

Summary
The current “isolate, inactivate, inject” vaccine development strategy has served the field of vaccinology well, and such empirical vaccine candidate development has even led to the eradication of smallpox. However, such an approach suffers from limitations, and as an empirical approach, does not fully utilize our knowledge of immunology and genetics. A more complete understanding of the biological processes culminating in disease resistance is needed. The advent of high-dimensional assay technology and “systems biology” along with a vaccinomics approach [1;2] is spawning a new era in the science of vaccine development. Here we review recent developments in systems biology and strategies for applying this approach and its resulting data to expand our knowledge base and drive directed development of new vaccines. We also provide applied examples and point out new directions for the field in order to illustrate the power of systems biology.
Keywords: system biology, bioinformatics, immune response, vaccines
Vaccines and the Promise of Systems Biology
Vaccines have been among the most successful public health interventions to date with most vaccine-preventable diseases having declined in the United States by 95-99% or more [3]. As we move into the 21st century; however, it is apparent that future vaccine development will be more difficult as more complex organisms become vaccine targets. To date, vaccine development has been empiric, often characterized by an “isolate, inactivate, inject” paradigm of development. Such an approach ignores both pathogen and host variability and as a result, significant limitations ensue such as inadequate immune protection, the inability to develop vaccines against hypervariable viruses (e.g. HIV, HCV, etc.), and an insufficient understanding of how protective immune responses develop and persist over time in response to vaccine antigens.
The last several years have seen an increasing emphasis on systems biology science that is expected to aid researchers in elucidating the pathways and networks involved in diverse biological processes. While the definition is evolving, systems biology has been described as “an interdisciplinary approach that systematically describes the complex interactions between all the parts in a biological system, with a view to elucidating new biological rules capable of predicting the behavior of the biological system” [4]. Biological systems are more than simple collections of genes/proteins; they are complex, intricately interacting sets of functional and sometimes redundant pathways that collectively produce coherent behaviors [5], of which the innate and adaptive immune responses are perfect examples. For this reason, vaccinologists in the 21st century must not only use increasingly high throughput technology to understand immune profiling after vaccination, but must also consider strategies designed to understand how such data can be harnessed toward new vaccine development. With the remarkable advances in technology it is appropriate to review how new technology, systems biology, and the analytic and bioinformatic approaches used to make sense of the data generated, can be best harnessed toward the goal of new vaccine development. We frame our review with a new paradigm to vaccine development with four phases: organize, analyze, utilize and immunize (Figure 1).
Figure 1. An iterative systems biology approach to vaccine development.

Movement from one phase to the next involves updating known biological knowledge with implications for study design, analytical strategies, study endpoints and laboratory techniques. Organize: Includes selecting the appropriate high-dimensional ‘omics’ technologies to interrogate the appropriate biological systems (DNA, RNA, protein, lipid, cell subset, etc…) as well as organizing and integrating a priori known knowledge regarding pathways and networks. Analyze: Includes strategies for study design and modeling methods to truly integrate data spanning each of the assayed biological systems. This step also includes statistical techniques to maximize power and minimize false discoveries while modeling the complex interactions and developing a greater understanding of both the host and pathogen biologies underlying the immune process. Utilize: Applying the new knowledge gained from the systems-level analysis to logically target areas for vaccine improvement. These could impact vaccine composition (an adjuvant driving appropriate Th1/Th2 balance), or efficacy testing (early immune signatures predictive of vaccine response). Immunize: Includes the physical steps necessary to implement the needed changes for novel vaccine development (moving from egg-based to cell-line based vaccine production) and to introduce the new vaccine into the population (clinical trials to confirm improved safety profiles or enhanced immunogenicity using newly discovered biomarkers).
Organize
Over the past decade or so, many high dimensional assays have become available to researchers allowing interrogation of thousands to millions of endpoints. These can be organized according to biological system or network within an organism. Importantly, these ‘omics’ technologies are available for the large-scale characterization of many of the essential components of biological systems such as: 1) DNA including: single nucleotide polymorphisms (SNPs), genetic insertions and deletions, chromosomal copy number variation (CNV), and DNA methylation, 2) RNA including: mRNA expression, microRNA expression, differential transcript detection, RNA interference screening, and 3) Protein including: protein expression and localization, protein-protein interaction using yeast 2-hybrid screening. The list will only grow with newly emerging fields, such as lipidomics, metabolomics, interactomics, localizomics, phosphoproteomics, and polychromatic flow cytometry made possible by newly available, high-throughput, high-dimensional technologies [6-11].
The resulting outputs from these technologies can be organized according to pathway or network knowledge. We have approached immune profiling of vaccine-induced immune responses through the “immune response network theory” [1;2]. This theory states that vaccine immune responses are the cumulative result of interactions driven by a host of genes. Further, these interactions are theoretically predictable. The basic elements of the network include genes which activate or suppress immune responses, the dominance profile of a given gene or polymorphism in relation to a specific antigen, epigenetic modifications of genes, the influence of signaling and other innate response genes, gene-gene interactions, and genes for other host response factors. By monitoring immune responses over time with this conceptual framework, we can begin to understand and “organize” the drivers of protective and non-protective immune responses to vaccine antigens, and, in turn, use this information to develop new vaccine strategies (Figure 2). For example, discovery of how a specific polymorphism of a viral receptor leads to measurable and quantifiable heterogenous innate, humoral, and/or cell-mediated immune responses not only advances our understanding of how vaccines work, but also informs strategies leading to new vaccine development [12].
