

SUMMARY
Missense mutations in the C-terminal B30.2 domain of pyrin cause familial Mediterranean fever (FMF), the most common Mendelian autoinflammatory disease. However, it remains controversial as to whether FMF is due to the loss of an inhibitor of inflammation or to the activity of a proinflammatory molecule. We generated both pyrin-deficient mice and “knockin” mice harboring mutant human B30.2 domains. Homozygous knockin, but not pyrin-deficient, mice exhibited spontaneous bone marrow-dependent inflammation similar to but more severe than human FMF. Caspase-1 was constitutively activated in knockin macrophages and active IL-1β was secreted when stimulated with lipopolysaccharide alone, which is also observed in FMF patients. The inflammatory phenotype of knockin mice was completely ablated by crossing with IL-1 receptor-deficient or adapter molecule ASC-deficient mice, but not NLRP3-deficient mice. Thus, our data provide evidence for a heretofore unrecognized ASC-dependent NLRP3-independent inflammasome in which gain-of-function pyrin mutations cause autoinflammatory disease.
INTRODUCTION
Familial Mediterranean fever (FMF, MIM249100) is the prototype and the most common of a class of disorders, termed Mendelian autoinflammatory diseases, that are characterized by recurring spontaneous episodes of fever and localized inflammation without high-titer autoantibodies or antigen-specific T cells (Galon et al., 2000; Kastner et al., 2010; Masters et al., 2009). There is a growing body of data supporting the view that these conditions represent genetic lesions of the innate immune system. FMF inflammation is mediated by a massive influx of polymorphonuclear leucocytes into the affected tissues, neutrophilia, and a rapid acute phase response. The gene responsible for FMF, designated MEFV, is composed of 10 exons encoding a 781-amino acid protein known as pyrin (also known as marenostrin) (French FMF Consortium, 1997; International FMF Consortium, 1997). So far more than 50 FMF-associated mutations have been identified within MEFV (Touitou et al., 2004). The carrier frequencies for FMF in the Middle East are as high as 1:3, suggesting that pyrin mutations may confer a selective advantage possibly related to their effects on innate immunity.
Pyrin is expressed predominantly in innate immune cells such as neutrophils, monocytes, and dendritic cells, but not in lymphocytes. In monocytes, the expression is variable and upregulated by proinflammatory cytokines, IFN-γ or TNF-α (Centola et al., 2000; Diaz et al., 2004). Pyrin is composed of at least five domains and each domain has a distinct role in interactions with several proteins that are related to inflammation (Chae et al., 2009). The most notable domain of pyrin is the N-terminal PYRIN domain (also called PYD, PAAD, or DAPIN). The PYRIN domain is also found in a large number of proteins implicated in the control of inflammation. PYRIN-domain containing proteins play a key role in the inflammasomes, cytoplasmic multiprotein complexes mediating the proteolytic activation of caspase-1, which are essential for the production of biologically active interleukin-1β (IL-1β).
We have reported a mouse model generated by targeted truncation of pyrin, which demonstrated that pyrin plays a role in the regulation of the inflammasome, mediated through PYRIN domain interactions (Chae et al., 2003). Nevertheless, the pyrin truncation mice did not show an overt phenotype related to FMF, suggesting that FMF may be caused by a gain of function induced by disease-associated missense changes in pyrin. Indeed no null FMF mutations have been identified to date. Most FMF-associated mutations are clustered in the C-terminal B30.2 domain of pyrin, and thus this domain has been the focus of current thinking of the pathogenesis of FMF. The B30.2 domain has a role in attenuating the activation of IL-1β through direct interaction with caspase-1 as well as other inflammasome components, such as the NACHT, LRR, and pyrin domain–containing proteins (NLRP or NALP) 1, 2, and 3 (Chae et al., 2006; Papin et al., 2007). Possibly due to technical differences, transfection studies have yielded contradictory results regarding the roles of wild type and mutant pyrin in IL-1 activation and inflammation more broadly (Chae et al., 2006; Chae et al., 2008; Papin et al., 2007; Yu et al., 2006; Yu et al., 2007).
In order to study the pathogenesis of FMF induced by mutations in the B30.2 domain in vivo, we have generated various gene insertion “knockin” (KI) mouse models with three frequent FMF-associated B30.2 mutations, M680I, M694V, and V726A, by inserting the B30.2 domain of human pyrin into mouse pyrin, which lacks a B30.2 orthologous domain. In contrast with the aforementioned truncation model, the KI mice showed severe spontaneous inflammatory phenotypes comparable to the inflammation of FMF patients. Using these KI mice, we explored the pathogenesis of FMF not only at the physiological level but also at the molecular level and found that the inflammatory phenotype of these mice is mediated by the ASC-dependent, NLRP3-independent production of IL-1β by bone marrow-derived cells.
RESULTS
Insertion of FMF-associated mutant B30.2 domains induces inflammation in mice
Although exon 10 of mouse Mefv has substantial homologies at the nucleotide level with exon 10 of human MEFV, which encodes the B30.2 domain, the mouse pyrin protein does not have the B30.2 domain due to frame-shift variations (Chae et al., 2000). Thus, to insert FMF-associated B30.2 mutations into mouse pyrin, exons 7–10 of mouse Mefv were replaced with exons 7–10 of human MEFV encoding wild-type (WT) or FMF-associated mutant B30.2 domains. The resultant fusion proteins were expected to express WT or one of three mutant human B30.2 domains, M680I, M694V, or V726A (Figures S1A and S1B; Figure 1A). Heterozygotes of all mutant KI mice (MefvM680II+, MefvM694V/+, and MefvV726A/+) were born from chimeric males and appeared normal without evidence of pathology. However, heterozygous WT B30.2 domain KI mice (MefvB30.2/+) were not born. A total of 44 chimeric WT B30.2 males were produced from five different positive embryonic stem (ES) cell clones, which had been screened by Southern blot and confirmed by long-range PCR. In spite of high amounts of coat color chimerism, none of the chimeric males produced heterozygotes. Homozygous mutant KI mice (MefvM680I/M680I, MefvM694V/M694V, and MefvV726A/V726A) were born from interbred heterozygotes in Mendelian ratios.
Figure 1. Pyrin with an FMF-associated mutant B30.2 domain induces inflammation in mice.

(A) Comparison of the schematic structure of pyrin proteins of human, mouse, and KI mice in which mouse pyrin is fused with the human B30.2 domains. Growth curves for (B) V726A mutant KI males (WT, n=5; heterozygotes, n=9; and homozygotes, n=4) and (D) M680I mutant KI females (WT, n=9; heterozygotes, n=10; and homozygotes, n=6). Data are as means ± s.d. (C) Hematoxylin and eosin (H&E) stained ear sections (200×) and liver sections (200×) from 8-week-old WT and MefvV726A/V726A mice, and H&E stained section (100×) (bottom) of ankle joints of 4-week-old MefvV726A/V726A mice. (E) H&E stained spleen sections (40×) from 8-week-old WT and MefvV726A/V726A mice. Additional information is provided in Figure S1 and Table S1.
