

Abstract
Nucleic acid-based diagnostics are highly sensitive and specific, but are easily disrupted by the presence of interferents in biological samples. In a laboratory or hospital setting, the influence of these interferents can be minimized using an RNA or DNA extraction procedure prior to analysis. However, in low resource settings, limited access to specialized instrumentation and trained personnel presents challenges that impede sample preparation. We have developed a self-contained nucleic acid extraction cassette suitable for operation in a low resource setting. This simple design contains processing solutions preloaded within a continuous length of 1.6 mm inner diameter Tygon tubing. Processing solutions are separated by air gaps and held in place during processing by the surface tension forces at the liquid-air interface, viz. surface tension valves. Nucleic acids preferentially adsorbed to silica-coated magnetic particles are separated from sample interferents by using an external magnet to transfer the nucleic acid biomarker through successive solutions to precipitate, wash and elute in the final cassette solution. The efficiency of the extraction cassette was evaluated using quantitative reverse transcriptase PCR (qRT-PCR) following extraction of respiratory syncytial virus (RSV) RNA. RNA was recovered from TE buffer or from lysates of RSV infected HEp-2 cells with 55 and 33% efficiency, respectively, of the Qiagen RNeasy kit. Recovery of RSV RNA from RSV infected HEp-2 cells was similar at 30% of the RNeasy kit. An overall limit of detection after extraction was determined to be nearly identical (97.5%) to a laboratory-based commercially available kit. These results indicate that this extraction cassette design has the potential to be an effective sample preparation device suitable for use in a low resource setting.
Keywords: sample preparation, low resource, nucleic acid diagnostics, RNA extraction, RNA-silica adsorption, RNA purification, respiratory syncytial virus, surface tension valve
1. Introduction
Recent research has focused on the development of nucleic acid-based detection for low resource settings.1 Nucleic acid-based detection systems, such as quantitative PCR (qPCR), are particularly attractive technologies for detection of pathogens because of their sensitivity, specificity and relatively rapid time-to-answer. The effectiveness of PCR is dependent on both the quality and quantity of nucleic acid template2 and the absence of interferents.3 For example, carbohydrates, proteins, lipids or other unidentified interferents present in clinical samples have all been shown to inhibit PCR and produce false negatives.4–6 In addition to various interferents, patient samples also contain nucleases, which directly reduce the number of nucleic acid targets present.5
To minimize false negatives and maximize the efficiency of nucleic acid-based diagnostics, nucleic acids are extracted and concentrated into an interferent-free buffer prior to testing. One classic laboratory method uses a phenol-chloroform cocktail.7 This method is highly effective, but is not as commonly utilized today because it is time consuming and requires the use of toxic organic chemicals. Several solid phase extraction kits are commercially available to purify DNA or RNA from patient samples. Many of these kits rely on selective nucleic acid binding to silica-coated surfaces in the presence of ethanol and a chaotropic agent such as guanidinium thiocyanate (GuSCN).8, 9 GuSCN also denatures protein contaminants including nucleases that may be present in the sample.10, 11 These kits are not cost effective for low resource use and often require the use of specialized laboratory equipment, such as a robot or centrifuge, and trained technicians that are unavailable in a low resource setting. Additionally, many involve multiple steps that increase the chance of contamination of both the sample and operator.
Microfluidics is one promising format for low resource nucleic acid-based diagnostics. Recently, there has been a growing interest in expanding microfluidic technologies for sample preparation.1, 12 Many of these devices are suitable for integrating with downstream nucleic acid amplification and detection technologies.13, 14 However, the small surface area of solid phase available for nucleic acid binding and the limited sample volume that can be flowed through the channels limit the total mass of nucleic acid recovered,1 and therefore negatively impact the limit of detection.
We have developed an alternative nucleic acid extraction cassette suitable for operation in a low resource setting. This self-contained extraction cassette is preloaded with processing solutions separated by air gaps, which we refer to as “surface tension valves.” In proof-of-principle RNA extraction studies, RSV infected cells are lysed and viral RNA is selectively adsorbed to silica-coated magnetic particles in the presence of GuSCN and ethanol. Individual processing solutions are preloaded into a single continuous length of Tygon tubing and are separated from one another and held in places by surface tension forces. Removal of interferents is achieved by selective RNA adsorption to silica-coated magnetic particles which are then pulled through each processing solution using an externally applied magnetic field. RNA is eluted from the surface of the magnetic particle in the final solution. This report describes the general characteristics of this approach and compares its performance to laboratory-based commercial kits.
2. Materials and Methods
2.1 Preparation of RSV N gene RNA standards
Because our laboratories have experience with respiratory syncytial virus (RSV) detection, we have chosen to develop our extraction cassette using RSV RNA. Escherichia coli DH5α transformed with the pGBKT7 vector containing RSV N gene was generously provided by the Crowe Laboratory at Vanderbilt University. E. coli were grown for 18 hours on kanamycin agar plates at 37 °C. A single colony was isolated and transferred into 25 mL of Miller’s LB broth with 50 ug/mL kanamycin antibiotics and grown overnight on a rotating rack at 37 °C to an optical density of 0.6–0.8 AU. The plasmid was extracted using a Qiagen Spin Miniprep Kit and linearized using the BssHII restriction enzyme. Linearization was confirmed by running both pre- and post-linearized plasmids on a 1% agarose gel. Linearized plasmid was recovered from the restriction digest by ethanol precipitation. The plasmid was then transcribed into RNA using a T7 MEGAscript transcription kit (Ambion, Austin, TX), and treated with DNase I. The expected RNA length was confirmed on a denaturing 2%-formaldehyde-1.2% agarose gel. The RNA was quantified by UV-Vis spectroscopy.