Figure 2. Application of systems biology to vaccine development.
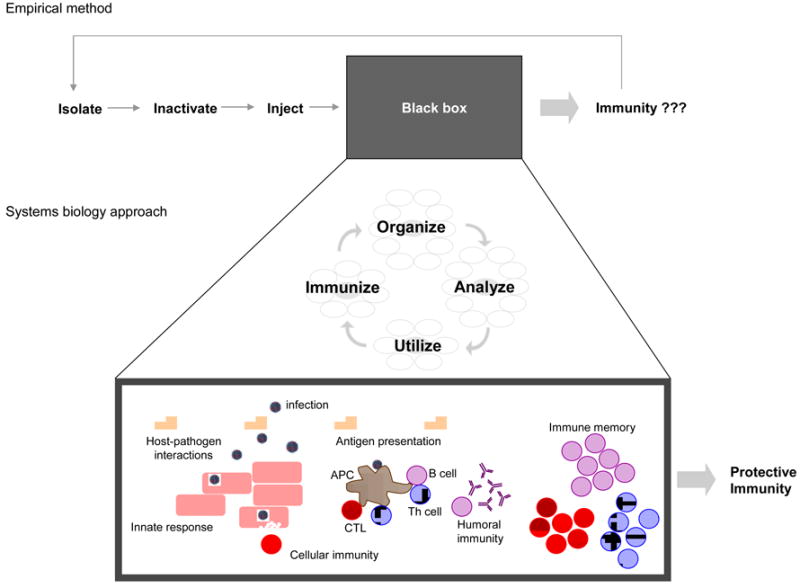
From Jenner's initial work with cowpox forward, vaccine development was an empirical science based on incomplete understanding of immune processes leading to protection. Pathogenic organisms were attenuated, inactivated, or killed and then injected. Success led to large-scale use of the vaccine, while failure meant repeating the process with a new pathogen strain or different inactivation procedure. The factors controlling success or failure were largely unknown. With a systems biology approach, modern high-dimensional data acquisition techniques allow researchers to comprehensively characterize the epigenetic, transcriptomic, proteomic, metabolomic, and other essential features of host-pathogen interactions and immune regulatory networks and processes in order to more fully elucidate the biological rules governing “immunity” enhancing our understanding of the “black box”. Cutting-edge bioinformatic algorithms and statistical methods are used to gain a deeper understanding of the data, which is then applied to develop next-generation vaccines which appropriately stimulate the key drivers of immune response.
Analyze
These high dimensional platforms pose challenges in the areas of experimental design, variable selection and modeling and data integration as discussed below.
Experimental Design
Most of the high dimensional assays produce abundance measures that are relative rather than absolute making the fundamental principals of randomization, replication and blocking critically important during the development of statistical experimental study designs. Direct application of these principals in order to minimize experimental effects such as batch effects and maximize use of patient and time resources in high throughput platforms has been recently described [13-15]. Considerations for subject selection, potential sources of bias and methods for avoiding false discoveries in marker discovery studies have been discussed at length and guidelines provided to ensure study conclusions are not influenced by extraneous systematic factors [15-18]. New testing and design strategies should be developed for vaccinology in order to achieve sample sizes large enough to ensure generalizability of results to the population. For example, Thomas et al. recently studied SNPs associated with risk of breast cancer in 9,770 cases and 10,799 controls via a three-stage testing strategy [19]. Applying such concepts to systems biology studies in vaccine development should help to minimize false discoveries and increase power, generalizability and reproducibility.
Variable Selection and Modeling
High dimensional data sets result in far more potential predictor variables (e.g., thousands of mRNAs) than subjects, and make proper modeling of the data a complicated task. Statistical modeling tools are being developed and continually improved to filter out non-informative data, select the most informative features for modeling purposes, incorporate a priori known biological knowledge in an effort to minimize false leads and perform some form of statistical model validation such as cross validation [20-24]. For example, gene set testing methods are becoming commonplace and Witten et al. are extending ridge and lasso regression to a full family of methods utilizing shrunken estimates to improve prediction [20]. As an applied example, we were recently funded by the NIH to use a systems biology approach to define immune profiles containing the key drivers of immune response to seasonal influenza vaccine in elderly subjects. In the organize phase we chose measures of humoral and cellular immunity and markers of immunosenescence together with high dimensional epigenetic, transcriptomic and proteomic assays. We will apply two complementary analysis strategies (Figure 3) in order to maximize power and minimize false discoveries. Our study design includes a replication cohort in addition to statistical model validation since it is important to verify that results discovered in an initial study can be repeated in a completely independent set of subjects. Formal replication is a complicated task, and guidelines exist for performing replication and to aid clinicians in judging the readiness of a model [25-27].