All homozygous mutant KI mice spontaneously developed inflammatory phenotypes, with MefvV726A/V726A the most severe. MefvV726A/V726A pups were runted with scales on the skin as early as 1 week of age. Some MefvV726A/V726A pups died around week 2 to 3, and their growth was severely retarded (Figure S1C; Figure 1B). Gross and histological examination of MefvV726A/V726A mice revealed severe spontaneous inflammation in multiple tissues such as skin, liver, joints, and bone marrow (BM) (Figure 1C). As in human FMF, a substantial infiltration of leukocytes, mainly neutrophils and macrophages, was also observed in all inflamed tissues. Moreover, homozygous KI mice showed anemia, lymphopenia, and granulocytosis in the peripheral blood (Table S1). Although MefvM680I/M680I and MefvM694V/M694V mice could not be distinguished from their WT or heterozygous littermates for several weeks after birth, milder but detectable illness developed after 4 to 8 weeks of age for MefvM680I/M680I mice (Figure 1D) and after more than 20 weeks birth for MefvM694V/M694V mice, with growth retardation, wasting, dermatitis, and arthritis, though less severe than that seen with homozygous with MefvV726A/V726A mice. Thus, the insertion of FMF-associated mutant B30.2 domains into mouse pyrin induces spontaneous inflammation that is similar to or more severe than the inflammation observed in FMF patients. Severity, as assessed by the age of onset, varies inversely with the severity of human phenotypes (MefvV726A/V726A > MefvM680I/M680I > MefvM694V/M694V).
The KI mice have increased CD11b+ cells in lymph node, spleen, and blood
The homozygous KI mice also showed lymphadenopathy and splenomegaly (Figure S1D). Histological examination of spleen from MefvV726A/V726A mice revealed the loss of normal germinal center architecture, and the infiltration of inflammatory cells (Figure 1E). Indeed, flow cytometric analysis of MefvV726A/V726A splenocytes revealed that CD11b+ cells were markedly expanded whereas T and B cells were substantially decreased (Figure 2A). The expansion in numbers of CD11b+ cells was also observed in LN (Figure S2A), BM, and peripheral blood as early as 4 weeks, and increasing with age. Subsequent analysis showed that the vast majority of the increased CD11b+ cells were Ly-6G+ neutrophils (granulocytes) (Figure 2A; Figure S2A), and the absolute number of F4/80+ cells (monocytes or macrophages) was also increased. In MefvM680I/M680I (Figure S2B) and MefvM694V/M694V mice (data not shown), the numbers of CD11b+ cells were also substantially increased, although not to the degree seen in MefvV726A/V726A mice.
Figure 2. Expansion of CD11b+ cells and gain of function B30.2 mutations with a gene dosage effect.

(A) Flow cytometry analyses of spleens from 4- and 12-week-old MefvV726A/V726A mice. Total splenocytes were analyzed for the relative frequencies of lymphoid (CD3 and CD19 for T cells and B cells, respectively, first row) and myeloid cells (CD11b, second row). Within CD11b+-gated myeloid cells, Ly-6G+ neutrophils and F4/80+ monocytes or macrophages were analyzed (third row). Numbers indicate percentage of total cells in respective quadrants or gates. Data are representative of three independent experiments. (B) Generation of hemizygous (MefvV726A/−) KI mice for the mutant B30.2 domain. Peripheral blood cells were analyzed for CD11b+ cells from their 8-week-old offspring. Data are representative of three independent experiments. (C) Body weights of six 8-week-old males of Mefv+/+, MefvV726A/+, MefvV726A/V726A, MefvV726A/−, and Mefv−/−. (D) Expression of pyrin in peritoneal macrophages from M680I-KI and pyrin-deficient mice following 24 h of culture. Additional information is provided in Figure S2.
The inflammation of KI mice is induced by a gain of function in the mutant B30.2 domain with dosage effect
Because, in the homozygous KI mice, the disease was induced by the modified pyrin with insertion (KI) of FMF-associated mutant B30.2 domain rather than disruption of WT pyrin, we hypothesized that FMF-associated pyrin mutations might lead to a gain of function. However heterozygotes showed normal growth without any inflammatory symptoms whereas homozygous KI mice were diseased, which is consistent with the recessive mode of inheritance for FMF and a loss-of-function model. To help clarify this issue, pyrin null (Mefv−/−) mice were generated (Figure S1E) and crossed with MefvV726A/+ mice to produce hemizygotes (MefvV726A/−) that expressed only mutant pyrin from a single allele. Mefv−/− mice were grossly normal, and MefvV726A/− mice also showed normal features of growth and no signs of disease (Figures 2B and 2C). These data indicate that missense mutations on both alleles are necessary for the induction of inflammation in KI mice, implying the importance of the amount of mutant pyrin in inducing the inflammation. Indeed, in macrophages, the LPS and IL-4-induced expression of pyrin from both alleles (Mefv+/+) were about two times of the pyrin expression from only single allele (Mefv+/−) (Figure 2D, right panel), and the expression of mutant pyrin in homozygotes was substantially higher than heterozygotes both before (Figure 2D, left panel) and after (Figure 2D, middle panel) stimulation of mouse pyrin expression with LPS and IL-4. Taken together, these results suggest that this mouse model of FMF can be explained by the gain of function and gene-dosage effect of mutant pyrin.
BM derived cells are necessary and sufficient for the induction of FMF inflammation
Because pyrin is expressed primarily in myeloid cells, and KI mice showed the expansion in numbers of CD11b+ cells in LNs, spleen, and blood, we hypothesized that BM derived cells might be critical for the pathogenesis of KI mice. When BM cells from MefvM680I/M680I mice were transferred to lethally irradiated WT mice, the recipient mice started losing body weight and developed dermatitis with spontaneous infiltration of inflammatory cells, whereas the control mice that had received WT BM cells showed normal growth without inflammatory phenotype (Figures 3A and 3B). Moreover, the expansion in numbers of the CD11b+ myeloid cells was also observed in the blood (Figure 3C), LNs, and spleen of the recipient mice as seen in unmanipulated MefvM680I/M680I mice, indicating that BM cells of KI mice transferred the KI phenotype into WT mice. Conversely, we also transferred congenic CD45.1 WT BM cells to CD45.2 MefvV726A/V726A mice suffering from severe inflammation. Most of the recipients showed normalized numbers of CD11b+ cells with gradual body weight gain to the normal body weight range of WT adult mice (Figures 3D and 3E). In contrast, we also observed that a few recipients failed to gain body weight and still had expanded CD11b+ cells in their blood. Subsequent cytometric analyses showed that the rescued mice were well reconstituted with WT donor cells as expected, but there were no reconstitution of donor cells in the sick recipients (Figure 3F). Due to a tendency of early death and small body size, we had given WT BM cells to 3 week old MefvV726A/V726A mice intraperitoneally rather than by intravenous injection, and had irradiated the recipients with two doses of 3.5G. As evidenced by the flow cytometric data, this regimen did not result in complete chimerism in all recipients. However, the data indicate a strong correlation between establishment of chimerism and phenotypic rescue.
