex specialized structures known as telomeres. Telomeres contain short DNA repeats and specifically associated proteins that distinguish natural chromosome ends from damaged DNA. Telomeres of human somatic cells carry 3–18 kb of double-stranded TTAGGG repeats, which gradually shorten with cell doublings (1–6). A compensatory mechanism is provided by the telomerase, a ribonucleoprotein complex containing a subunit with reverse transcriptase motifs that adds TTAGGG repeats onto the 3′ DNA end of chromosomes using its RNA component as a template (7–10). Telomerase, usually absent in somatic cells, is present in germline and immortalized cells. Reactivation of the telomerase reverse transcriptase subunit, operated by the oncogene myc (11–13), is considered a fundamental step in cell immortalization and oncogenesis (14–17).
The primary role of telomeres is to protect the ends of linear chromosomes from being recognized as damaged DNA, which would lead to chromosome recombination and fusion. Furthermore, they must allow access to telomerase to add telomere repeats in order to maintain their length. In fact, all conventional DNA polymerases require a primer and can synthesize DNA only in the 5′→3′ direction, leaving a gap at the ends of the newly synthesized strands after removal of the last RNA primer (18). Telomere structure is conserved in almost all eukaryotes. The typical G-rich short repeat (TTGGGG in Tetrahymena, TTTTGGGG in Oxytricha and TTAGGG in vertebrates) always runs with the G-rich strand 5′→3′ toward a chromosome end. Therefore, while the C-rich DNA is copied by leading strand synthesis, the G-rich strand is copied by lagging strand synthesis. Consequently, a terminal G-rich 3′-overhang, at least as long as the removed RNA primer, is expected at all ends. The presence of a 3′-overhang tail is a general feature of eukaryotic chromosomes (19). The exact length of the G-rich strand in Oxytrichia is 14 nt, longer than the C-strand (20). In Saccharomyces cerevisiae, although most of the G-rich overhangs are <30 nt, long single-stranded regions have been observed in late S phase (21).
In human chromosomes long terminal protrusions of single-stranded G-rich sequence variable from 100 to 280 nt were detected (22–24). Long TTAGGG single-stranded sequence, besides being an effective substrate for telomerase, may form several non-canonical structures. It has been described that in vitro oligonucleotide of single-stranded TTGGGG repeats can form a structure of four-stranded parallel or anti-parallel helices in which four guanines in a planar alignment to each other form a quadruplex. Alternatively, a single-stranded G-rich DNA may associate with double-stranded G:C-rich DNA to form a triple helix via G:G:C pairing (25–27). Recently, an electron microscopy observation revealed that up to 42% of the telomere ends form a large loop in a reaction that depends on the presence of a 3′-overhang G-rich strand and the telomeric single-stranded binding protein TRF2 (28,29). The free 3′-end was tucked back inside the double-stranded DNA at the loop junction. Furthermore a 3′-G tail of 100 nt length seems to be required for t-loop stabilization. This previously undescribed structure, named the t-loop, could explain how the extreme tip of a chromosome is distinguished from a DNA break, preventing an inappropriate DNA damage response and avoiding G-rich strand degradation or end-to-end fusion of chromosomes (30). However, the discovery of the t-loop poses new questions on how telomerase accesses the hidden 3′ terminus. On the other hand, the now base-paired 3′-end could theoretically be elongated by conventional polymerases, rendering the telomerase action superfluous.
Due to the crucial role of the structure of the extreme end of telomeres in function, we addressed the task of detecting and analyzing the length of the single-stranded telomeric sequences with a simple new approach. Basically, (CCCTAA)n oligonucleotides were phosphorylated to completion and labeled with [γ-32P]ATP at high specific activity (31). Oligonucleotide ligation assay (OLA) was performed, hybridizing telomeric repeat oligonucleotide under stringent conditions to undenatured DNA in the presence of a ligase. Oligonucleotides will be ligated to each other only if natural single-stranded DNA of perfect base-pairing matching is present. The ligated products are then analysed by running them in a denaturing sequencing gel.
MATERIALS AND METHODS
DNA extraction
DNA was obtained from three different cell line cultures and from peripheral blood lymphocytes. Human foreskin fibroblasts were cultured in BME medium plus 10% FCS. HeLa cells were grown in DMEM plus 10% FCS and U937 cells in RPMI plus 10% FCS. Lymphocytes were isolated by centrifugation on Ficoll gradient from a buffy coat. A method for obtaining high molecular weight DNA from these cells was followed.