Figure 3. Two pronged systems biology approach to understanding influenza vaccine response in the elderly.

Our primary biology to gene approach is a deductive approach relying on known biological information to construct gene sets known to be involved in key immune processes. Integrated transcriptomic/proteomic/cellular data from our profiling assays will be used to develop immunologic profiles related to defined immune response outcomes as described. Our secondary gene to biology approach is an inductive, evidence based approach which will rely on individual variables. Modules in this approach are genes with co-regulated gene expression. This has historically been the primary analytical approach in the gene expression literature.
Data Integration
Perhaps the largest analytical obstacle to systems biology approaches is the logical integration of diverse data types in order to fully understand and interconnect relationships between genes, transcripts, proteins, metabolites, and epigenetic regulators. To address this, the analysis and modeling tools necessary to make sense of the immense volumes of data being generated are becoming increasingly sophisticated. One such example is the use of model-based analysis for flow cytometry data to supplement traditional gating-based analysis [28]. Another is the use of “omics” data repositories such as the Gene Expression Omnibus, the Open Proteomics Database, or the Biomolecular Network Database, and increasing implementation of algorithms and software packages designed to meld heterogenous data types [6;29].
Statistical strategies for modeling these diverse high dimensional data types in a truly integrated fashion are beginning to be developed, but many more are needed. For example, Reif et al. evaluate performance of variable selection techniques within high dimensional SNP and proteomic data sets separately versus in the two combined and concluded that combined analysis was generally preferable [30;31].
Bioinformatics
Bioinformatics advances in vaccine development can be grouped into three general areas: pathogen biology, host biology, and the interaction between the two. As the focus of bioinformatics evolved from a single gene/single target paradigm to systems biology, so has the approach to vaccine development. From the classic reverse vaccinology (viral genome to targets), we are now building targeted approaches and extensive in silico screening. In this section, we will describe some recent advances associated with this evolution.
In pathogen biology, deep bioinformatics analyses of Brucella [32] and flavivirus [33] have identified new candidates for rational vaccine design. These approaches start by identifying a set of highly immunogenic genes based on prior vaccine data, structural and localization predictions, and comparative cross-species/strain virulence. Selected immunogenic protein subsequences are then manufactured and experimentally validated. Next-generation epitope prediction and identification techniques are becoming increasingly sophisticated, taking advantage of the growing genomic and proteomic datasets [34]. Better epitope prediction based on immunogenicity in the context of host response has been developed, for example, in silico docking [35], consensus epitope prediction [36], and multiple epitope coverage [37]. These methods rely on novel algorithms, as well as aggregation methods, and curated databases. Currently they are limited by the quantity and quality of curated reference data due to the use of machine learning or pattern recognition algorithms.
Although pathogen evolution from vaccination was described more than a decade ago, recent sequencing data and bioinformatics analysis, such as allele dynamic plots can map the population drifts of the rapidly evolving genes [38;39], and will likely prove useful as we systematically tune our vaccine development in response to specific challenges.
To improve our understanding of the interplay between pathogen and host, modeling of large-scale protein-protein interactions, and RNAi knockdown screening techniques are increasingly being used to identify virulence factors, critical host pathways involved in pathogenesis, and candidate genes essential for productive infections [40;41]. For almost all of these methods, the ability to use shared databases to train advanced learning algorithms is critical for successful outcome.
Innate immune response pathways that involve pattern recognition receptors (PRR) and its subfamilies, e.g. toll-like receptors (TLRs), have been expanded considerably over the last few years. These new discoveries, coupled with detailed gene expression studies, have created systems models that are predictive for innate immune response. In fact, systems biology analyses are beginning to shed new light on the important role that innate pathways play in subsequent adaptive immunity [31;42]. Refinement of these approaches would allow creation of individualized immune profiles that have the potential for customizing adjuvants as well as exploring the possible development of vaccines for auto-immune diseases with adaptive response [43].
Adaptive immune response has also undergone a systems modeling approach [44] to improve our understanding of host biology. In addition to epitope binding predictions described above, Love et al. used deep sequencing of escape-prone epitopes of SIV to model the effect on CD8+ T cell response [45]. A significant upstream component of vaccine development is to monitor the different pathogen strains in circulation in order to select the appropriate ones for vaccine targeting. The application of bioinformatics and microarrays to strain monitoring is becoming more prevalent [46]. Modeling host population with social structures has gained insight into the transmission of pathogens [47;48]. Finally, understanding allelic differences between individuals and the effect on immune response has gained significant ground from the point of pharmacogenomics and vaccine development [2;49;50].
The development of the bioinformatics approaches described above is the direct result of available high dimensional data afforded by biotechnology and statistical analysis. Without these partner disciplines, the expansion of bioinformatics analysis from simple gene targeting to host-pathogen modeling will be limited. However, we are seeing continued and faster use of system complexity in vaccine development, indicating a successful amalgamation of these paradigms.