Figure 3. The inflammatory phenotypes of KI mice are induced from cells of the hematopoietic lineage.

(A–C) BM cells from KI mice induce inflammatory phenotypes in WT mice. WT mice were lethally irradiated and given BM cells from either MefvM680I/M680I or WT mice intravenously. (A) Body weight after bone marrow transplantation (BMT). Data are shown in percent relative body weight changes as means ± s.d. of duplicate measurements from four of each recipient. (B) H&E stained ear sections (100×) from both recipients (WT BM cells into WT and MefvM680I/M680I BM cells into WT) after six weeks of BMT. (C) Flow cytometry analyses of peripheral blood cells from both recipients after 18 weeks of BMT. Data are representative of three independent experiments. (D–F) BM cells from congenic CD45.1 WT mice cure the inflammatory phenotypes of CD45.2 KI mice. MefvV726A/V726A mice were irradiated and given BM cells from either WT or MefvV726A/V726A mice intraperitoneally. (D) Flow cytometry analyses of peripheral blood cells after 10 weeks of BMT. (E) Body weight after BMT. Data are shown in percent relative body weight changes of individual recipients as indicated. (F) The degree of chimerism measured concomitantly by staining the congenic marker of donor cells (CD45.1). (G) The expansion of CD11b+ cell numbers is mediated by soluble factors. Congenic CD45.1+ WT BM cells were pre-mixed with either CD45.2+ WT BM cells (upper panels) or CD45.2+ MefvM680I/M680I BM cells (lower panels) and injected into lethally irradiated CD45.1+/CD45.2+ WT mice. Flow cytometry analyses of peripheral blood after 6 weeks of BMT for the chimerism (dot plot) and relative CD11b+, CD3+, and CD19+ cells according to the origin of each cell population (histogram). Data are representative of three independent experiments.
The expansion of myeloid cell numbers is induced by soluble factors
Next we investigated whether the expansion of CD11b+ cell numbers in KI mice was due to the direct involvement of the FMF-associated mutant B30.2 domain in the proliferation of the inflammatory cells or initiated by soluble factors secreted from the cells expressing mutant pyrin. We used mixed BM transplants in which CD45.1+ WT BM cells were premixed with CD45.2+ MefvM680I/M680I or CD45.2+ WT BM cells and transferred into lethally irradiated CD45.1+CD45.2+ heterozygous WT recipients. We observed that within both recipients of CD45.1+ WT-CD45.2+ WT BM cells and of CD45.1+ WT-CD45.2+MefvM680I/M680I BM cells, each donor accounted for between 40–50% of total blood cells at 6 weeks after BM transplant. Subsequent analysis of blood cell lineages differentiated from each donor showed that CD11b+ cells comprised over 50% of both CD45.1+ and CD45.2+ cells in the recipients of mixed CD45.1+ WT-CD45.2+MefvM680I/M680I BM cells, whereas CD11b+ cells comprised only about 20% of each lineage in mixed WT chimeras (Figure 3G). These data suggest that the expansion of myeloid cell numbers is mediated by soluble factors, such as cytokines, abnormally secreted from mutant cells rather than a direct effect of mutant pyrin in the cells expressing it.
The disease of KI mice is induced by IL-1β
In order to find the soluble factors that mediate inflammation in the KI mice, various cytokines were measured from the sera of MefvV726A/V726A mice. We observed a significant elevation of multiple cytokines, chemokines, and hematopoietic factors in the sera of MefvV726A/V726A mice compared to the WT or heterozygous littermates (Figure 4A; Figure S3A). Of the elevated cytokines, we primarily focused on IL-1β because it has been demonstrated that pyrin has a role in the regulation of IL-1β secretion (Chae et al., 2003; Chae et al., 2006; Chae et al., 2008; Papin et al., 2007; Yu et al., 2006). The functionally active 17 kDa IL-1β is secreted mainly from innate immune cells after proteolytic cleavage of 34 kDa pro-IL-1β by activated caspase-1. For the activation of caspase-1, the 45 kDa pro-caspase-1 must be cleaved into 22 and 10 kDa catalytic subunits (p20 and p10, respectively). Thus the activation and secretion of both IL-1β and caspase-1 was examined by immunoblot from the cell culture supernatants of the BM CD11b+ cells and BM derived macrophages (BMDMs). WT and MefvV726A/+ cells secreted IL-1β and p10 only after stimulation with LPS followed by ATP treatment, whereas the cells stimulated with LPS alone did not secrete IL-1β and p10 despite high pro-IL-1β expression. In contrast, IL-1β and p10 were highly secreted from BM CD11b+ cells and BMDM of MefvV726A/V726A mice by LPS stimulation alone without ATP treatment (Figure 4B; Figure S3B). Moreover, we could also observe the secretion of p10 from the untreated MefvV726A/V726A BM CD11b+ cells, indicating that the inflammasome is constitutively activated in the myeloid cells of homozygous KI mice.
Figure 4. Inflammation in KI mice is induced by IL-1β.
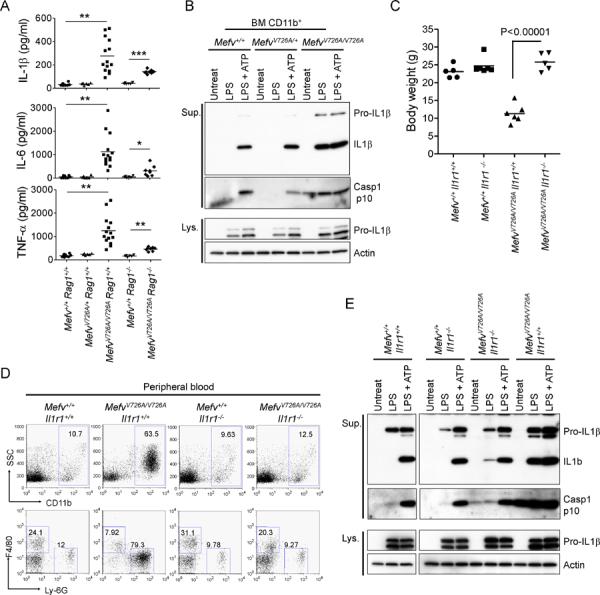
(A) Luminex analysis of serum cytokines of 8-week-old WT, MefvV726A/+, MefvV726A/V726A Rag1−/−, and MefvV726A/V726A Rag1−/− mice. *, P < 0.05; **, P < 0.0001; ***, P < 0.000001. (B) CD11b+ cells from BM of WT, MefvV726A/+, and MefvV726A/V726A mice were stimulated with LPS or no stimulus for 3 hr followed by treatment with or without ATP. Cell culture supernatants (Sup.) and cell lysates (Lys.) were analyzed by immunoblotting. (C–E) Analyses of MefvV726A/V726A mice on the Il1r1−/− background. (C) Body weights of 8-week-old males. (D) Flow cytometry analyses of peripheral blood cells from 8-week-old mice. Data are representative of three independent experiments. (E) Inflammasome activation in MefvV726A/V726A mice withIl1r1−/− background. Culture supernatants and lysates from BM CD11b+ cells were collected and subjected to immunoblot as described in (B). Additional information is provided in Figure S3.