Oligonucleotide ligation assays
Oligonucleotides were end-labeled and completely phosphorylated by T4 polynucleotide kinase in a forward reaction aimed to reach very high specificity activity (31). A mix of 50 µl containing 0.16 µM of oligonucleotide and 1.6 µM of [γ-32P]ATP (3000 Ci/mmol, 10 mCi/ml), 70 mM Tris pH 7.6, 10 mM MgCl2, 5 mM DTT and 20 U of T4 polynucleotide kinase was incubated for 40 min at 37°C. Then 1 µl of 0.1 M ATP and a further 10 U kinase were added and the reaction was prolonged for 20 min. After heat inactivation of the enzyme for 20 min at 65°C, oligonucleotides were precipitated with ethanol and dissolved in an appropriate volume of water. Hybridization and ligation were conducted in a volume of 20 µl containing 5 µg of undenatured DNA, 0.5 pmol of oligonucleotide, 20 mM Tris pH 7.6, 25 mM potassium acetate, 10 mM magnesium acetate, 10 mM DTT, 1 mM NAD, 0.1% Triton X-100. To ensure completion of the oligonucleotide annealing to naturally occurring single-stranded DNA sequences and to avoid long enzymatic incubation, all ingredients (except ligase) were placed into 0.5 ml PCR tubes and incubated at the desired temperature (30°C for 12mer, 50°C for 18mer, 65°C for 24mer) for 12–14 h. Subsequently, 20 U thermostable Taq ligase was added and the reaction was prolonged for 5 h. Reactions were ended by adding 30 µl of water and by phenol–chloroform extraction. Samples were precipitated with ethanol and dissolved in 6 µl of TE.
Reaction product electrophoresis and analysis
Reaction products were analyzed both on denaturating 6% acrylamide sequencing gel and in 1% agarose native gel. For analysis of ligated products under denaturating conditions, half of the volume was mixed with 4 µl of formamide dye. Samples were heated at 90°C and immediately quenched on ice before loading an amount of 2 µl onto the gel. The remaining 3 µl was diluted in water and in a glycerol loading buffer to a volume of 10 µl and run in 1% agarose native gel for 4 h at 90 V in TAE buffer. The gels were dried on Whatman filter paper and exposed to autoradiography film. The images were acquired by 1D Image Analysis Software (Kodak Digital Science). The value of the intensity (background subtracted) of each band of the ladder was normalized by dividing for the number of concatenated oligonucleotide probes in the band. This value was then normalized to the total intensity and plotted as relative frequency of the 3′-overhang length. This analysis was performed for at least three different experiments for each cell line and the mean relative frequencies were compared by the t-test.
Solution hybridization experiments
The non-denaturing hybridization assay was carried out as described (22). Briefly, [γ-32P]ATP-labeled (TTAGGG)3, (CCCTAA)3, (CGACCTCGAGATCCYRGCTCACTGCAA) and (CA)9 oligonucleotide probes were used. Aliquots of 5 µg undigested genomic DNA or DpnII-, AluI- or Bal31-digested DNA were dissolved in hybridization buffer (50 mM Tris–HCl pH 8.0, 50 mM NaCl, 1 mM EDTA), added to labeled probe (0.5 pmol) in a volume of 20 µl and incubated 16 h at 50°C. Hybridized samples were size-fractionated on 1% agarose gel in 1× TAE at 45 V for 3 h. The gels were dried on Whatman filter paper and exposed to autoradiography film.
Enzymatic DNA modification
Exonuclease I. For removal of the 3′ single-stranded DNA, Escherichia coli Exonuclease I was used. DNA was incubated in appropriate buffer (10 mM Tris–HCl pH 8.0, 1 mM EDTA, 10 mM MgCl2, 20 mM KCl and 10 mM 2-mercaptoethanol) and digested using 1 U/µl of enzyme ExoI (US Biochemicals) for 24 h at 37°C. The reactions were stopped, the mixtures were extracted with phenol–chloroform and the DNA was precipitated and dissolved in H2O.
Exonuclease III. For cleaving 3′ recessed but not 3′ protruding ends of DNA, Exonuclease III was used. DNA was incubated in appropriate buffer (66 mM Tris–HCl pH 8.0, 6.6 mM MgCl2, 5 mM DTT, 50 µg/ml BSA) with 1 U/µl of Exonuclease III at 37°C for 30 min.
T7 (Gene6) Exonuclease. For removal of the 5′-single-stranded DNA, the DNA was incubated with T7 (Gene6) Exonuclease (0.6 U/µl) (US Biochemicals) in 50 mM Tris–HCl pH 8.1, 5 mM MgCl2, 20 mM KCl and 5 mM 2-mercaptoethanol at 37°C for 20 min.