2.2 Preparation of RSV infected and uninfected HEp-2 cell lysates
Uninfected HEp-2 cell lysates were prepared from a confluent monolayer of HEp-2 cells from a T-150 flask. The cells were harvested by scraping from the T150 flask and centrifuging at 500 × g for 5 minutes. The pellet was resuspended into 8 mL denaturing solution (4 M guanidinium thiocyanate, 25 mM sodium citrate [pH 7.0] 0.5% N-laurosylsarcosine [Sarkosyl], 0.1 M 2-mercaptoethanol) and passed through a pipette tip 10 times. The cell lysates were stored at a concentration of approximately 3 × 106 lysed cells per mL in 1 mL aliquots at −80 °C.
Infected HEp-2 cell lysates were prepared by infecting confluent monolayer of HEp-2 cells in two T150 flasks with RSV strain A2. After 4 days, one flask was harvested as described above and used to perform RSV RNA extractions from HEp-2 lysates. A plaque assay was performed on the second flask to quantify the concentration of infectious particles. To prepare the assay, cells were scraped from the T150 flask and centrifuged at 500 × g for 5 minutes. The pellet was resuspended into 8 mL of media, and cells were lysed by 3 cycles of freezing in an ethanol and dry ice slurry and thawing in a 37 °C water bath. The cell lysate was centrifuged at 100 × g for 5 minutes, and the supernatant was stored at −80 °C in 1 mL aliquots.
One hundred uL of the lysed cells was serially diluted, and each dilution added in triplicate to a confluent monolayer of HEp-2 cells in a 24-well plate. Plates were incubated at 37 °C for 1 hour. One mL of sterile 0.75% methyl cellulose (w/v) was then added to each well and the plate was placed at 37 °C for an additional 4 days. The infected HEp-2 cells were fixed in 80% methanol at −20 °C for 1 hour, washed 3 times with PBS, and blocked with a 5% milk solution for 1 hour. One hundred fifty μL of 30 μg/mL anti-F protein primary antibody in 5% powdered milk solution was added to each well. After 1 hour, wells were washed 3 times with PBS, and 150 μL of 0.5 μg/mL anti-mouse IgG HRP conjugate secondary antibody (Promega, Madison, WI) in 5% powdered milk solution was added for 1 hour. Wells were washed 5 times with PBS and 150 μL of TrueBlue peroxidase substrate (KLP, Gaithersburg, MD) added for 20 minutes at room temperature. Punctate blue plaques were counted and averaged, and the plaque forming units (pfus) were determined by multiplying by the dilution factor.
2.3 Conversion of RSV N gene RNA copy number to pfu/mL reported in clinical literature
The number of pfu/mL of the RSV infected HEp-2 cell lysates was converted to RSV N gene copies/uL by comparing qRT-PCR and plaque assay results on each half of a split culture of infected Hep-2 cells harvested 4 days post-infection, the point at which peak titers are reached. The concentration of N gene RNA in copies/uL was determined by RT-PCR using a standard curve following RNA extraction with the RNeasy kit. A calculated extraction efficiency of 18.1% (see section 3, Figure 4B) was assumed to determine the total number of RNA copies present in the sample prior to extraction. The number of N gene copies/pfu was approximated by dividing the total number of RNA copies/uL by the number of pfu/mL and determined to be ~104 copies/pfu.
Figure 4.

Comparison of the percent of RSV RNA recovered after addition to TE buffer (A) or HEp-2 cell lysates (B) using the extraction cassette (left bars), RNeasy kit (middle bars), or no extraction (right bars) (mean ± s.d., n = 9). The recovery efficiency of the cassette was 55% and 42% of the RNeasy kit from TE buffer and HEp-2 cell lysates, respectively.
2.4 Quantitative RT-PCR
An 82-bp fragment of the RSV N gene was amplified using forward primer 5′-GCTCTTAGCAAAGTCAAGTTGAAATGA-3′ and reverse primer 5′-TGCTCCGTTGGATGGTGTATT-3′.15 Reactions were performed in a 25 μL volume using 5 μL of RNA template and the Clontech one-step RT-PCR kit according to manufacturer’s instructions. Thermal cycling consisted of 48 °C for 20 minutes to synthesize cDNA, 95 °C for 3 minutes to inactivate the reverse transcriptase and activate QTaq DNA polymerase, and 40 cycles of 95 °C for 15 s and 60 °C for 60 s using a Rotor-Gene Q thermal cycler (Qiagen, Germantown, MD). Product specificity was confirmed using melting curve analysis and gel electrophoresis. Data was collected and Ct values recorded by Rotor-Gene Q Software (Qiagen, Germantown, MD) and converted to number of copies of RNA per μL using a standard curve.