To further investigate the role of IL-1β in the pathogenesis of KI mice in vivo, we crossed MefvV726A/V726A mice with IL-1 receptor-deficient (Il1r1−/−) mice to generate MefvV726A/V726AIl1r1−/− mice. The MefvV726A/V726AIl1r1−/− mice were grossly normal without any inflammatory phenotypes such as myeloid cell number expansion (Figures 4C and 4D). However the inflammasome of the BM CD11b+ cells was still activated and IL-1β was secreted from the cells with LPS alone (Figure 4E), suggesting that constitutive inflammasome activation may be a primary event in FMF KI mice. Taken together, these results demonstrate that IL-1β is a key soluble factor for inducing the inflammation in these mice and that IL-1β blockade ameliorates the disease.
The inflammasome is highly activated in the macrophages of FMF patients
Consistent with several case reports demonstrating the marked amelioration of symptoms in FMF patients upon treatment with recombinant IL-1 receptor antagonist (Belkhir et al., 2007; Bilginer et al., 2009; Calligaris et al., 2008; Chae et al., 2006; Kuijk et al., 2007; Moser et al., 2009; Roldan et al., 2008), the aforementioned mouse data suggest that the inflammatory symptoms of human FMF may also be triggered by IL-1β. However, there has been no direct evidence showing the constitutive activation of the inflammasome and subsequent high IL-1β production from myeloid cells of FMF patients. Thus, we first examined IL-1β secretion from peripheral blood mononuclear cells (PBMCs) of FMF patients who had mutations in the B30.2-domain of pyrin. Similar to the FMF KI mice, we could observe that, without ATP treatment, various Toll-like receptor (TLR) ligands could induce IL-1β secretion from PBMCs of most of the FMF patients tested (Figures S4A–S4C). However, some of the healthy controls also secreted IL-1β in response to LPS alone. Moreover, PBMCs from a few FMF patients secreted less IL-1β than a healthy control, though most FMF patients secreted higher amounts of IL-1β than healthy controls (Figure S4D). This inconsistency may be due to differences in pyrin expression that may in turn result from genetic or environmental factors, including treatment (Booty et al., 2009; Chae et al., 2008).
In order to minimize the variation coming from treatments and environmental factors, the PBMCs were first differentiated into macrophages in vitro, and IL-1β secretion was then measured (Figures 5A and 5B). In these experimental conditions, marked IL-1β and p20 secretion were observed in FMF patients but not healthy controls when the macrophages were stimulated with LPS followed by ATP treatment, whereas they were not secreted or secreted at low amounts by LPS alone. We also examined inflammasome activation from the macrophages in which pyrin expression was highly induced by IFN-γ treatment for 16 hrs prior to LPS stimulation. The increased expression of pyrin was verified in the IFN- γ treated macrophages from both patients and healthy controls, but, similar to the inflammasome activation in the FMF KI mice, IL-1β and p20 were secreted only from patients' macrophages by LPS stimulation alone without ATP treatment. These results suggest that mutant pyrin is involved in inflammasome activation to a much greater degree than WT pyrin.
Figure 5. Inflammasome activation in FMF patients.

Macrophages differentiated from PBMCs of (A) two healthy donors (control 8 and 9), two FMF patients (FMF 9, homozygote of M694V; FMF 10, compound heterozygote of V726A/E148Q), (B) two healthy donors (control 10 and 11), and two FMF patients (FMF 11, heterozygote of V726A; FMF 12, compound heterozygote of V726A/P369S) were treated with IFN-γ or no stimulus for 16 hr. Macrophages were treated as described in Figure 4B, and cell culture supernatants (Sup.) and cell lysates (Lys.) were analyzed by immunoblotting. Additional information is provided in Figure S4.
The adaptive immune system has no major role in the KI phenotype
Figures 2 and 3 indicate that the inflammatory phenotype of KI mice is initiated by hematopoietic stem cells, and that the myeloid lineage is probably critical for the pathogenesis of FMF. However, in KI mice, we could also observe an increased fraction of activated memory T cells (Figure S5), though the overall numbers of lymphocytes were relatively low in the LN and spleen of homozygous KI mice. Moreover the cytokines IL-2, IL-9, IL-17, and IFN-γ, which are known to be primarily produced by T cells, were also significantly increased in the sera of KI mice (Figure S3A). Thus, to test whether T cells have a role in the pathogenesis of the KI phenotype, we produced MefvV726A/V726A mice on the RAG1 -deficient background (MefvV726A/V726ARag1−/−). MefvV726A/V726ARag1−/− mice were runted with inflammatory phenotypes similar to MefvV726A/V726ARag1+/+ mice, and showed constitutive inflammasome activation in their myeloid cells comparable to what was observed in cells from MefvV726A/V726ARag1+/+ mice (Figures 6A–6D). Nevertheless, activated T-cells may still amplify the knockin phenotype, because several cytokines were significantly decreased in MefvV726A/V726ARag1−/− mice as compared to MefvV726A/V726ARag1+/+ mice, though the amounts were still higher than in WT mice (Figure S3).
Figure 6. Analyses ofMefvV726A/V726A mice on aRag1−/− background.

(A and B) Flow cytometry analyses of peripheral blood cells (A) and splenocytes (B) from 8-week-old mice analyzed as described in Figure 3C. Data are representative of three independent experiments. (C) Body weights of 8-week-old males. (D) Inflammasome activation in MefvV726A/V726A mice with Rag1−/− background. Culture supernatants and lysates from BM CD11b+ cells were analyzed by immunoblotting. Additional information is provided in Figure S5.
Inflammation in FMF KI mice is ASC-dependent but NLRP3-independent
Finally, we investigated the underlying mechanism for the inflammasome activation of the FMF KI mice in vivo. Because apoptosis-associated speck-like protein with a CARD (ASC) is the essential adaptor molecule among the components of most inflammasomes, and pyrin itself can interact with ASC through cognate PYRIN domain interactions, we crossed our MefvV726A/V726A mice with ASC-deficient mice (Asc−/−). MefvV726A/V726AAsc−/− mice showed normal growth without any inflammatory phenotype or myeloid cell expansion (Figures 7A and 7B). We could also observe the complete ablation of IL-1β secretion and caspase-1 activation from the BM CD11b+ cells of MefvV726A/V726AAsc−/− mice after LPS stimulation, even with ATP treatment, as seen with the cells from Asc−/− mice (Figure 7C), indicating that ASC is critical for the caspase-1 activation mediated by FMF-associated pyrin mutations.