Utilize and Immunize
The ultimate aim of vaccinology is to develop safe and effective vaccines to protect susceptible populations, thus the goal of a systems biology approach must be to provide a comprehensive understanding of the biologic processes necessary for development of effective immune responses, which in turn must be adapted to the development of better vaccines (Figure 2). In fact, the increasing power to quickly characterize host and pathogen responses at the genetic, transcriptomic, and proteomic level, all on a global scale, complemented by novel bioinformatics approaches, is having a critical and growing influence on new vaccine development. Immunogenetic studies performed by us and others have demonstrated that by understanding critical genetic determinants of immune response, we may reveal the basis of vaccine low- or non-response, or susceptibility to adverse events [30;51-53]. This information may allow a more individualized approach to vaccination in order to enhance immune response in vaccine non-responders, or to elicit protective immunity without complications. For example, recent systems biology and bioinformatics studies of the yellow fever vaccine have greatly enhanced our understanding of both innate immunity, and have provided predictive models of CD8+ T cell responses [31;54;55]. In this regard, systems biology (and vaccinomics) may provide essential information regarding the key drivers of immunity, knowledge that can be exploited in the appropriate selection of adjuvant, antigen dose, and even route of administration in order to elicit optimal immunity [1;2]. Similarly, identification of critical immune epitopes may spur the development of safe and effective subunit based vaccines, such as the protein-based HBV and papilloma virus vaccines [56;57], or even peptide-based vaccines, which, when combined with new knowledge regarding HLA haplotypes and super-types, may be targeted broadly or to a specific population most at risk [12;58-60]. Yet another potential benefit of systems biology is the development of predictive models that may allow us to identify early biomarkers of vaccine efficacy or even warn of imminent adverse reactions. Similar to the interferon signature in systemic lupus erythematosus, [61-63] an immune profile indicative of ineffective response or adverse reaction may indicate targets for improved adjuvant usage or even therapeutic intervention. Predictive biomarkers may also streamline vaccine efficacy testing, allowing for cheaper and faster preclinical development.
Thus the promise of systems biology is to allow a deeper understanding of the complex biological processes and interactions necessary for protective immunity after vaccination. Such characteristics may lead to new vaccine candidates that induce long-lasting, population-level immunity and the ability to eradicate infectious diseases (Box 1).
Box 1. Characteristics of Vaccine Development Approaches.
| Paradigm | Characteristics | |
|---|---|---|
|
| ||
| Empirical | Isolate Inactivate Inject |
Trial and error experimentation |
| Many notable successes (smallpox, rabies, polio, HBV) | ||
| Several limitations: | ||
| ||
|
| ||
| Systems Biology | Organize Analyze Utilize Immunize |
Defined correlates of protection |
| Functional understanding of immune processes | ||
| Insights into molecular basis of memory formation | ||
| Detailed view of host-pathogen interactions | ||
| New vaccines for problematic pathogens | ||
| Improved adjuvants | ||
| Long-lived protective immunity | ||
Biomarkers of: non-response
| ||
| Safer vaccines | ||
| Avoidance of inflammation/autoimmunity | ||
Conclusions
A new era of vaccine development is apparent and is leading to much excitement in the field of vaccinology. We have characterized this as the “second golden age of vaccinology” [1]. Current challenges in vaccinology are important as vaccines represent the only medical intervention delivered to virtually every human on earth. Moving from the empirical strategy to a systems biology vaccinomics strategy (Box 1) is associated with many challenges. Addressing these will require multidisciplinary teams including clinicians and laboratory scientists with biological subject matter knowledge, epidemiologists with an understanding of bias and populations, statisticians with an understanding of experimental design and modeling, bioinformaticians with an understanding of biology, and computational tools and public databases. We anticipate the reward of meeting these challenges to bring the field of vaccinology to new frontiers, and the benefit to human kind to be immense.
Highlights.
-
•
This paper demonstrates a basis for the genetic contribution to vaccine-induced humoral and cellular immune responses.
-
••
This paper provides a brilliant review of issues relating to vaccine pharmacogenomics.
-
•
This manuscript demonstrates the pervasiveness of batch effects in high dimensional technology and proposes strategies to minimize the impact on research.
-
•
This paper demonstrates and evaluates different analytical integration strategies of two high dimensional data types.
-
••
This article uses a systems biology approach to identify predictive biomarkers associated with immune responses to the yellow fever vaccine and is an excellent example of the methods and processes described in this review.
-
•
This paper presents an interesting visualization for viral strain evolution.
-
•
This paper represents the typical use of networks, in this case, the discovery of infection-related genes.
-
•
This paper simulates the spread of disease using social network models and the effects of selected vaccination.
-
•
This chapter provides a complementary review of the bioinformatics resources available for immunology.
Acknowledgments
We acknowledge support from the National Institutes of Health grants AI-33144, AI-48793, AI-89859, and HHSN272201000025C for this work.