2.5 RNA extraction using prototype capillary extraction cassette
A prototype extraction cassette (Figure 1) was constructed from glass capillary tubes and pipette tips. Glass capillary chambers (2 mm i.d.) were cut from stock tubing into 80 mm lengths, and the ends were flared outward. Six capillary chambers were aligned linearly on the top of a horizontal aluminum stage using machined aluminum mounts. A 1000 μL pipette tip was placed as a spacer in between each capillary chamber with the wide end of the pipette tip around the preceding capillary chamber and the narrow end resting inside the flared region of the next capillary chamber. Thus successive processing chambers were separated from one another by air spacers within the pipette tips. The first capillary chamber was reserved for the RNA sample and was initially left empty. The remaining chambers were preloaded with the processing reagents supplied in the MagAttract RNA Cell Mini M48 kit (Qiagen, Germantown, MD) as follows: 200 μL of “Buffer MW” wash buffer, 200 μL “Buffer RPE” wash buffer, 200 μL “Buffer RPE” wash buffer, 200 μL RNase/DNase free water, 30 μL RNase/DNase free water heated to 65 °C for elution of RNA. Thirty μL of sample was added to 150 μL of “Buffer RLT” and homogenized by passage through a 20-gauge needle five times. Twenty μL of the MagAttract bead solution (Qiagen, Germantown, MD) was added to the homogenized sample, vortexed, and placed on a rotary mixer for 5 minutes at room temperature. The sample was then pipetted into the first chamber, shown on the left in Figure 1. A 2.54 cm cube of grade 40 NdFeB magnet (National Imports, Vienna, VA) was placed adjacent to the first capillary chamber and slowly pulled parallel to the chambers at a rate of ~4 mm/second to pull the magnetic beads through each of the processing chambers. The total pull-through time was ~2 minutes. After reaching the final elution chamber, the beads were held to one side by the magnet and the eluent was collected.
Figure 1.
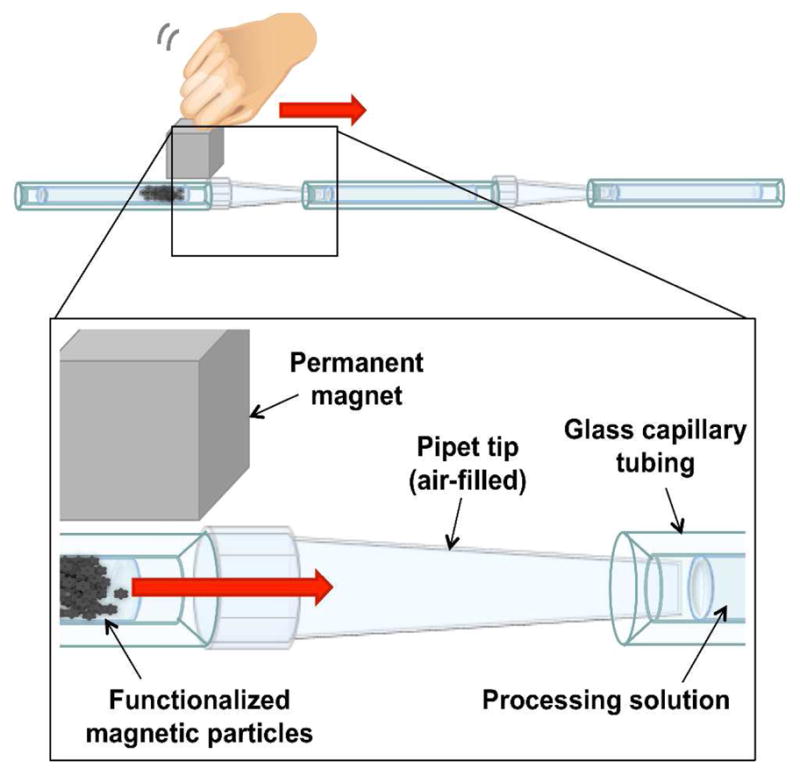
Design of the prototype extraction method showing the processing solutions held in place in glass tubing and separated by air-filled pipette tips. RNA is adsorbed to silica-coated magnetic particles which are pulled left to right through successive processing chambers using an external magnet. Following processing, the RNA is eluted in a final water chamber.
This initial design was used to perform proof-of-principle studies using 14 frozen de-identified nasal wash samples provided by Dr. John Williams’ lab (Vanderbilt University Hospital, Nashville, TN). Use of specimens was approved by Vanderbilt University’s IRB. At the time of collection, nasal swabs were placed in opti-MEM media (Invitrogen, Oslo, Norway) and frozen at −80 °C. Each sample was characterized by Williams’ lab for respiratory syncytial virus (RSV) using RT-PCR after an extraction using Roche Total Nucleic Acid Extraction Kit (Basel, Switzerland). RT-PCR was performed using Roche LC Magna Pure machine (Basel, Switzerland). We obtained samples which tested positive for RSV as determined by a calculated cycle threshold (Ct). We also obtained samples which tested negative for RSV as determined by no calculated cycle threshold value within the cycles that were performed. Seven samples characterized as RSV positive and seven as RSV negative were selected at random. Frozen samples were briefly thawed, divided and refrozen as 100 μL aliquots to facilitate comparison across different RNA extraction methods.