Figure 7. FMF-associated B30.2 mutations activate an ASC-dependent but NALP3 independent pyrin inflammasome.

(A–C) Analyses of the FMF KI (MefvV726A/V726A) mice with Asc−/− background. (A) Flow cytometry analyses of peripheral blood cells from 8-week-old mice analyzed as in Figure 3C. Data are representative of three independent experiments. (B) Body weights of 8-week-old males. (C) Inflammasome activation in MefvV726A/V726A mice with Asc−/− background. Culture supernatants and lysates from BM CD11b+ cells were analyzed by immunoblotting. (D–E) Analyses of the FMF KI (MefvV726A/V726A) mice with Nlrp3−/− background. . (D) Inflammasome activation in MefvV726A/V726A mice with Nlrp3−/− background. Culture supernatants and lysates from BM CD11b+ cells were analyzed by immunoblotting. (E) Flow cytometry analyses of peripheral blood cells from 8-week-old mice analyzed as in Figure 3C. Data are representative of three independent experiments Additional information is provided in Figure S6.
Given the association of ASC with NLRP3 in the NLRP3 inflammasome, we asked whether the deficiency of NLRP3 would affect the FMF KI phenotype. As a control, we observed that the CD11b+ cells from NLRP3-deficient (Nlrp3−/−) mice were not able to secrete IL-1β in response to the LPS and ATP stimulation in vitro (Figure 7D). However, the NLRP3 deficient KI mice (MefvV726A/V726ANlrp3−/−) were runted and showed similar inflammatory phenotypes to the symptoms of KI mice expressing Nlrp3 (MefvV726A/V726ANlrp3+/+) (Figures 7B and 7E). Moreover we observed that the LPS stimulated CD11b+ cells of MefvV726A/V726ANlrp3−/− actively secreted IL-1β and p10, even without ATP treatment, whereas the cells of Mefv+/+Nlrp3−/− did not secrete IL-1β after LPS stimulation with ATP treatment (Figure 7D). These results demonstrate that NLRP3 is not necessary for the caspase-1 activation of the FMF KI mice.
We also examined other NLRP3-independent inflammasomes by introducing Salmonella typhimurium or double-stranded DNA into BMDM to interrogate NLRC4 (IPAF, ICE protease-activating factor) or AIM2 (absent in melanoma 2) inflammasome activation, respectively (Figures S6A–S6C). IL-1β secretion in these short-term assays was similar among WT, pyrin-deficient, and pyrin KI mice, which suggests that the NLRC4 and AIM2 inflammasomes do not markedly contribute to caspase-1 activation of the FMF KI mice. Conversely, data from pyrin-deficient mice demonstrate that pyrin is dispensable for NLRC4 and AIM2 inflammasome activation as well as NLRP3 activation by LPS and ATP. Nevertheless, macrophages from pyrin-deficient mice produced somewhat higher amounts of mature IL-1β compared with WT mice when stimulated with LPS alone for 1 to 3 days, as seen in our previous study of pyrin-truncation mice (Chae et al., 2003) (Figure S6D). Taken together with previously reported findings on the direct interaction of pyrin with ASC, our results provide evidence for a heretofore unrecognized ASC-dependent, pyrin inflammasome, which is activated in the presence of the FMF-associated mutations of pyrin.
DISCUSSION
In this manuscript we described the profound phenotype established by introducing FMF-associated mutations into the mouse germline. Consistent with the conception of FMF as an autoinflammatory disorder, the cells and molecules of the innate immune system are strongly implicated in the pathogenesis of disease in these animals. BM-derived cells are both necessary and sufficient to confer the phenotype, which includes a marked expansion of granulocyte numbers in the blood and peripheral lymphoid organs that persists on a Rag1−/− background. Extending earlier data implicating IL-1β in the pathogenesis of FMF, the disease phenotype is markedly attenuated in mice deficient for the IL-1 receptor, and additional crosses provided genetic evidence for an ASC-dependent, NLRP3-independent pyrin inflammasome.
Characterization of these pyrin KI mice provides yet another step in a paradigm-shift in our understanding of the genetics of FMF. Based on segregation analysis in Sephardi Jewish families with severe disease (Sohar et al., 1967), FMF has long been considered an autosomal recessive illness, and in fact the positional cloning studies leading to the identification of MEFV were based on this assumption. However, the availability of genetic testing has led both to the recognition of a biochemical phenotype in asymptomatic heterozygotes (Lachmann et al., 2006) and to the diagnosis of FMF in patients with relatively mild clinical phenotypes. As many as 30% of the patients diagnosed with FMF have only a single demonstrable mutation despite sequencing of the entire MEFV genomic region and several other known autoinflammatory genes (Booty et al., 2009; Marek-Yagel et al., 2009; Ozen, 2009). This observation, taken together with the fact that nearly all FMF-associated mutations are missense rather than null mutations, has led to a reconsideration of the simple loss-of-function recessive model of FMF inheritance.
The data presented here add substantially to the case for a gain-of-function model. In the current study, we have shown that the insertion of three different FMF-associated human B30.2 domains into mouse pyrin induced inflammatory phenotypes similar to or more severe than those seen in FMF patients, whereas deleting mouse pyrin produced no overt inflammatory phenotype. Neither heterozygous nor hemizygous KI mice exhibited the cardinal features seen in homozygotes, indicating a gene dosage effect. The fact that pyrin expression is cytokine-inducible suggests a possible feed-forward mechanism in which homozygous KI cells may express more pyrin at baseline, which in turn leads to inflammatory cytokine production and further increases in pyrin expression. Such a gene-dosage effect may also be operative in humans, in that higher pyrin protein expression has been observed in the leukocytes of FMF patients, compared with healthy controls (Booty et al., 2009; Chae et al., 2008).
It is possible that the disease phenotype observed in FMF KI mice is due not to the expression of the specific FMF-associated B30.2 pyrin mutations but rather to the expression of any pyrin that chimerizes the N-terminal mouse pyrin with the C-terminal human B30.2 domain. Absent the generation of knockin mice for the wild type human B30.2 domain, this will always remain formally possible. However, against this hypothesis, there is a genotype-phenotype relationship in the mice, with the severity of the phenotype in the mice inversely proportionate to the severity of the phenotype associated with the corresponding genotype in humans. A similar observation has recently been reported for NLRP3 KI mice (Brydges et al., 2009). Of possible relevance in the case of pyrin, mice do not express a C-terminal B30.2 domain (Chae et al., 2000). Thus, it is not totally surprising that mice harboring the wild type human B30.2 domain may express a phenotype even more severe (embryonic lethality) than the V726A homozygous genotype, which is associated with the mildest FMF phenotype in humans.