Footnotes
Conflicts of Interest: The authors declare no conflicts of interest relevant to this topic.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Poland GA. Pharmacology, vaccinomics, and the second golden age of vaccinology. Clin Pharmacol Ther. 2007;82:623–626. doi: 10.1038/sj.clpt.6100379. [DOI] [PubMed] [Google Scholar]
- •2.Poland GA, Ovsyannikova IG, Jacobson RM, Smith DI. Heterogeneity in vaccine immune response: the role of immunogenetics and the emerging field of vaccinomics. Clin Pharmacol Ther. 2007;82:653–664. doi: 10.1038/sj.clpt.6100415. This paper demonstrates a basis for the genetic contribution to vaccine-induced humoral and cellular immune responses. [DOI] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention. Reported vaccine-preventable diseases--United States, 1993, and the childhood immunization initiative. MMWR. 1994;43:57–60. [PubMed] [Google Scholar]
- 4.Pulendran B, Li S, Nakaya HI. Systems vaccinology. Immunity. 2010;33:516–529. doi: 10.1016/j.immuni.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kitano H. Computational systems biology. Nature. 2002;420:206–210. doi: 10.1038/nature01254. [DOI] [PubMed] [Google Scholar]
- 6.Joyce AR, Palsson BO. The model organism as a system: integrating ‘omics’ data sets. Nat Rev Mol Cell Biol. 2006;7:198–210. doi: 10.1038/nrm1857. [DOI] [PubMed] [Google Scholar]
- 7.Ng A, Bursteinas B, Gao Q, Mollison E, Zvelebil M. Resources for integrative systems biology: from data through databases to networks and dynamic system models. Brief Bioinform. 2006;7:318–330. doi: 10.1093/bib/bbl036. [DOI] [PubMed] [Google Scholar]
- 8.Luber CA, Cox J, Lauterbach H, Fancke B, Selbach M, Tschopp J, Akira S, Wiegand M, Hochrein H, O'Keeffe M, Mann M. Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity. 2010;32:279–289. doi: 10.1016/j.immuni.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 9.Nita-Lazar A. Quantitative analysis of phosphorylation-based protein signaling networks in the immune system by mass spectrometry. Wiley Interdiscip Rev Syst Biol Med. 2010 doi: 10.1002/wsbm.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bakal C, Perrimon N. Realizing the promise of RNAi high throughput screening. Dev Cell. 2010;18:506–507. doi: 10.1016/j.devcel.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aleksic J, Russell S. ChIPing away at the genome: the new frontier travel guide. Mol Biosyst. 2009;5:1421–1428. doi: 10.1039/B906179G. [DOI] [PubMed] [Google Scholar]
- ••12.Poland GA, Ovsyannikova IG, Jacobson RM. Application of pharmacogenomics to vaccines. Pharmacogenomics. 2009;10:837–852. doi: 10.2217/PGS.09.25. This paper provides a brilliant review of issues relating to vaccine pharmacogenomics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oberg AL, Vitek O. Statistical design of quantitative mass spectrometry-based proteomic experiments. J Proteome Res. 2009;8:2144–2156. doi: 10.1021/pr8010099. [DOI] [PubMed] [Google Scholar]
- 14.Auer PL, Doerge RW. Statistical design and analysis of RNA sequencing data. Genetics. 2010;185:405–416. doi: 10.1534/genetics.110.114983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •15.Leek JT, Scharpf RB, Bravo HC, Simcha D, Langmead B, Johnson WE, Geman D, Baggerly K, Irizarry RA. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet. 2010;11:733–739. doi: 10.1038/nrg2825. This manuscript demonstrates the pervasiveness of batch effects in high dimensional technology and proposes strategies to minimize the impact on research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simon RM, Paik S, Hayes DF. Use of archived specimens in evaluation of prognostic and predictive biomarkers. J Natl Cancer Inst. 2009;101:1446–1452. doi: 10.1093/jnci/djp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ransohoff DF, Gourlay ML. Sources of bias in specimens for research about molecular markers for cancer. J Clin Oncol. 2010;28:698–704. doi: 10.1200/JCO.2009.25.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Subramanian J, Simon R. Gene expression-based prognostic signatures in lung cancer: ready for clinical use? J Natl Cancer Inst. 2010;102:464–474. doi: 10.1093/jnci/djq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas G, Jacobs KB, Kraft P, Yeager M, Wacholder S, Cox DG, Hankinson SE, Hutchinson A, Wang Z, Yu K, Chatterjee N, Garcia-Closas M, Gonzalez-Bosquet J, Prokunina-Olsson L, Orr N, Willett WC, Colditz GA, Ziegler RG, Berg CD, Buys SS, McCarty CA, Feigelson HS, Calle EE, Thun MJ, Diver R, Prentice R, Jackson R, Kooperberg C, Chlebowski R, Lissowska J, Peplonska B, Brinton LA, Sigurdson A, Doody M, Bhatti P, Alexander BH, Buring J, Lee IM, Vatten LJ, Hveem K, Kumle M, Hayes RB, Tucker M, Gerhard DS, Fraumeni JF, Jr, Hoover RN, Chanock SJ, Hunter DJ. A multistage genome-wide association study in breast cancer identifies two new risk alleles at 1p11.2 and 14q24.1 (RAD51L1) Nat Genet. 