The number of extracted RSV N gene RNA copies/μL was calculated for the 7 RSV positive and 7 RSV negative nasal wash samples after 4 different extraction methods using a standard curve. The recovery efficiency of RNA extraction was compared to the RNeasy Mini kit (Qiagen, Germantown, MD), Dynabeads mRNA Direct kit (Invitrogen, Oslo, Norway) used according to manufacturer’s protocols, as well as the MagAttract RNA Cell Mini M48 kit (Qiagen, Germantown, MD) performed manually instead of with the Qiagen M48 BioRobot which was unavailable for these studies. The results were also compared to the calculated copy numbers of RSV N gene RNA detectable in each sample directly amplified by RT-PCR prior to extraction. For each extraction method utilized, RNA was eluted in a 50 μL volume to ensure that RT-PCR analysis was comparable across all extraction methods tested.
2.6 RNA extraction from TE buffer and HEp-2 cell lysates using continuous tubing extraction cassette
The initial prototype design of Figure 1 was further simplified into a continuous tubing design shown in Figure 2. In this design, 8 processing solutions were preloaded within ~61 cm length of Tygon tubing (1.6 mm i.d.). These solutions were chaotropic wash buffer (300 μL of 4 M guanidine hydrochloride, 25 mM sodium citrate, pH 7.0), two sections containing RNA precipitation buffer (300 μL of 80% ethanol, 5 mM potassium phosphate, pH 8.5,), three sections containing a water wash (100 μL of molecular grade water), and RNA elution (50 μL of molecular grade water). The 50 μL elution volume was chosen so that the RT-PCR input would be comparable to other extraction methods such as the RNeasy kit. Each solution was separated from the next by an air gap ~2 mm in length. Three types of extraction test samples were prepared: 5 μL of RSV N gene standard RNA in TE buffer at a concentration of 1 × 106 copies/μL, 20 μL of HEp-2 cell lysates (2 × 103 cells/μL) spiked with 5 μL of RNA standard, or 20 μL of RSV infected HEp-2 cell lysates. Cell lysate samples were homogenized by passage through a 25 gauge needle five times. Prior to extraction, samples were added to 230 μL of RNA-silica binding buffer (230 μL of 2 M guanidine thiocyanate, 25 mM sodium citrate, pH 7.0, 50% ethanol) and 20 μL of silica-coated 1 μm diameter magnetic particles (3 × 106 particles/μL) (Bioneer Inc., Alameda, CA) and placed on a rotating mixer for 5 minutes at room temperature. After mixing, each sample was loaded into the tubing, and the tubing ends were capped. The particles were collected in the first chamber by the external magnet and pulled through the surface tension valves and each successive chamber at ~4 mm/second using ~5 cm diameter neodymium ring magnet (Emovendo LLC, Petersburg, WV) as depicted in Figure 2. Particles were dispersed in the chaotropic wash and RNA precipitation solutions by rapidly moving the magnet back and forth before being recollected. In the water wash solutions, the particles were moved at ~8 mm/second to minimize RNA loss by elution during the wash. Finally, the particles were dispersed in the final elution chamber and incubated at room temperature for 5 minutes before removal. Although it was utilized in the prototype design, the elution of RNA at 65 °C was not performed in this final design because it would be impractical in most low resource settings. The final chamber contents were collected for RT-PCR analysis. Each RNA extraction was completed in ~15 minutes.
Figure 2.

Design of the continuous tubing extraction cassette showing individual processing solutions separated by surface tension valves. An external magnet is used to pull RNA adsorbed to silica-coated magnetic particles through each processing solution. Following processing, the RNA is eluted in a final water chamber.
2.7 Continuous tubing extraction cassette limit of detection
The extraction cassette limit of detection was determined by calculating the minimum quantity of target RNA that must be added to RSV negative cell lysates to be detectable by RT-PCR following extraction. This value was compared to the limit of detection calculated for the RNeasy kit. Twenty uL of uninfected HEp-2 cell lysate was spiked with 5 μL of RNA in TE buffer containing 0, 5 × 103, 5 × 104, 1 × 105, 5 × 105, 1 × 106, and 5 × 106 copies of RSV N gene RNA standard and extracted by both methods as described in section 2.6. After extraction, the RNA was quantified by RT-PCR. The limit of detection was defined as 3 s.d. above the mean value obtained for control extractions containing no RNA.
2.8 Post-extraction RNA distribution analysis
Extraction test samples were prepared using 5 μL of RSV RNA standard in TE buffer added to 230 μL of silica binding buffer. Twenty μL of magnetic particles were added to the sample and mixed for 5 minutes. RNA was extracted using the extraction cassette as described in section 2.6. After extraction, each chamber solution was removed by cutting the tubing with a razor blade and collecting in a separate tube. Each solution was purified with the RNeasy Mini kit according to manufacturer’s protocol in order to remove PCR inhibitors. To account for RNA loss during the secondary RNA purification step, a control containing 5 μL of RSV RNA standard was purified from TE buffer using the RNeasy kit. The purified RNA was quantified by RT-PCR analysis and normalized to the TE buffer control to account for loss during this second extraction. RNA remaining on magnetic particles after extraction was determined by recollecting the particles post-elution in 100 μL of nuclease free water. The particles were placed on a rotary mixer for 12 hours at 4 °C. Particles were removed and RNA in solution was purified with the RNeasy Mini kit and quantified by RT-PCR.