We present several lines of evidence that IL-1 plays a crucial role in the phenotype observed in mice homozygous for FMF-associated pyrin mutations. To date there are four different macromolecular inflammasomes, nucleated by NLRP1, NLRP3, NLRC4, and AIM2. The NLRP3 inflammasome has been shown to be activated by a wide range of pathogen-associated or danger-associated molecular patterns (PAMPs, DAMPs) and murine Nlrp3−/− cells do not produce any detectable amount of IL-1β in response to LPS and ATP in vitro (Mariathasan et al., 2006; Sutterwala et al., 2006). Nevertheless, we have found that the NLRP3 inflammasome has no role in the inflammation of pyrin. KI mice. In addition, we did not observe any differences in IL-1β secretion among WT, pyrin-deficient, and KI macrophages induced by double-stranded DNA or S. typhimurium, suggesting that the AIM2 or NLRC4 inflammasome is not involved in the inflammation of KI mice. The necessity of ASC for the inflammation of KI mice also excludes the NLRP1 inflammasome from the pathogenesis of FMF inflammation, because murine NLRP1 orthologs are predicted to be unable to interact with ASC due to lack of functional PYRIN domains. Indeed, ASC is dispensable for NLRP1-dependent caspase-1 activation in mouse macrophages (Hsu et al., 2008). Taken together, these data suggest a previously unrecognized ASC-dependent inflammasome in which mutant pyrin induces IL-1β activation.
An inhibitory role of pyrin in caspase-1 activation has been proposed in our previous study with a mouse model expressing a truncated form of pyrin (Chae et al., 2003). However, this interpretation has been debated since the pyrin-truncation mice express a functional N-terminal PYRIN domain that can interact with ASC. In the current study, we have shown that pyrin null macrophages produced higher amounts of mature IL-1β when stimulated with LPS alone for 1 to 3 days, although they did not show evidence of increased IL-1β activation in short-term assays of inflammasome activation. Moreover, neither pyrin-truncation nor deficient mice exhibit a spontaneous inflammatory phenotype. These results suggest a more subtle inhibitory role of pyrin that may be limited to non-inflammasome mediated IL-1β activation.
To test the hypothesis that pyrin mutations cause IL-1β activation in FMF patients, we cultured patient or control PBMCs for 7 days to minimize the effect of the patients' colchicine treatment, and then induced pyrin with interferon γ (Centola et al., 2000). In 4/4 FMF patients, but 0/4 controls, we observed caspase-1 activation and IL-1β secretion even without an ATP second signal. These data provide a scientific basis for IL-1 inhibition in the treatment of FMF, particularly in patients with life-threatening amyloid deposition.
Our data also established a conceptual basis for the possible development of hematopoietic stem cell therapy for refractory FMF. In reciprocal BM transfer experiments, we found that cells of the hematopoietic lineage are both necessary and sufficient for the FMF KI phenotype. In a patient with FMF who underwent bone marrow transplantation for congenital dyserythropoietic anemia, FMF symptoms completely resolved off colchicine treatment (Milledge et al., 2002). While the morbidity of allogeneic bone marrow transplantation generally makes this an unattractive alternative, the possibility of gene-correction with inducible pleuripotent stem cells may eventually modify this calculus.
FMF KI mice may also shed light on the role of pyrin variants in human history and evolution. The extraordinarily high carrier frequency of FMF-associated MEFV mutations in certain populations, coupled with the fact that different mutations predominate in geographically proximate ethnic groups, strongly suggests that heterozygous FMF mutations may confer a selective advantage against some as-yet unknown pathogen. Moreover, sequence comparisons of the C-terminal B30.2 doman of pyrin with non-human primates suggest episodic positive selection (Schaner et al., 2001). The fact that several disease-associated mutations are clustered around a putative binding pocket in the C-terminal B30.2 domain provides a possible structural basis for this hypothesis (Masters et al., 2009). It is thus possible that one of the functions of pyrin is to serve as an intracellular receptor for one or more PAMPs. FMF KI mice may serve as the experimental platform to test such hypotheses, thereby allowing us to probe these most fundamental questions regarding the role of pyrin and its mutants in human health and disease.
EXPERIMENTAL PROCEDURES
Mice
Generation of FMF KI mice
Using pPNT-double loxP as a construct backbone, the targeting constructs were generated by inserting a 6.0 kb KpnI/PvuII genomic fragment encompassing the end of exon 2 to exon 6 (5' arm) and a 2.5 kb PstI/SpeI genomic fragment (3' arm) (Figure S1A). For the KI of WT human B30.2 domain, a 1.7 kb genomic fragment of human MEFV encompassing exon 7 to 10 was inserted upstream of the 3' arm. The mutant genomic fragments with M680I, M694V, or V726A were also generated by site directed mutagenesis using QuickChange XL site-directed mutagenesis kit (Stratagene) and inserted in the upstream segment of the 3' arm. Linearized constructs were introduced into 129/SvEvTac-derived TC-1 embryonic stem (ES) cells. G418- and gancyclovir-resistant clones were screened for homologous recombination by long-range PCR and Southern blotting (Figure S1B). Heterozygotes generated from chimeras were crossed with EIIa-Cre transgenic mice (Jackson Laboratory) to remove the neo cassette and backcrossed with C57BL/6 mice at least 6 generations.
Generation of Pyrin-deficient mice
As shown in Figure S1C, the targeting constructs were generated by inserting a 4.7 kb genomic fragment upstream of exon 1 (5' arm) and a 6.0 kb genomic fragment encompassing exon 3 to exon 7 (3' arm). Using the linearized construct, pyrin KO mice were generated as described in the generation of KI mice, and backcrossed with C57BL/6 mice at least 6 generations.
Other mouse strains
Il1r1−/− and Rag1−/− mice were from the Jackson Laboratory. Asc−/− and Nlrp3−/− mice were a gift from Dr. Vishva M. Dixit (Genentech Inc.).
All animal studies were performed according to National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee of National Institute of Arthritis and Musculoskeletal and Skin Diseases.
Flow cytometry analysis
RBC-lysed single cell suspensions were obtained from LNs, spleen, BM, and peripheral blood, and stained with fluorochrome-conjugated antibodies in Pharmingen Stain Buffer (BSA) (BD Pharmingen, San Diego, CA). CD3 APC, CD19 FITC, CD11b PE, Ly-6G FITC, CD3 PerCP-Cy5.5, CD4 FITC, CD8a APC, CD25 PE, CD44 PE, CD62L PE, CD69 PE were purchased from BD Pharmingen. F4/80 Alexa647 Ab was purchased from Invitrogen (Carlsbad, CA). CD45.1 (Alexa488, Alexa647) and CD45.2 (Alexa488, Alexa647) Abs were purchased from Biolegend (San Diego, CA). Cells were analyzed on a FACSCaliber flow cytometer (BD Biosciences, San Jose, CA) and analyzed using FlowJo software (Three Star, Ashland, OR).