2009;41:579–584. doi: 10.1038/ng.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Witten DM, Tibshirani R. Covariance-regularized regression and classification for high-dimensional problems. J R Stat Soc Series B Stat Methodol. 2009;71:615–636. doi: 10.1111/j.1467-9868.2009.00699.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis NA, Crowe JE, Jr, Pajewski NM, McKinney BA. Surfing a genetic association interaction network to identify modulators of antibody response to smallpox vaccine. Genes Immun. 2010 doi: 10.1038/gene.2010.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKinney BA, Crowe JE, Guo J, Tian D. Capturing the spectrum of interaction effects in genetic association studies by simulated evaporative cooling network analysis. PLoS Genet. 2009;5:e1000432. doi: 10.1371/journal.pgen.1000432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao Y, Simon R. Development and validation of predictive indices for a continuous outcome using gene expression profiles. Cancer Inform. 2010;9:105–114. doi: 10.4137/cin.s3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi L, Campbell G, Jones WD, Campagne F, Wen Z, Walker SJ, Su Z, Chu TM, Goodsaid FM, Pusztai L, Shaughnessy JD, Jr, Oberthuer A, Thomas RS, Paules RS, Fielden M, Barlogie B, Chen W, Du P, Fischer M, Furlanello C, Gallas BD, Ge X, Megherbi DB, Symmans WF, Wang MD, Zhang J, Bitter H, Brors B, Bushel PR, Bylesjo M, Chen M, Cheng J, Cheng J, Chou J, Davison TS, Delorenzi M, Deng Y, Devanarayan V, Dix DJ, Dopazo J, Dorff KC, Elloumi F, Fan J, Fan S, Fan X, Fang H, Gonzaludo N, Hess KR, Hong H, Huan J, Irizarry RA, Judson R, Juraeva D, Lababidi S, Lambert CG, Li L, Li Y, Li Z, Lin SM, Liu G, Lobenhofer EK, Luo J, Luo W, McCall MN, Nikolsky Y, Pennello GA, Perkins RG, Philip R, Popovici V, Price ND, Qian F, Scherer A, Shi T, Shi W, Sung J, Thierry-Mieg D, Thierry-Mieg J, Thodima V, Trygg J, Vishnuvajjala L, Wang SJ, Wu J, Wu Y, Xie Q, Yousef WA, Zhang L, Zhang X, Zhong S, Zhou Y, Zhu S, Arasappan D, Bao W, Lucas AB, Berthold F, Brennan RJ, Buness A, Catalano JG, Chang C, Chen R, Cheng Y, Cui J, Czika W, Demichelis F, Deng X, Dosymbekov D, Eils R, Feng Y, Fostel J, Fulmer-Smentek S, Fuscoe JC, Gatto L, Ge W, Goldstein DR, Guo L, Halbert DN, Han J, Harris SC, Hatzis C, Herman D, Huang J, Jensen RV, Jiang R, Johnson CD, Jurman G, Kahlert Y, Khuder SA, Kohl M, Li J, Li L, Li M, Li QZ, Li S, Li Z, Liu J, Liu Y, Liu Z, Meng L, Madera M, Martinez-Murillo F, Medina I, Meehan J, Miclaus K, Moffitt RA, Montaner D, Mukherjee P, Mulligan GJ, Neville P, Nikolskaya T, Ning B, Page GP, Parker J, Parry RM, Peng X, Peterson RL, Phan JH, Quanz B, Ren Y, Riccadonna S, Roter AH, Samuelson FW, Schumacher MM, Shambaugh JD, Shi Q, Shippy R, Si S, Smalter A, Sotiriou C, Soukup M, Staedtler F, Steiner G, Stokes TH, Sun Q, Tan PY, Tang R, Tezak Z, Thorn B, Tsyganova M, Turpaz Y, Vega SC, Visintainer R, von FJ, Wang C, Wang E, Wang J, Wang W, Westermann F, Willey JC, Woods M, Wu S, Xiao N, Xu J, Xu L, Yang L, Zeng X, Zhang J, Zhang L, Zhang M, Zhao C, Puri RK, Scherf U, Tong W, Wolfinger RD. The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nat Biotechnol. 2010;28:827–838. doi: 10.1038/nbt.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Subramanian J, Simon R. What should physicians look for in evaluating prognostic gene-expression signatures? Nat Rev Clin Oncol. 2010;7:327–334. doi: 10.1038/nrclinonc.2010.60. [DOI] [PubMed] [Google Scholar]
- 26.Steyerberg E. Clinical Prediction Models: A Practical Approach to Development, Validation, and Updating, Ed 2009. Statistics for Biology and Health; Springer: 2009. [Google Scholar]
- 27.Altman DG, Vergouwe Y, Royston P, Moons KG. Prognosis and prognostic research: validating a prognostic model. Br Med J. 2009;338:b605. doi: 10.1136/bmj.b605. [DOI] [PubMed] [Google Scholar]
- 28.Frelinger J, Ottinger J, Gouttefangeas C, Chan C. Modeling flow cytometry data for cancer vaccine immune monitoring. Cancer Immunol Immunother. 2010;59:1435–1441. doi: 10.1007/s00262-010-0883-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Myers CL, Chiriac C, Troyanskaya OG. Discovering biological networks from diverse functional genomic data. Methods Mol Biol. 2009;563:157–175. doi: 10.1007/978-1-60761-175-2_9. [DOI] [PubMed] [Google Scholar]