3. Results
RNA extraction from aliquots of frozen nasal wash samples using the prototype extraction cassette, shown in Figure 1, recovered on average 510 ± 800 RSV RNA copies per μL (Figure 3 dark bars). Using aliquots from the same sample, the commercial RNeasy Mini kit, Dynabeads mRNA Direct kit and MagAttract RNA Cell Mini M48 kit recovered 4,400 ± 10,000, 750 ± 1,300, and 940 ± 1,000 copies per μL, respectively. In an unextracted RSV positive nasal wash sample, 3 ± 3 RSV RNA copies were detected. In samples previously classified as RSV negative, an average of less than 1 copy of RSV RNA was detectable per μL for all methods (Figure 3, light bars).
Figure 3.

The number of copies of RNA per μL extracted from RSV positive (black bars) and RSV negative (gray bars) nasal wash samples using five extraction methods were compared. Extractions were performed using prototype extraction cassette, RNeasy Mini kit, Dynabeads mRNA Direct kit, and the MagAttract RNA Cell Mini M48 kit (mean ± s.d., n = 7).
Motivated by these encouraging preliminary results using the relatively crude prototype shown in Figure 1, a more thorough study was undertaken using a simplified continuous tubing design shown in Figure 2. Due to the high sample-to-sample variability and limited number of the frozen nasal wash samples available, further testing was done with RSV infected HEp-2 cell lysates, which unlike the unknown patient samples had no sample-to-sample variation. Using the continuous tubing extraction cassette shown in Figure 2, extraction of an RSV N gene standard added to TE buffer was recovered at an efficiency of 22.5 ± 19% (1.1 ± 0.95 × 106 copies) (Figure 4A). Recovery efficiency was calculated by dividing the total number of copies extracted by the initial number of copies present in the sample and multiplying by 100%. In TE buffer, the RNeasy kit recovered 41 ± 19% (2.1 ± 0.95 × 106 copies). TE buffer does not contain PCR interferents so, as expected, the detection of unextracted standard RNA was 100% (Figure 4A, right bar).
In the more complex uninfected HEp-2 cell lysate sample matrix, the recovery efficiency of RNA was 7.6 ± 4.8% (3.8 ± 0.24 × 105 copies) using the extraction cassette, and 18.1 ± 2.4% (9.1 ± 1.2 × 105 copies) using the RNeasy kit (Figure 4B). The spiked cell lysates evidently contained RT-PCR interferents since there was no amplification of the unextracted spiked sample by RT-PCR (Figure 4B, right bar).
Using the continuous tubing extraction cassette, RSV RNA extracted from RSV infected HEp-2 cell lysates containing 4.6 × 105 pfu/mL recovered 3.6 ± 0.09 × 105 RNA copies per μL from the elution chamber compared to 1.2 ± 0.07 × 106 copies per μL using the RNeasy kit (Figure 5, black bars). Less than 100 copies/μL was reported in extractions obtained from uninfected cell lysates (Figure 5, gray bars), and RNA was not detectable for infected or uninfected cell lysates which were not extracted prior to RT-PCR (Ct > 40) (Figure 5, “Unextracted”).
Figure 5.

Comparison of RNA extracted from RSV infected (black bars) and uninfected (gray bars) HEp-2 cell lysates using the extraction cassette and RNeasy kit. Unextracted samples failed to report RSV RNA in either sample (mean ± s.d, n = 3).
For all methods, RNA loss during extraction was significant. A post-extraction examination of the distribution of RNA in the processing solutions was successful in accounting for some of this loss. In a separate series of experiments using the continuous tubing extraction cassette, we found that only 59.5% (3.0 × 106 copies) of RNA could be accounted for in a post-processing distribution analysis of RSV N gene standard added to TE buffer (Figure 6). Similar to the results found in Figure 4A, 28 ± 4.5% (1.4 ± 0.23 × 106 copies) of the RNA was recovered in the elution. Significant RNA was recovered in the water wash solutions, which contained 21.7 ± 4.6% (1.1 ± 0.23 × 106 copies) of the initial RNA. An additional 7.8 ± 3.5% (3.9 ± 1.8 × 105 copies) of the RNA was recovered from the silica particles after 12 hours of further elution at 4 °C. Less than 2% of the RNA was recovered in the RNA-silica binding, chaotropic wash, and RNA precipitation solutions. The tube wall was also checked for RNA binding by washing with water post-extraction, and no detectable RNA could be recovered (data not shown). Approximately 40.5% (2.0 × 106 copies) was either lost or degraded during processing.
Figure 6.

The post-extraction distribution of RNA in each processing solution after RNA extraction from TE buffer is shown. Insignificant amounts were recovered in the first three steps, but the water wash and silica particles contained significant RNA (mean ± s.d., n = 3).
The post-extraction limit of detection was established for the continuous tubing extraction cassette by determining how many RSV RNA copies had to be added to a HEp-2 cell lysate to produce a detectable signal following RNA extraction and RT-PCR. Five thousand copies of RSV RNA spiked into HEp-2 cell lysates (e.g. 5 uL of RNA at 1,000 copies/uL into 20 uL of cell lysate) was the lowest concentration detectable by RT-PCR after sample extraction using both the continuous tubing extraction cassette and RNeasy kit (Figure 7). For the extraction cassette, 197 ± 8.5 copies were reported in the sample containing no RSV RNA, resulting in a 3 s.d. limit of detection of roughly 222 copies. Cell lysates spiked with 5,000 copies prior to extraction reported a value of 228 ± 58.5 copies per PCR reaction. Similarly, 202 ± 9.5 copies were reported in the sample containing no RSV RNA, and 312 ± 26.8 copies from lysates spiked with 5,000 copies and extracted with the RNeasy kit.