S1 nuclease. For selective removal of single-stranded DNA, S1 nuclease (Pharmacia) was used. Genomic DNA was incubated with 1 U/µl of S1 nuclease in appropriate buffer (30 mM sodium acetate pH 4.6, 50 mM NaCl, 1 mM ZnCl2 and 5% glycerol). The reaction was incubated at 37°C for 10 min, extracted with phenol–chloroform and the DNA ws precipitated and dissolved in H2O.
Mung bean nuclease. Genomic DNA was incubated with 0.26 U/µl Mung Bean Nuclease (Pharmacia) at 30°C for 30 min in nuclease buffer (30 mM sodium acetate pH 4.5, 10 mM NaCl, 1 mM ZnCl and 5% glycerol). The mixture was extracted and the DNA was precipitated.
Bal31 nuclease. For the Bal31 nuclease experiment, undigested genomic DNA was incubated at 30°C for 15 min with 3 U of Bal31 nuclease (Promega) in 100 µl buffer containing 120 mM NaCl, 2.4 mM CaCl2, 2.4 mM MgCl2, 4 mM Tris–HCl, 0.2 mM EDTA pH 8.0. The samples were inactivated by incubation for 10 min at 65°C. Bal31-treated DNA samples were extracted with phenol–chloroform, precipitated with ethanol and dissolved in H2O.
RESULTS
Experimental design
The OLA is a well established method for distinguishing alleles that differ by a single point mutation. Briefly, two oligonucleotides are designed to hybridize in exact juxtaposition to the target DNA sequences permitting their covalent joining by a DNA ligase. Successful ligation can be monitored by a variety of means including visualizing the ligation product after separation of different size fragments by gel electrophoresis. Since human telomeres are composed of repetitions of a 6 nt sequence, in-frame hybridized complementary oligonucleotide of a telomeric sequence is expected to produce a ladder of concatenated oligonucleotides whose lengths are comparable to the target sequences. If single-stranded telomeric regions are present in native DNA, they will be replenished by in-frame oligonucleotides that will be covalently joined by ligase. OLA with telomeric repeat oligonucleotide (T-OLA) was performed under stringent non-denaturating conditions using thermostable ligase. The oligonucleotides were labeled to very high specific activity and phosphorylated to completion. The concatenated products were released from the target DNA by heat denaturation and separated by denaturating PAGE. Visualization was obtained by autoradiography of radioactively labeled ligated products. In addition, samples of the reactions were run in non-denaturating agarose gels to check for DNA quantity, relationship and consistency between observed hybridization signal to high molecular weight DNA and T-OLA products.
Telomeric-oligonucleotide ligation assays
T-OLA measures the length of the naturally occurring single-stranded telomeric DNA. A ladder of ligated products was observed in reactions with oligonucleotides containing two, three and four telomeric repeats. Since similar results were obtained with these types of oligonucleotides, the 18mer was selected for most of the experiments described because experimental conditions were easier to control. Results obtained with (CCCTAA)3 oligonucleotide and DNA extracted from peripheral blood lymphocytes, along with a series of control reactions performed to check for sensitivity and specificity, are shown in Figure 1. DNA pre-treatment with Bal31, S1 or Mung Bean exonucleases completely abolished the oligonucleotide ligation reaction. Several other oligonucleotides expected to give negative results were employed as well. A 24mer telomeric repeat containing three mismatches (CCCTTACCCTTACCCTTACCCTAA) was also used. This oligonucleotide is frequently used in the standard TRAP assay (32) to detect telomerase activity. No signal was observed either with this oligonucleotide or with (CCCTAA)3G, a 19mer designed to introduce a mismatch in the ligation frame. Other negative results were observed for single-stranded DNA of the CGG and CTG repeat family. In contrast, a short, but very distinct, ladder was produced by the oligonucleotide complementary to microsatellites of the CA repeat class. A very weak ladder of ligated products was produced by OLA performed with (TTAGGG)3 oligonucleotide. The product length was comparable with those obtained with (CCCTAA)3 oligonucleotide. The telomere end standard model predicts a recessive 5′-C strand. Since ligase catalyses phosphodiester bonds between adjacent 5′-phosphate and 3′-hydroxyl residues, it could happen that products would be retained if juxtaposition were occurring between oligonucleotide and 5′-ends. In some experiments, DNA was dephosphorylated by calf intestinal phosphatase (CIAP). A small reduction of the ladder intensity resulted from reaction with (CCCTAA)3 CIAP-treated DNA. As expected, most of the products of T-OLA that migrated with large molecular weight DNA under non-denaturating conditions migrated as a ladder of products in denaturating gels, confirming that only a small fraction of T-OLA products were unreleased from high molecular weight DNA. Very similar results were observed using three repeat telomeric oligonucleotides of different frame.