- •30.Reif DM, Motsinger-Reif AA, McKinney BA, Rock MT, Crowe JE, Jr, Moore JH. Integrated analysis of genetic and proteomic data identifies biomarkers associated with adverse events following smallpox vaccination. Genes Immun. 2009;10:112–119. doi: 10.1038/gene.2008.80. This paper demonstrates and evaluates different analytical integration strategies of two high dimensional data types. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••31.Querec TD, Akondy RS, Lee EK, Cao W, Nakaya HI, Teuwen D, Pirani A, Gernert K, Deng J, Marzolf B, Kennedy K, Wu H, Bennouna S, Oluoch H, Miller J, Vencio RZ, Mulligan M, Aderem A, Ahmed R, Pulendran B. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat Immunol. 2009;10:116–125. doi: 10.1038/ni.1688. This article uses a systems biology approach to identify predictive biomarkers associated with immune responses to the yellow fever vaccine and is an excellent example of the methods and processes described in this review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He Y, Xiang Z. Bioinformatics analysis of Brucella vaccines and vaccine targets using VIOLIN. Immunome Res. 2010;6 1:S5. doi: 10.1186/1745-7580-6-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koraka P, Martina BE, Osterhaus AD. Bioinformatics in new generation flavivirus vaccines. J Biomed Biotechnol. 2010;2010:864029. doi: 10.1155/2010/864029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heinz E, Tischler P, Rattei T, Myers G, Wagner M, Horn M. Comprehensive in silico prediction and analysis of chlamydial outer membrane proteins reflects evolution and life style of the Chlamydiae. BMC Genomics. 2009;10:634. doi: 10.1186/1471-2164-10-634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khan JM, Ranganathan S. pDOCK: a new technique for rapid and accurate docking of peptide ligands to Major Histocompatibility Complexes. Immunome Res. 2010;6 1:S2. doi: 10.1186/1745-7580-6-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanchez-Burgos G, Ramos-Castaneda J, Cedillo-Rivera R, Dumonteil E. Immunogenicity of novel Dengue virus epitopes identified by bioinformatic analysis. Virus Res. 2010;153:113–120. doi: 10.1016/j.virusres.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 37.Larsen MV, Lelic A, Parsons R, Nielsen M, Hoof I, Lamberth K, Loeb MB, Buus S, Bramson J, Lund O. Identification of CD8+ T cell epitopes in the West Nile virus polyprotein by reverse-immunology using NetCTL. PLoS ONE. 2010;5:e12697. doi: 10.1371/journal.pone.0012697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •38.Steinbruck L, McHardy AC. Allele dynamics plots for the study of evolutionary dynamics in viral populations. Nucleic Acids Res. 2010 doi: 10.1093/nar/gkq909. This paper presents an interesting visualization for viral strain evolution. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chong YL, Padhi A, Hudson PJ, Poss M. The effect of vaccination on the evolution and population dynamics of avian paramyxovirus-1. PLoS Pathog. 2010;6:e1000872. doi: 10.1371/journal.ppat.1000872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang A, Johnston SC, Chou J, Dean D. A systemic network for Chlamydia pneumoniae entry into human cells. J Bacteriol. 2010;192:2809–2815. doi: 10.1128/JB.01462-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •41.Guo X, Xu Y, Bian G, Pike AD, Xie Y, Xi Z. Response of the mosquito protein interaction network to dengue infection. BMC Genomics. 2010;11:380. doi: 10.1186/1471-2164-11-380. This paper represents the typical use of networks, in this case, the discovery of infection-related genes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumagai Y, Akira S. Identification and functions of pattern-recognition receptors. J Allergy Clin Immunol. 2010;125:985–992. doi: 10.1016/j.jaci.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 43.Jan M, Meng S, Chen NC, Mai J, Wang H, Yang XF. Inflammatory and autoimmune reactions in atherosclerosis and vaccine design informatics. J Biomed Biotechnol. 2010;2010:459798. doi: 10.1155/2010/459798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahlers JD, Belyakov IM. New paradigms for generating effective CD8+ T cell responses against HIV-1/AIDS. Discov Med. 2010;9:528–537. [PubMed] [Google Scholar]