Cell culture and Immunoblots
Peritoneal macrophages were obtained by washing the peritoneum with RPMI medium. Mouse CD11b+ cells were isolated from BM using CD11b MicroBeads (Miltenyi Biotech, Auburn, CA). BMDM were obtained by differentiating BM cells with 10 ng/ml of M-CSF (Peprotech, Rocky Hill, NJ) for 7 days. Peritoneal macrophages were treated with 1 μg/ml of LPS (ultrapure, Invivogen, San Diego, CA), 12.5 ng/ml of IL-4 (Peprotech), or LPS/ IL-4. After 24 hr, cell lysates were collected. CD11b+ cells or BMDM were stimulated with LPS (1 μg/ml) or no stimulus for 3 hr. Cell culture media were changed with fresh media containing 5 mM ATP or media only. After 30 min, cell culture supernatants and cell lysates were collected.
Blood specimens from healthy controls and FMF patients were drawn after obtaining informed consent under a protocol approved by the NIAMS Institutional Review Board. PBMCs were isolated by Ficoll-Hypaque centrifugation, and also differentiated into macrophages using 800 units/ml of GM-CSF (Peprotech). Macrophages were treated with 20 ng/ml of IFN-γ (Peprotech) or no stimulus for 16 hr. Macrophages or PBMCs were stimulated with TLR ligands, LPS (1 μg/ml), Pam3CSK4 (1 μg/ml), or Gardiquimod™ (1 μg/ml) (Invivogen). After 3 hr, cell culture media were changed with fresh media, cells were incubated for 30 min, and cell culture supernatants and cell lysates were collected. Cell lysates or cell culture supernatants were subjected to SDS-PAGE for Western blot analysis using primary antibodies: anti-mouse pyrin Ab (Chae et al., 2003); anti-human pyrin Ab (Chae et al., 2006); anti-human IL-1β and mouse IL-1β Abs (R&D Systems, Minneapolis, MN); anti-mouse caspase-1, human caspase-1, and actin Abs (Santa Cruz Biotechnology, Santa Cruz, CA).
BMT
For routine BMT experiments, six to eight week old female mice were lethally (10 Gy) irradiated the day before BMT, and reconstituted by intravenous injection of 2μ106 (WT) or 5×106 (KI) RBC-lysed BM cells, unless otherwise specified. For the transfer of KI phenotypes into WT recipients, CD45.1+/+ C57BL/6 mice were used as recipients. For the rescue of inflammatory phenotypes of KI mice with WT BM cells, 10×106 BM cells from CD45.1+/+ C57BL/6 mice were transferred intraperitoneally into 3 to 4 week old MefvV726A/V726A mice that had been irradiated with two 3.5 Gy (2 hr interval, finished 2 hr before i.p. injection). For the mixed BMT, 2×106 CD45.1+/+ WT BM cells were pre-mixed with either 2×106 CD45.2+/+ WT or 5×106 CD45.2+/+ KI (MefvM680I/M680I) BM cells immediately before injection, and transferred to recipients heterozygous for both CD45.1 and CD45.2 antigens.
Measurement of cytokines, chemokines, and hematopoietic factors
Sera were obtained from 8-week-old mice and analyzed by Bio-Plex Mouse Cytokine 23-plex Assay (Bio-Rad, Hercules, CA) according to the manufacturer's instructions.
Statistical Analyses
Statistical analyses were performed with the Student's t-test.
Supplementary Material
Highlights
Pyrin mutant knockin, but not pyrin-deficient mice exhibit marked neutrophilic inflammation
There is constitutive inflammasome activation in knockin mice and FMF patients
Autoinflammation in knockin mice is abrogated on the Il1r1−/− background
Deficiency in ASC, but not NLRP3 or RAG1, abrogates autoinflammation in knockin mice
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Program of NIAMS, NCI, and NHGRI, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Belkhir R, Moulonguet-Doleris L, Hachulla E, Prinseau J, Baglin A, Hanslik T. Treatment of familial Mediterranean fever with anakinra. Ann. Intern. Med. 2007;146:825–826. doi: 10.7326/0003-4819-146-11-200706050-00023. [DOI] [PubMed] [Google Scholar]
- Bilginer Y, Ayaz NA, Ozen S. Anti-IL-1 treatment for secondary amyloidosis in an adolescent with FMF and Behcet's disease. Clin. Rheumatol. 2009 doi: 10.1007/s10067-009-1279-8. [DOI] [PubMed] [Google Scholar]
- Booty MG, Chae JJ, Masters SL, Remmers EF, Barham B, Le JM, Barron KS, Holland SM, Kastner DL, Aksentijevich I. Familial Mediterranean fever with a single MEFV mutation: where is the second hit? Arthritis Rheum. 2009;60:1851–1861. doi: 10.1002/art.24569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brydges SD, Mueller JL, McGeough MD, Pena CA, Misaghi A, Gandhi C, Putnam CD, Boyle DL, Firestein GS, Horner AA, et al. Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity. 2009;30:875–887. doi: 10.1016/j.immuni.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calligaris L, Marchetti F, Tommasini A, Ventura A. The efficacy of anakinra in an adolescent with colchicine-resistant familial Mediterranean fever. Eur. J. Pediatr. 2008;167:695–696. doi: 10.1007/s00431-007-0547-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centola M, Wood G, Frucht DM, Galon J, Aringer M, Farrell C, Kingma DW, Horwitz ME, Mansfield E, Holland SM, et al. The gene for familial Mediterranean fever, MEFV, is expressed in early leukocyte development and is regulated in response to inflammatory mediators. Blood. 2000;95:3223–3231. [PubMed] [Google Scholar]
- Chae JJ, Aksentijevich I, Kastner DL. Advances in the understanding of familial Mediterranean fever and possibilities for targeted therapy. Br. J. Haematol. 2009;146:467–478. doi: 10.1111/j.1365-2141.2009.07733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae JJ, Centola M, Aksentijevich I, Dutra A, Tran M, Wood G, Nagaraju K, Kingma DW, Liu PP, Kastner DL. Isolation, genomic organization, and expression analysis of the mouse and rat homologs of MEFV, the gene for familial mediterranean fever. Mamm Genome. 2000;11:428–435. doi: 10.1007/s003350010082. [DOI] [PubMed] [Google Scholar]
- Chae JJ, Komarow HD, Cheng J, Wood G, Raben N, Liu PP, Kastner DL. Targeted disruption of pyrin, the FMF protein, causes heightened sensitivity to endotoxin and a defect in macrophage apoptosis. Mol. Cell. 2003;11:591–604. doi: 10.1016/s1097-2765(03)00056-x. [DOI] [PubMed] [Google Scholar]