Figure 7.

The limit of detection of RNA detectable by RT-PCR after extraction from HEp-2 cell lysates spiked with known amounts of RSV RNA using either the continuous tubing extraction cassette (●) or the RNeasy kit (○) (mean ± s.d, n=3). When a sample containing no copies of RNA was extracted, 197 ± 8.5 RNA copies were detected with the extraction cassette and 202 ± 9.5 copies were detected with the RNeasy kit. The limit of detection is shown for the continuous tubing extraction cassette (dotted line). Comparable data from Figs 4B ( ) and 5 (△), which were performed at a single concentration are also included.
4. Discussion
Preparation of patient samples is necessary to avoid false negative prior to nucleic acid-based testing.6 Sample preparation techniques mirroring the simple low resource nucleic acid-based diagnostic devices currently being developed are necessary to make diagnosis practical at the point-of-care. Unfortunately, the operation of most existing commercial kits appropriate for RNA extraction and concentration require specialized laboratory equipment and trained laboratory personnel not available in a low resource setting.
In agreement with previous studies6, we found that without an initial extraction step, only purified RNA in solutions containing no interferents (e.g. TE buffer) can be directly detected by RT-PCR (Figure 4A). Direct amplification of viral RNA by RT-PCR prior to RNA extraction failed to detect viral RNA in RSV infected clinical nasal wash samples (Figure 3), HEp-2 cell lysates spiked with RSV RNA (Figure 4B) and RSV infected HEp-2 cell lysates (Figure 5). Therefore, RSV false negatives are likely to be obtained when the extraction step is omitted prior to RT-PCR, and sample preparation is necessary to make an accurate diagnosis.
A post-extraction limit of detection study was performed using spiked cell lysate samples, and the results suggest that the proposed continuous tubing extraction cassette and the RNeasy kit have a similar limit of detection of ~200 copies per μL. The limit of detection of current clinical diagnostics is ~104 pfu/mL.16 By dividing an RSV infected culture into two parts and measuring pfu/mL by traditional methods and by copies/uL RT-PCR (see section 2.3), it was determined that 104pfu/mL corresponds to ~105 copies/uL. Therefore, RT-PCR following extraction yields ~50-fold improvement in the limit of detection. In combination with a point-of-care nucleic acid-based diagnostic, the proposed extraction cassette would be ideal in a low resource setting, and with only a two-fold improvement in the extraction process or optimization of the RT-PCR, lower limits at or below the RSV infectious dose 50 (dose that will infect 50% of subjects, or ~100 copies per μL)16 are likely achievable.
Currently, there are no commercially available low resource nucleic acid extraction devices for comparison to the proposed method. However, several laboratory-based commercial kits are available, and we compared the proposed low resource method to these approaches. We have utilized a quantitative method for evaluating and directly calculating the efficiency of the extraction cassette in order to simplify comparison of the device to the other methods used in this study. Despite its simplicity, the extraction cassette isolated between 30 and 55% of the Qiagen RNeasy kit. Therefore, using RT-PCR, the Ct values for the extraction cassette would fall within ~1 cycle of the RNeasy kit. As shown in Figure 4, ~22.5% of the total RNA input is recovered by the current design under idealized conditions (spiked TE buffer). More complex sample matrices such as cell lysates or nasal wash samples evidently contain components that inhibit RT-PCR and/or make RNA recovery more difficult. All extraction methods tested had lower extraction efficiencies when used to extract RNA from cell lysates. For example, compared to extraction from TE buffer, the recovery from spiked cell lysates using the extraction cassette was reduced by 65%. Similarly, the recovery using the RNeasy kit was reduced by 57%.
The prototype design was tested with a small subset of previously collected de-identified nasal wash samples. These samples were labeled RSV positive or negative during the collection process (not part of this study) using a commercial laboratory RNA extraction process and RT-PCR. The testing of these samples was not designed as a blinded study and served as a simple validation of the basic extraction design. The evaluation of these samples with our prototype device indicated that the basic design performed similarly to commercially available kits (Figure 3), but in general recovered less than the other kits tested. All of the extraction methods used correctly classified the RSV positive and negative samples. However, the amount of RSV RNA present in these samples was quite variable as indicated by the coefficient of variation (s.d./mean) obtained with all of the extraction methods. The coefficients of variation were 157% (extraction prototype), 227% (RNeasy), 173% (Dynabeads), and 106% (MagAttract). This high variation was the primary reason for using the HEp-2 cell lysates as a more controllable clinical sample analogue for further device development and testing. The error obtained with known RNA input is more indicative of variation inherent in the methods themselves. As Figure 5 indicates, under these more controlled conditions, the coefficient of variation is substantially reduced for both the extraction cassette at 6% and the RNeasy kit at 13%.