Figure 1.
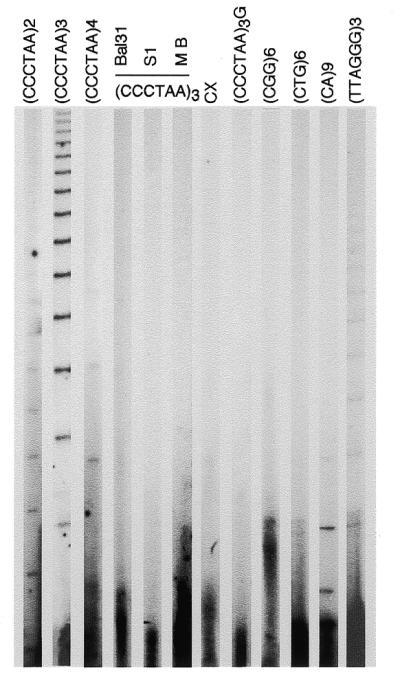
Oligonucleotide ligation assay to non-denaturated DNA. Ladders of ligated oligonucleotide were obtained by running the samples on 6% denaturating polyacrylamide gel. An aliquot of 5 µg of undenaturated DNA from peripheral blood lymphocytes was hybridized with 32P end-labeled oligonucleotide. OLA was performed as described in the text. Oligonucleotide probes consisting of two, three or four CCCTAA telomeric repeats were used as indicated. DNA pre-treated with Bal31, S1 and Mung Bean (MB) Exonucleases was used for OLA to the (CCCTAA)3 probe. Other probes were: CCCTTACCCTTACCCTTACCCTAA (CX), (CCCTAA)3G,(CGG)6, (CTG)6, (CA)9 and (TTAGGG)3. Each lane was loaded with 2 µl of formamide containing samples representing 20% of the OLA reaction. For autoradiography, gels were exposed for 5 days at –70°C to Biomax film and a Biomax intensifying screen.
Solution hybridization experiments
Since telomeric G-tails have been most frequently detected by solution hybridization and native agarose gel electrophoresis, we performed a non-denaturing hybridization assay probing for highly repeated DNA sequences such as the dinucleotide CA repeat class and the Alu family, as well as for telomeric repeats. Results of hybridization with undigested and DpnII-, AluI- or Bal31-digested DNA are shown in Figure 2. The localization of the signal relative to digested DNA and the sensitivity to Bal31 exonuclease support evidence for specificity, confirming the presence of relatively abundant G-rich telomeric single-stranded DNA. It should be considered that CA repeats comprises 0.5% (33) and the Alu family comprises ~2% of the whole genomic DNA. This means that these DNA families contain ∼20 and 100 times the amount of telomeric DNA. On the other hand, we were unable to detect any single-stranded DNA relative to GCC and GAT repeats that are ~0.05% of the genome. Hybridization signals obtained with probe for Alu and CA-repeated DNA families suggest that unpaired DNA throughout the genome can be evident for highly repetitive DNA.
Figure 2.
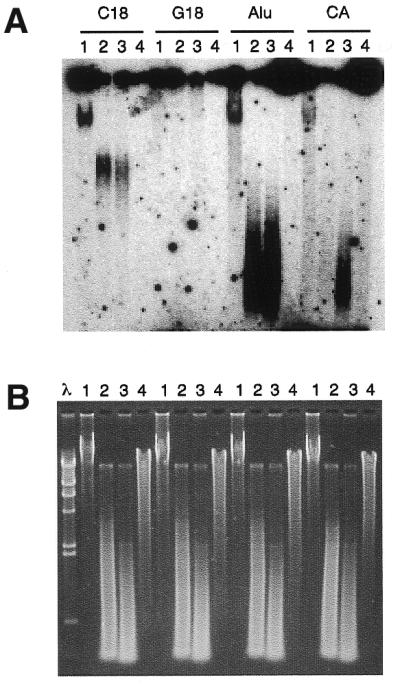
Non-denaturing solution hybridization analysis. Solution hybridization of non-denaturating fibroblast DNA is shown. All DNA samples were hybridized to 32P-labeled probes [C18 for (CCCTAA)3, G18 for (TTAGGG)3, Alu for CGACCTCGAGATCCYRGCTCACTGCAA and CA for (CA)9]. After overnight hybridization at 50°C, the samples were separated by electrophoresis in 1% agarose and detected by autoradiography. (A) For each probe, lanes 1 show reactions with undigested DNA. Hybridizations to digested DNA with DpnII (lanes 2), with AluI (lanes 3) and with Bal31 Exonuclease (lanes 4) are shown. In all cases, 5 µg of DNA was used. (B) Ethidium bromide staining of the gel shown in (A). The λ–HindIII molecular marker is also shown.