- Chae JJ, Wood G, Masters SL, Richard K, Park G, Smith BJ, Kastner DL. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc. Natl. Acad. Sci. USA. 2006;103:9982–9987. doi: 10.1073/pnas.0602081103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae JJ, Wood G, Richard K, Jaffe H, Colburn NT, Masters SL, Gumucio DL, Shoham NG, Kastner DL. The familial Mediterranean fever protein, pyrin, is cleaved by caspase-1 and activates NF-kappaB through its N-terminal fragment. Blood. 2008;112:1794–1803. doi: 10.1182/blood-2008-01-134932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz A, Hu C, Kastner DL, Schaner P, Reginato AM, Richards N, Gumucio DL. Lipopolysaccharide-induced expression of multiple alternatively spliced MEFV transcripts in human synovial fibroblasts: a prominent splice isoform lacks the C-terminal domain that is highly mutated in familial Mediterranean fever. Arthritis Rheum. 2004;50:3679–3689. doi: 10.1002/art.20600. [DOI] [PubMed] [Google Scholar]
- French FMF Consortium A candidate gene for familial Mediterranean fever. Nat. Genet. 1997;17:25–31. doi: 10.1038/ng0997-25. [DOI] [PubMed] [Google Scholar]
- Galon J, Aksentijevich I, McDermott MF, O'Shea JJ, Kastner DL. TNFRSF1A mutations and autoinflammatory syndromes. Curr. Opin. Immunol. 2000;12:479–486. doi: 10.1016/s0952-7915(00)00124-2. [DOI] [PubMed] [Google Scholar]
- Hsu LC, Ali SR, McGillivray S, Tseng PH, Mariathasan S, Humke EW, Eckmann L, Powell JJ, Nizet V, Dixit VM, Karin M. A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc. Natl. Acad. Sci. USA. 2008;105:7803–7808. doi: 10.1073/pnas.0802726105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International FMF Consortium Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell. 1997;90:797–807. doi: 10.1016/s0092-8674(00)80539-5. [DOI] [PubMed] [Google Scholar]
- Kastner DL, Aksentijevich I, Goldbach-Mansky R. Autoinflammatory disease reloaded: a clinical perspective. Cell. 2010;140:784–790. doi: 10.1016/j.cell.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijk LM, Govers AM, Frenkel J, Hofhuis WJ. Effective treatment of a colchicine-resistant familial Mediterranean fever patient with anakinra. Ann Rheum. Dis. 2007;66:1545–1546. doi: 10.1136/ard.2007.071498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann HJ, Sengul B, Yavuzsen TU, Booth DR, Booth SE, Bybee A, Gallimore JR, Soyturk M, Akar S, Tunca M, Hawkins PN. Clinical and subclinical inflammation in patients with familial Mediterranean fever and in heterozygous carriers of MEFV mutations. Rheumatology (Oxford) 2006;45:746–750. doi: 10.1093/rheumatology/kei279. [DOI] [PubMed] [Google Scholar]
- Marek-Yagel D, Berkun Y, Padeh S, Abu A, Reznik-Wolf H, Livneh A, Pras M, Pras E. Clinical disease among patients heterozygous for familial Mediterranean fever. Arthritis Rheum. 2009;60:1862–1866. doi: 10.1002/art.24570. [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu. Rev. Immunol. 2009;27:621–668. doi: 10.1146/annurev.immunol.25.022106.141627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milledge J, Shaw PJ, Mansour A, Williamson S, Bennetts B, Roscioli T, Curtin J, Christodoulou J. Allogeneic bone marrow transplantation: cure for familial Mediterranean fever. Blood. 2002;100:774–777. doi: 10.1182/blood-2002-02-0651. [DOI] [PubMed] [Google Scholar]
- Moser C, Pohl G, Haslinger I, Knapp S, Rowczenio D, Russel T, Lachmann HJ, Lang U, Kovarik J. Successful treatment of familial Mediterranean fever with anakinra and outcome after renal transplantation. Nephrol. Dial. Transplant. 2009;24:676–678. doi: 10.1093/ndt/gfn646. [DOI] [PubMed] [Google Scholar]
- Ozen S. Changing concepts in familial Mediterranean fever: is it possible to have an autosomal-recessive disease with only one mutation? Arthritis Rheum. 2009;60:1575–1577. doi: 10.1002/art.24565. [DOI] [PubMed] [Google Scholar]
- Papin S, Cuenin S, Agostini L, Martinon F, Werner S, Beer HD, Grutter C, Grutter M, Tschopp J. The SPRY domain of Pyrin, mutated in familial Mediterranean fever patients, interacts with inflammasome components and inhibits proIL-1beta processing. Cell Death Differ. 2007;14:1457–1466. doi: 10.1038/sj.cdd.4402142. [DOI] [PubMed] [Google Scholar]
- Roldan R, Ruiz AM, Miranda MD, Collantes E. Anakinra: new therapeutic approach in children with Familial Mediterranean Fever resistant to colchicine. Joint Bone Spine. 2008;75:504–505. doi: 10.1016/j.jbspin.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Schaner P, Richards N, Wadhwa A, Aksentijevich I, Kastner D, Tucker P, Gumucio D. Episodic evolution of pyrin in primates: human mutations recapitulate ancestral amino acid states. Nat. Genet. 2001;27:318–321. doi: 10.1038/85893. [DOI] [PubMed] [Google Scholar]
- Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- Sohar E, Gafni J, Pras M, Heller H. Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am. J. Med. 1967;43:227–253. doi: 10.1016/0002-9343(67)90167-2. [DOI] [PubMed] [Google Scholar]
- Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ, Galan JE, Askenase PW, Flavell RA. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24:317–327. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Touitou I, Lesage S, McDermott M, Cuisset L, Hoffman H, Dode C, Shoham N, Aganna E, Hugot JP, Wise C, et al. Infevers: an evolving mutation database for auto-inflammatory syndromes. Hum. Mutat. 2004;24:194–198. doi: 10.1002/humu.20080. [DOI] [PubMed] [Google Scholar]
- Yu JW, Fernandes-Alnemri T, Datta P, Wu J, Juliana C, Solorzano L, McCormick M, Zhang Z, Alnemri ES. Pyrin activates the ASC pyroptosome in response to engagement by autoinflammatory PSTPIP1 mutants. Mol. Cell. 2007;28:214–227. doi: 10.1016/j.molcel.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JW, Wu J, Zhang Z, Datta P, Ibrahimi I, Taniguchi S, Sagara J, Fernandes-Alnemri T, Alnemri ES. Cryopyrin and pyrin activate caspase-1, but not NF-kappaB, via ASC oligomerization. Cell Death Differ. 2006;13:236–249. doi: 10.1038/sj.cdd.4401734. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.