The prototype design was refined into the extraction cassette by loading processing solutions into a single length of tubing. The surface tension in the small diameter tubing holds each solution in place, and individual solutions remain undisturbed when magnetic particles pass through the surface tension valves from one solution to the next. This is an agreement with previous studies using a filament-antibody recognition assay which found that high capillary forces held solutions within small diameter capillary tubes even in the presence of a moving filament.17 Other research groups have analogously demonstrated that an immiscible hydrophobic liquid can effectively separate nucleic acid processing solutions. Work by Sur and coworkers demonstrated that silica-coated magnetic beads are able to carry nucleic acids through a lipophilic barrier floating on the surface of two separate processing solutions to isolate RNA from RT-PCR interferents.18 Subsequently, Barry and coworkers adapted the idea to separate solutions in a horizontal format, relying on the surface tension forces at the immiscible interface to prevent the processing solutions from mixing while allowing the transport of magnetic beads through the lipophilic barrier.19 For the development of a low resource RNA extraction device, the use of surface tension valves in a continuous length of tubing offers several advantages over the reported nucleic acid isolation technologies using lipophilic barriers.
One of the advantages of the extraction cassette design is the inherent flexibility and simplicity which provides a unique format for the development of other sample processing cassettes. The scale of the cassette is similar to that of laboratory-based commercially available kits and can be modified to incorporate relatively large sample volumes. The simple design also makes it suitable for large-scale manufacturing. Additionally, it is possible to adapt the cassette format to perform many other solid phase based assays. By functionalizing the magnetic beads with the appropriate capture moiety and loading the cassette with the appropriate processing solutions, assays could be developed for low resource processing of a variety of biomarkers, including DNA, proteins or carbohydrates. The cassette also has the potential to be coupled with downstream platforms for integrated sample preparation, signal amplification and detection. For example, by simply coupling the end of the extraction cassette tubing onto a thermocycler tube or onto the input port of a thermocycler, it is possible to elute the RNA into a prepared RT-PCR buffer without extra handling or risk of contamination.
The simple format of the extraction cassette also improves the reliability of the device. The utilization of surface tension valves in the continuous tubing design allows the extraction process to be fully self-contained. Individual processing solutions are preloaded into the tubing during manufacturing, eliminating the need for sample handling and pipetting during the extraction process. This is advantageous as it minimizes the potential for contamination of the wash solutions, the extracted RNA, or the operator. The continuous diameter of the tubing minimizes particle loss during sample pull-through by eliminating locations where the particles can become trapped, a limitation of the original prototype design shown in Figure 1. The surface tension valves also minimize interferent carryover by preventing diffusion down the tubing, and separating the water wash into three successive steps also helped to minimize carryover.
Fluid stability and continued separation during processing are key to this design. The surface tension at the valve interface is affected by the surface properties of the tubing and the properties of the air/liquid interface. Consequently, the overall stability of the solution chambers within the cassette is dependent on each of these surface properties.19 Some preliminary studies have been performed to estimate liquid carryover of processing solution across a valve. Each of the four processing solutions was mixed with sodium fluorescein to create a fluorescein concentration of 2.7 μM. Twenty μL of beads (approximately 5.6 × 107 beads) were transported from each of these labeled solutions across a valve into 80 μL of unlabeled water. The fluorescence of the water downstream of the valve was measured and a standard curve used to estimate the volume of solution transferred. The volumes of processing solution carried over for the RNA-silica binding buffer, chaotropic wash buffer, RNA precipitation buffer and water wash were 7.61 μL ± 0.59, 9.88 μL ± 0.27, 5.03 μL ± 0.26, and 5.41 μL ± 0.39, respectively. If we assume that each downstream chamber is perfectly mixed, then the final RNA elution could contain a maximum of ~8.0 × 10−4% ethanol and ~25 nM guanidine salt, two known RT-PCR contaminants. These values are far below the amounts experimentally determined to affect RT-PCR, which we determined to be ~ 1% ethanol or ~ 100 μM guanidine salt (data not shown). Further, ongoing studies are directed at better understanding these properties and the influence of magnetic particle properties such as diameter, density, magnetic susceptibility, and surface chemistry on particle transport through a surface tension valve.
Nucleotide extractions from biologically relevant samples are never 100% efficient, neither in a lab laboratory environment, nor in low resource settings. In fact, the gold standard RNeasy kit only recovered 18.1 ± 2.4% of the RSV N gene RNA spiked into HEp-2 cell lysates. The performance of the extraction cassette would be improved by reducing the overall loss of RNA during the extraction process. Unlike commercially available extraction kits, all of the required components in this study are based on previously published strategies for RNA extraction. It is likely that further modifications to individual processing solutions will lead to an increase in the recovery efficiency. An estimation of the RNA distribution within each wash chamber allows us to identify variables for optimization (Figure 6). Significant quantities of RNA were lost during the water wash steps, which are necessary to remove the ethanol in the absence of centrifugation. For downstream RNA detection by RT-PCR, the ethanol must be removed prior to amplification; however, other nucleic acid-based detection strategies may not be inhibited by the presence of ethanol. In these cases, the water wash chambers could be reduced or eliminated and the recovery efficiency of the extraction cassette improved. Approximately 8% of the RNA still remained on the silica particles after a 5 minute elution in water. By increasing the elution time, the overall yield of the device could be improved by up to 8% in 12 hours, but the total extraction time would be dramatically increased. Future studies will explore potential methods to minimize this loss, including the possibility of direct amplification of the RNA bound to the bead surface without an elution step. Minimal RNA was detected within the RNA-silica binding, chaotropic wash and RNA precipitation solutions. The post-extraction RNA distribution in the processing solutions suggests that RNA may be irreversibly bound to tubing or particle surfaces, degraded during processing, or located on particles that become trapped in the surface tension valves during magnetic pull-through.