T-OLA with enzymatically modified DNA
T-OLA may determine the length of single-stranded telomeric DNA. Enzymatic treatment of DNA prior to T-OLA enables a series of predictions to be drawn. A 3′-overhang tail is expected to be specifically digested by E.coli Exonuclease I but is insensible to Exonuclease III. A 5′-exonuclease such as T7 (Gene 6) Exonuclease should increase the amount of single-stranded DNA, lengthening the G-rich tail of the 3′ overhang structure. The effect of enzymatic treatment on DNA from fibroblast, U937 and HeLa cells and lymphocytes probed either for GGGTTA or CA repeats were evaluated by directly comparing ladder products from treated and untreated DNA. The results, an example of which is shown in Figure 3, confirmed the 3′-overhang structure of single-stranded DNA detected by T-OLA. Nevertheless, a partial resistance to Exonuclease I, mostly in U937 and HeLa cells, for longer products were obtained. T7 (Gene 6) Exonuclease treatment differentially altered the T-OLA products in fibroblast and lymphocytes versus U937 and HeLa cells as well. The ladder was stronger and richer in longer products in fibroblasts and lymphocytes compared to U937 and HeLa cells. Results of experiments conducted on enzymatically modified DNA probed with CA or GT oligonucleotide were consistent with unpaired internal DNA regions. In fact, Bal31, S1 or Mung Bean Exonuclease are efficient in completely abolishing the ladder products. Exonuclease I and Exonuclease III have a reducing effect while T7 (Gene6) Exonuclease has a small increasing effect.
Figure 3.
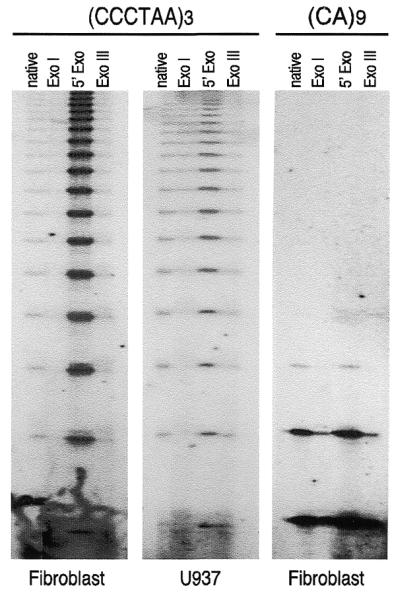
Effects of enzymatic treatments of DNA on OLA. Treatments were performed as reported in the text. In all reactions, 5 µg of treated DNA were analyzed by OLA, either probing with (CCCTAA)3, or with (CA)9 oligonucleotide. Since no difference has been observed between fibroblasts and lymphocytes or between HeLa and U937 cells probing for telomeric repeats, and for all kinds of cell probing for CA repeats, only one kind of cell type for each category is shown. The ladders of ligated products obtained by autoradiography of denaturing gels are shown. Native, undigested DNA; ExoI: Exonuclease I; 5′ Exo: T7 (Gene6) Exonuclease; Exo III: Exonuclease III.
Length of single-stranded telomeric DNA
In order to determine the length distribution of the G-rich single-stranded DNA, repeated experiments were performed on DNA extracted from human fibroblasts, U937, HeLa cell lines and from peripheral blood lymphocytes. In all experiments a ladder ranging from 24 nt, the minimum length appreciable with our method, to 360 nt was consistently obtained. On long exposure of experiments conducted with fibroblasts and with lymphocytes, fragments >400 nt can also be seen. The distribution of the percentage of G-rich tails obtained normalizing for the number of oligonucleotide probes present in each band is shown in Figure 4. In all cell types, 37% of the 3′ G-rich tail was in the class ranging from 108 to 270 nt long. In fibroblasts and lymphocytes, 56% of G-rich tails were <90 nt. A higher relative percentage (62%) of this class of lengths was present in HeLa and U937 cells.
Figure 4.

Size distribution of G-rich 3′ overhangs in four human cell types. The percentages of G-rich tails of different lengths are reported for fibroblasts, lymphocytes, HeLa and U937 cells. The frequency of overhangs of different length is obtained by normalizing the intensity of each single band to the sum of intensity of all the bands of the ladder. On the x-axis the number of oligonucleotides ligated by the OLA are reported.