- 45.Love TM, Thurston SW, Keefer MC, Dewhurst S, Lee HY. Mathematical modeling of ultradeep sequencing data reveals that acute CD8+ T-lymphocyte responses exert strong selective pressure in simian immunodeficiency virus-infected macaques but still fail to clear founder epitope sequences. J Virol. 2010;84:5802–5814. doi: 10.1128/JVI.00117-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaewpongsri S, Sukasem C, Srichunrusami C, Pasomsub E, Zwang J, Pairoj W, Chantratita W. An integrated bioinformatics approach to the characterization of influenza A/H5N1 viral sequences by microarray data: Implication for monitoring H5N1 emerging strains and designing appropriate influenza vaccines. Mol Cell Probes. 2010;24:387–395. doi: 10.1016/j.mcp.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 47.Romano CM, de Carvalho-Mello IM, Jamal LF, de Melo FL, Iamarino A, Motoki M, Pinho JR, Holmes EC, de Andrade Zanotto PM. Social networks shape the transmission dynamics of hepatitis C virus. PLoS ONE. 2010;5:e11170. doi: 10.1371/journal.pone.0011170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •48.Salathe M, Jones JH. Dynamics and control of diseases in networks with community structure. PLoS Comput Biol. 2010;6:e1000736. doi: 10.1371/journal.pcbi.1000736. This paper simulates the spread of disease using social network models and the effects of selected vaccination. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pappalrado F, Zhang P, Halling-Brown M, Basford K, Scalia A, Shepherd A, Moss D, Motta S, Brusic V. Computational Simulations of the Immune systems for personalized medicine: State of the art and challenges. Current Pharmacogenomics and Personalized Medicine. 2008;6:260–271. [Google Scholar]
- •50.Yan Q. Immunoinformatics and systems biology methods for personalized medicine. Methods Mol Biol. 2010;662:203–220. doi: 10.1007/978-1-60761-800-3_10. This chapter provides a complementary review of the bioinformatics resources available for immunology. [DOI] [PubMed] [Google Scholar]
- 51.Poland GA, Ovsyannikova IG, Jacobson RM, Vierkant RA, Jacobsen SJ, Pankratz VS, Schaid DJ. Identification of an association between HLA class II alleles and low antibody levels after measles immunization. Vaccine. 2001;20:430–438. doi: 10.1016/s0264-410x(01)00346-2. [DOI] [PubMed] [Google Scholar]
- 52.Ovsyannikova IG, Jacobson RM, Dhiman N, Vierkant RA, Pankratz VS, Poland GA. Human leukocyte antigen and cytokine receptor gene polymorphisms associated with heterogeneous immune responses to mumps viral vaccine. Pediatrics. 2008;121:e1091–e1099. doi: 10.1542/peds.2007-1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McKinney BA, Reif DM, Rock MT, Edwards KM, Kingsmore SF, Moore JH, Crowe JE., Jr Cytokine expression patterns associated with systemic adverse events following smallpox immunization. J Infect Dis. 2006;194:444–453. doi: 10.1086/505503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pulendran B. Learning immunology from the yellow fever vaccine: innate immunity to systems vaccinology. Nat Rev Immunol. 2009;9:741–747. doi: 10.1038/nri2629. [DOI] [PubMed] [Google Scholar]
- 55.Gaucher D, Therrien R, Kettaf N, Angermann BR, Boucher G, Filali-Mouhim A, Moser JM, Mehta RS, Drake DR, III, Castro E, Akondy R, Rinfret A, Yassine-Diab B, Said EA, Chouikh Y, Cameron MJ, Clum R, Kelvin D, Somogyi R, Greller LD, Balderas RS, Wilkinson P, Pantaleo G, Tartaglia J, Haddad EK, Sekaly RP. Yellow fever vaccine induces integrated multilineage and polyfunctional immune responses. J Exp Med. 2008;205:3119–3131. doi: 10.1084/jem.20082292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zahradnik JM, Couch RB, Gerin JL. Safety and immunogenicity of a purified hepatitis B virus vaccine prepared by using recombinant DNA technology. J Infect Dis. 1987;155:903–908. doi: 10.1093/infdis/155.5.903. [DOI] [PubMed] [Google Scholar]
- 57.Lowy DR, Schiller JT. Prophylactic human papillomavirus vaccines. J Clin Invest. 2006;116:1167–1173. doi: 10.1172/JCI28607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ovsyannikova IG, johnson KL, Bergen IIIHR, Poland GA. Mass spectrometry and peptide-based vaccine development. Clin Pharmacol Ther. 2007;82:644–652. doi: 10.1038/sj.clpt.6100389. [DOI] [PubMed] [Google Scholar]
- 59.Ovsyannikova IG, Pankratz SV, Vierkant R, Jacobson RM, Poland GA. Human leukocyte antigen haplotypes in the genetic control of immune response to measles-mumps-rubella vaccine. J Infect Dis. 2006;193:655–663. doi: 10.1086/500144. [DOI] [PubMed] [Google Scholar]
- 60.Ovsyannikova IG, Jacobson RM, Vierkant RA, Pankratz VS, Poland GA. HLA supertypes and immune responses to measles-mumps-rubella viral vaccine: Findings and implications for vaccine design. Vaccine. 2007;25:3090–3100. doi: 10.1016/j.vaccine.2007.01.020. [DOI] [PubMed] [Google Scholar]
- 61.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, Behrens TW. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bauer JW, Baechler EC, Petri M, Batliwalla FM, Crawford D, Ortmann WA, Espe KJ, Li W, Patel DD, Gregersen PK, Behrens TW. Elevated serum levels of interferon-regulated chemokines are biomarkers for active human systemic lupus erythematosus. PLoS Med. 2006;3:e491. doi: 10.1371/journal.pmed.0030491. [DOI] [PMC free article] [PubMed] [Google Scholar]