The extraction cassette investigated here can potentially be utilized for sample preparation in a low resource setting. It is relatively inexpensive to produce at less than $1.00 per extraction. A rough cost estimate based on current catalog prices of the chemicals and materials required for the continuous tubing design suggests that the most expensive items are the magnetic particles (about $0.50) and the Tygon tubing (about $0.30). In its current form, the recovery efficiency of this device is acceptable. However, the continuous tubing design can likely be further improved by solution and surface optimization studies. Its major advantages are that, unlike other commercially available methods, it can be performed without a laboratory centrifuge or access to a pipetter and without the skills necessary to operate these laboratory devices. Thus, this approach is an attractive low resource alternative to commercially available methods.
5. Conclusion
We have shown that our self-contained extraction cassette performs effectively and offers many advantages over similar reported devices and commercially available kits. The simplicity and flexibility of the cassette make it an robust sample preparation tool suitable for use in low resource settings where nucleic acid-based diagnostics must be utilized without specialized equipment and/or trained personnel. The extraction cassette is an ideal format for coupling with downstream nucleic acid amplification and detection modalities. Additionally, the technology is readily adaptable for the isolation of other potential biomarkers of interest, including DNA, proteins or carbohydrates.
Acknowledgments
This work was supported in part by a Vanderbilt University IDEAS Award, NIH R21 EB009235, and N.A. was supported by NIH Training Grant T32 HL007751 from the Heart, Lung, and Blood Institute.
References
- 1.Niemz A, Ferguson TM, Boyle DS. Trends Biotechnol. 2011;29(5):240–250. doi: 10.1016/j.tibtech.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beuselinck K, van Ranst M, van Eldere J. Journal of Clinical Microbiology. 2005;43(11):5541–5546. doi: 10.1128/JCM.43.11.5541-5546.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Radstrom P, Knutsson R, Wolffs P, Lovenklev M, Lofstrom C. Mol Biotechnol. 2004;26(2):133–46. doi: 10.1385/MB:26:2:133. [DOI] [PubMed] [Google Scholar]
- 4.Monteiro L, Bonnemaison D, Vekris A, Petry KG, Bonnet J, Vidal R, Cabrita J, Megraud F. Journal of Clinical Microbiology. 1997;35(4):995–998. doi: 10.1128/jcm.35.4.995-998.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson IG. Applied and Environmental Microbiology. 1997;63(10):3741–3751. doi: 10.1128/aem.63.10.3741-3751.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coiras MT, Perez-Brena P, Garcia ML, Casas I. J Med Virol. 2003;69(1):132–44. doi: 10.1002/jmv.10255. [DOI] [PubMed] [Google Scholar]
- 7.Chomczynski P, Sacchi N. Analytical Biochemistry. 1987;162(1):156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 8.Avison MB. Measuring gene expression. Taylor & Francis; New York; Abingdon [England]: 2007. p. 324. [Google Scholar]
- 9.Yamada O, Matsumoto T, Nakashima M, Hagari S, Kamahora T, Ueyama H, Kishi Y, Uemura H, Kurimura T. Journal of Virological Methods. 1990;27(2):203–209. doi: 10.1016/0166-0934(90)90136-4. [DOI] [PubMed] [Google Scholar]
- 10.Chirgwin JM, Przybyla AE, Macdonald RJ, Rutter WJ. Biochemistry. 1979;18(24):5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- 11.MacDonald RJ, Swift GH, Przybyla AE, Chirgwin JM, Shelby LB, Allan RK. Methods in Enzymology. Vol. 152. Academic Press; 1987. Isolation of RNA using guanidinium salts; pp. 219–227. [DOI] [PubMed] [Google Scholar]
- 12.Price CW, Leslie DC, Landers JP. Lab Chip. 2009;9(17):2484–2494. doi: 10.1039/b907652m. [DOI] [PubMed] [Google Scholar]
- 13.Chen DF, Mauk M, Qiu XB, Liu CC, Kim JT, Ramprasad S, Ongagna S, Abrams WR, Malamud D, Corstjens PLAM, Bau HH. Biomed Microdevices. 2010;12(4):705–719. doi: 10.1007/s10544-010-9423-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hagan KA, Reedy CR, Uchimoto ML, Basu D, Engel DA, Landers JP. Lab Chip. 2011;11(5):957–61. doi: 10.1039/c0lc00136h. [DOI] [PubMed] [Google Scholar]
- 15.Hu AZ, Colella M, Tam JS, Rappaport R, Cheng SM. Journal of Clinical Microbiology. 2003;41(1):149–154. doi: 10.1128/JCM.41.1.149-154.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins PL, Crowe JE. Respiratory Syncytial Virus and Metapneumovirus. Lippencott Williams and Wilkins; Philadelphia: 2007. [Google Scholar]
- 17.Stone GP, Wetzel JD, Russ PK, Dermody TS, Haselton FR. Annals of Biomedical Engineering. 2006;34(11):1778–1785. doi: 10.1007/s10439-006-9199-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sur K, McFall SM, Yeh ET, Jangam SR, Hayden MA, Stroupe SD, Kelso DM. J Mol Diagn. 2010;12(5):620–8. doi: 10.2353/jmoldx.2010.090190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berry SM, Alarid ET, Beebe DJ. Lab Chip. 2011;11(10):1747–53. doi: 10.1039/c1lc00004g. [DOI] [PMC free article] [PubMed] [Google Scholar]