In order to calibrate our method, we analyzed human umbilical vein endothelial cells (HUVEC) by T-OLA. The 3′-overhang of these cells has been previously analyzed both by a strand-replacement technique (PENT) (22) and by electron microscopic examination (24). Using PENT, G-rich overhangs averaging 130–210 bases in length have been detected in these cells as well as in the other cells tested. On the contrary, it has been shown by electron microscopic observation that overhangs of HUVEC are twice as long as those present in fibroblasts and they reach up to 650 nt in length. T-OLA was also able to detect 3′-overhangs up to 650 nt. Nevertheless, the length distribution was very similar to that of all the other cells we analyzed, the predominant class being <90 nt in length.
DISCUSSION
Chromosome stability in eukaryotic cells depends on the maintenance of a functional tract of telomeric repeats (34–37). The actual telomere length seems to be determined by a complex mechanism that includes lengthening as well as shortening activities. The ubiquitous G-rich tails are probably necessary for maintenance of chromosome end structure and function, such as protection against degradation and inter-chromosome fusion. Makarov et al. (22) and Wright et al. (24), following two different approaches, reported the presence of long G tails in human cells. In the first report using PENT, the authors reported that >80% of the telomeres have long G-rich overhangs averaging 130–210 nt. In the second approach, telomeres were purified and analyzed. Electron microscopic observation of those telomeres showed long overhangs of 200 (±75) nt. Both approaches very likely underestimated the frequency of shorter G tails.
We have measured the G-rich strand of naturally occurring unpaired telomeric DNA in lymphocytes, primary fibroblast, HeLa and U937 human cells, as well as HUVEC by a novel approach. In contrast to previous reports, we show that a significant amount of G-rich tails are <90 nt; at the same time G-rich regions >400 nt have been detected. Several technical considerations have to be discussed at this point. Reproducibility and accuracy of our protocol for measuring G-rich tails depends on the completion of several reactions. Unphosphorylated oligonucleotides might not be ligated, leading to the accumulation of short partially concatenated products. The same result would be seen if the hybridization and ligation reactions were incomplete. This does not seem to be the case since we followed the labeling reaction, which itself produced >90% γ-32P-labeled oligonucleotide, with a large amount of ATP. T-OLA was conducted with a large excess of oligonucleotide for times as long as 72 h without observing changes in the length distribution.
Recently it has been reported that human cells may be quite different in the length distribution of the G-rich single-stranded 3′-overhang and that the rate of shortening during cell doubling is dependent on the size of the overhang. In fact, the rate of telomere shortening of HUVEC was double that of fibroblasts that had a G-rich tail half as long, confirming the critical role of the length of the G-rich tail (38). Nevertheless, even in this report it is likely that the short G-tails were underestimated. Our data confirm that in HUVEC 3′-overhangs as long as twice that of fibroblasts are present, but we show that the length distribution is very similar to that of fibroblasts and other cells tested. It has been proposed that exonuclease erosion of the telomere produced by the leading strand synthesis results in relatively small 3′-overhang G-rich tails. A much larger 3′-overhang would be a consequence of the inability of the last RNA primer to position itself near the 3′-terminus of lagging strand synthesis. According to our data, most of the G-rich tails have been generated by 5′ erosion, mostly in cells presenting telomerase activity. On the other end, relatively longer overhangs are present in cells lacking telomerase.
Our experimental approach for measuring the 3′-overhang was unable to determine per se the exact structure of the single-stranded DNA we were detecting. In any case, the results obtained with enzyme-treated DNA are consistent with a structure that is prevalently 3′-overhang. The relative inefficiency of Exonuclease I in removing the telomeric 3′-overhang has been reported (24). The difference of T7 (Gene 6) Exonuclease in increasing the amount and quality of the T-OLA product in fibroblast and lymphocytes versus U937 and HeLa cells has been noted. These observations may well be explained by the presence of a secondary structure such as the D-loop. Although we can start sizing from a minimum of 24 nt, the high proportion of short fragments and the trend of length distribution suggest that very short G-rich tails, which should not be able to promote a t-loop structure, are present. The length distributions in HeLa and U937 cell lines were very similar, as were those of the fibroblasts and of lymphocytes. Nevertheless, very long regions of single-stranded G-rich DNA were present only in normal diploid cells that lack telomerase.
Elongation of the 3′-end by telomerase, degradation of the 5′-ends probably by an exonuclease, as well as sequestration of the 3′-end by the D-loop or other non-canonical structures are all mechanisms possibly involved in regulating telomere length in proliferating cells and controlling the lifespan of human cells. Further analysis of the terminal structure of the telomere and telomere length homeostasis should lead to a better understanding of the underlying mechanism. Methods aimed at increasing the rate of shortening can be exploited in the treatment of cancer. On the other hand, reducing the rate of shortening should increase the replicative lifespan of cells.
Acknowledgments
ACKNOWLEDGEMENT
We thank FARMM onlus for financially supporting this project.
References
- 1.Muller H.J. (1938) The remaking of chromosomes. The Collecting Net, Woods Hope (USA), 13, 181–195.
- 2.McClintock B. (1939) The behavior in successive nuclear divisions of a chromosome broken at meiosis. Proc. Natl Acad. Sci. USA, 25, 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blackburn E.H. (1991) Structure and function of telomeres. Nature, 350, 569–573. [DOI] [PubMed] [Google Scholar]
- 4.Blackburn E.H. (1994) Telomeres: no end in sight. Cell, 77, 621–623. [DOI] [PubMed] [Google Scholar]
- 5.Zakian V.A. (1995) Telomeres: beginning to understand the end. Science, 270, 1601–1607. [DOI] [PubMed] [Google Scholar]
- 6.Greider C.W. (1996) Telomere length regulation. Annu. Rev. Biochem., 65, 337–365. [DOI] [PubMed] [Google Scholar]
- 7.Greider C.W. and Blackburn,E.H. (1989) A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature, 337, 331–337. [DOI] [PubMed] [Google Scholar]
- 8.Morin G.B. (1989) The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell, 59, 521–529. [DOI] [PubMed] [Google Scholar]
- 9.Counter C.M., Avilion,A.A., LeFeuvre,C.E., Stewart,N.G., Greider,C.W., Harley,C.B. and Bacchetti,S. (1992) Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J., 11, 1921–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lingner J., Hughes,T.R., Shevchenko,A., Mann,M., Lundblad,V. and Cech,T.R. (1997) Reverse transcriptase motifs in the catalytic subunit of telomerase. Science, 276, 561–567. [DOI] [PubMed] [Google Scholar]
- 11.Wang J., Xie,L.Y., Allan,S., Beach,D. and Hannon,G.J. (1998) Myc activates telomerase. Genes Dev., 12, 1769–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu K.J., Grandori,C., Amacker,M., Simon-Vermot,N., Polack,A., Lingner,J. and Dalla-Favera,R. (1999) Direct activation of TERT transcription by c-MYC. Nature Genet., 21, 220–224. [DOI] [PubMed] [Google Scholar]
- 13.Falchetti M.L., Falcone,G., D’Ambrosio,E., Verna,R., Alema,S. and Levi,A. (1999) Induction of telomerase activity in v-myc-transformed avian cells. Oncogene, 18, 1515–1519. [DOI] [PubMed] [Google Scholar]
- 14.Counter C.M., Meyerson,M., Eaton,E.N., Ellisen,L.W., Caddle,S.D., Haber,D.A. and Weinberg,R.A. (1998) Telomerase activity is restored in human cells by ectopic expression of hTERT (hEST2), the catalytic subunit of telomerase. Oncogene, 16, 1217–1222. [DOI] [PubMed] [Google Scholar]
- 15.Bodnar A.G., Ouellette,M., Frolkis,M., Holt,S.E., Chiu,C.P., Morin,G.B., Shay,J.W., Lichtsteiner,S. and Wright,W.E. (1998) Extension of life-span by introduction of telomerase into normal human cells. Science, 279, 349–352. [DOI] [PubMed] [Google Scholar]
- 16.Hahn W.C., Counter,C.M., Lundberg,A.S., Beijersbergen,R.L., Brooks,M.W. and Weinberg,R.A. (1999) Creation of human tumour cells with defined genetic elements. Nature, 400, 464–468. [DOI] [PubMed] [Google Scholar]
- 17.Herbert B., Pitts,A.E., Baker,S.I., Hamilton,S.E., Wright,W.E., Shay,J.W. and Corey,D.R. (1999) Inhibition of human telomerase in immortal human cells leads to progressive telomere shortening and cell death. Proc. Natl Acad. Sci. USA, 96, 14276–14281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watson J.D. (1972) Origin of concatameric T7 DNA. Nat. New Biol., 239, 197–201. [DOI] [PubMed] [Google Scholar]
- 19.Henderson E.R. and Blackburn,E.H. (1989) An overhanging 3′ terminus is a conserved feature of telomeres. Mol. Cell. Biol., 9, 345–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klobutcher L.A., Swanton,M.T., Donini,P. and Prescott,D.M. (1981) All gene-sized DNA molecules in four species of hypotrichs have the same terminal sequence and or unusual 3′ terminus. Proc. Natl Acad. Sci. USA, 78, 3015–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wellinger R.J., Wolfe,A.J. and Zakian,V.A. (1993) Saccharomyces telomeres acquire single-strand TG1-3 tails late in S phase. Cell, 72, 51–60. [DOI] [PubMed] [Google Scholar]
- 22.Makarov V., Hirose,Y. and Langmore,J.P. (1997) Long G tails at both ends of human chromosomes suggest a C strand degradation mechanism for telomere shortening. Cell, 88, 657–666. [DOI] [PubMed] [Google Scholar]
- 23.McElligott R. and Wellinger,R.J. (1997) The terminal DNA structure of mammalian chromosomes. EMBO J., 16, 3705–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wright W.E., Tesmer,V.M., Huffman,K.E., Levene,S.D. and Shay,J.W. (1997) Normal human chromosomes have long G-rich telomeric overhangs at one end. Genes Dev., 11, 2801–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henderson E.R., Hardin,C.C., Walk,S.K., Tinoco,I.,Jr and Blackburn,E.H. (1987) Telomeric DNA oligonucleotides form novel intramolecular structures containing guanine-guanine base pair. Cell, 51, 899–908. [DOI] [PubMed] [Google Scholar]
- 26.Sundquist W.I. and Klug,A. (1989) Telomeric DNA dimerizes by formation of guanine tetrads between hairpin loops. Nature, 342, 825–829. [DOI] [PubMed] [Google Scholar]
- 27.Sen D. and Gilbert,W. (1990) A sodium-potassium switch in the formation of four-stranded G4-DNA. Nature, 344, 410–414. [DOI] [PubMed] [Google Scholar]
- 28.Van Steensel B., Smogorzewska,A. and de Lange,T. (1998) TRF2 protects human telomeres from end-to-end fusions. Cell, 92, 401–413. [DOI] [PubMed] [Google Scholar]
- 29.Griffith J.D., Comeau,L., Rosenfield,S., Stansel,R.M., Bianchi,A., Moss,H. and de Lange,T. (1999) Mammalian telomeres end in a large duplex loop. Cell, 97, 503–514. [DOI] [PubMed] [Google Scholar]
- 30.Karleseder J., Broccoli,D., Dai,Y., Hardy,S. and de Lage,T. (1999) p53 and ATM dependent apoptosis induced by telomeres lacking TRF2. Science, 283, 1321–1325. [DOI] [PubMed] [Google Scholar]
- 31.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Labelling of synthetic oligonucleotides by phosphorylation with bacteriophage T4 polynucleotide kinase. Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 11.31–11.32.
- 32.Kim N.W., Piatyszek,M.A., Prowse,K.R., Harley,C.B., West,M.D., Ho,P.L., Coviello,G.M., Wright,W.E., Weinrich,S.L. and Shay,J.W. (1994) Specific association of human telomerase activity with immortal cells and cancer. Science, 266, 2011–2015. [DOI] [PubMed] [Google Scholar]
- 33.Tautz D. and Renz,M. (1984) Simple sequences are ubiquitous components of eukaryotic genomes. Nucleic Acids Res., 12, 4127–4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lundblad V. and Szostak,J.W. (1989) A mutant with a defect in telomere elongation leads to senescence in yeast. Cell, 57, 633–643. [DOI] [PubMed] [Google Scholar]
- 35.Zakian V.A. (1996) Structure, function and replication of Saccharomyces cerevisiae telomeres. Annu. Rev. Genet., 30, 141–172. [DOI] [PubMed] [Google Scholar]
- 36.Lee H.W., Blasco,M.A., Gottlieb,G.J., Horner,J.W.,II, Greider,C.W. and DePinho,R.A. (1998) Essential role of mouse telomerase in highly proliferative organs. Nature, 392, 569–574. [DOI] [PubMed] [Google Scholar]
- 37.Blasco M.A., Gasser,S.M. and Lingner,J. (1999) Telomeres and telomerase. Genes Dev., 13, 2353–2359. [DOI] [PubMed] [Google Scholar]
- 38.Huffman K.E., Levene,S.D., Tesmer,V.M., Shay,J.W. and Wright,W.E. (2000) Telomere shortening is proportional to the size of the G-rich telomeric 3′-overhang. J. Biol. Chem., 275, 19719–19722. [DOI] [PubMed] [Google Scholar]