


Abstract
We have developed a novel allele-specific primer elongation protocol using a DNA polymerase on oligonucleotide chips. Oligonucleotide primers carrying polymorphic sites at their free 3́end were covalently bound to glass slides. The generation of single-stranded targets of genomic DNA containing single nuclotide polymorphisms (SNPs) to be typed was achieved by an asymmetric PCR reaction or exonuclease treatment of phosphothioate (PTO)-modified PCR products. In the presence of DNA polymerase and all four dNTPs, with Cy3-dUTP replacing dTTP, allele-specific extension of the immobilized primers took place along a stretch of target DNA sequence. The yield of elongated products was increased by repeated reaction cycles. We performed multiplexed assays with many small DNA targets, or used single targets of up to 4.4 kb mitochondrial DNA (mtDNA) sequence to detect multiple SNPs in one reaction. The latter approach greatly simplifies preamplification of SNP-containing regions, thereby providing a framework for typing hundreds of mtDNA polymorphisms.
INTRODUCTION
Single nucleotide polymorphisms (SNPs) are the most frequent type of DNA sequence variation among individuals. They are defined by the presence of two alternative bases at a particular position in a DNA sequence and occur at about one per 500–1000 bp in the human genome (1). The recent completion of the first reference sequence of the human genome has provided a basis for comprehensive analysis of sequence variation in man. The identification and dense mapping of SNPs is of considerable significance for association studies of complex diseases (1–5), pharmacogenetics (6,7), population genetics (8,9) and physical mapping (10). Their use as genetic markers is favored by their high abundance, low mutation rate and the easy automation of typing. With the development of DNA microarrays, a highly parallel, large-scale method for DNA sequence comparison is available.
To date, different approaches for the detection of single nucleotide variations by DNA microarrays have been reported. Sequence-specific hybridization of immobilized allele-specific oligonucleotides (ASO) to labeled DNA targets relies on the influence of single base mismatches on the stability of DNA duplexes (11–15), which is, however, strongly dependent on the nucleotide sequence context of SNPs and on the reaction conditions (16,17). To increase the specificity of this assay, a redundant set of oligonucleotides per SNP was used, which allowed for the evaluation of hybridization signals in a variety of sequence contexts. Other attempts to improve the discrimination of hybridization mismatches included electronically manipulated DNA microchips, which enable an adjustment of hybridization stringency through a variable electric field at individual sites of immobilized oligonucleotides (18,19), or the use of modified nucleosides and chaotropic agents (20). The ability of DNA ligases, reverse transcriptases and DNA polymerases to distinguish terminal mismatches in DNA–DNA or DNA–RNA duplexes has also been utilized to increase detection accuracy. Gunderson et al. (21) used a DNA target ligation reaction to immobilized oligonucleotides for mutation detection and DNA resequencing. A single nucleotide primer extension reaction mediated by a DNA polymerase in conjunction with labeled dideoxytriphosphates as terminators, was applied by Pastinen et al. (22) and yielded >10-fold better discrimination power than simple ASO hybridization. Most recently, an allele-specific primer extension system was described (23), which relies on a sequence-specific elongation of immobilized allele-specific primers along an RNA target by reverse transcriptase. In contrast to single nucleotide primer extension protocols, a single fluorophore and only two immobilized primers instead of four are required, and a longer elongation product results. Comparison of the allele-specific with the single nucleotide primer extension system revealed a similar specificity, as also reported previously for assays performed in a polyacrylamide gel support (24).
Here we describe a novel polymerase extension method using oligonucleotide microarrays to detect SNPs in human mitochondrial DNA (mtDNA). Oligonucleotide primers covalently bound to glass slides at their 5′-ends contain a variable base at the terminal 3′ position. Allele-specific elongation in the presence of all four dNTPs (one of which is Cy3-dUTP) takes place along single-stranded DNA (ssDNA) targets generated either by asymmetric PCR or exonuclease treatment of phosphothioate (PTO)-modified PCR products. To increase the yield of elongated labeled products at perfectly matched DNA duplexes, cycled reactions consisting of denaturation, annealing and extension steps were performed on the microarray. Sites of polymerase extension reactions were detected by fluorescence scanning of the labeled products.
We used human mtDNA as a system to test this novel method. Hundreds of mtDNA polymorphisms are known (http://infinity.gen.emory.edu/mitomap.html) and the analysis of sequence variations has important applications in the fields of population genetics (25), medical genetics (25,26,27) and forensic science (28).
MATERIALS AND METHODS
Oligonucleotides and DNA isolation
HPLC-purified oligonucleotides were obtained from Metabion (Martinsried, Germany) and MWG Biotech AG (Ebersberg, Germany). Conventional oligonucleotides were used for PCR; allele-specific primers on the array were 5′-amino-modified oligonucleotides. Lyophilized allele-specific primers were dissolved in 400 mM Na2CO3 pH 9.0 to a final concentration of 100 µM and stored frozen at –20°C until use.
For the generation of ssDNA targets by exonuclease digestion of PCR products, PTO-modified oligonucleotides were utilized. These primers had three PTO-bonds introduced into the 5′-end.
DNA was isolated from whole blood by means of an extraction kit (QIAamp DNA Blood Mini Extraction Kit, Qiagen, Hilden, Germany) and was used as a template for PCR amplification of targets.
Preparation of microarrays
Aminosilane-coated In Situ PCR Glass Slides from Perkin Elmer (Weiterstadt, Germany) were used in early experiments. Superior results were obtained with standard glass slides silanized as follows: after cleaning with 1 M NaOH (Merck, Darmstadt, Germany) overnight, slides were rinsed three times in H2O followed by a neutralization step in 1% HCl (Merck) and a washing step in H2O. Silanization with 1% 3-aminopropyl-trimethoxysilane (Sigma, Taufkirchen, Germany) was performed by incubation for 30 min followed by two washing steps in acetone (Merck). Subsequently, slides were baked at 110°C for 45 min. The slide surface was activated to allow covalent binding of amino-modified oligonucleotide primers according to Guo et al. (11). Solutions of 5′-terminal amino-modified oligonucleotides were transferred into 96-well microtiter plates in volumes of 5 µl and spotted using a commercially available robot (Beecher Instruments, Silver Springs, MD) which deposits ∼5 nl at each spotting site, resulting in spots of ∼200 µm in diameter. The humidity during spotting was 70% and the temperature kept at ∼21°C. After spotting, slides were incubated for another 2 h under the same conditions and stored at room temperature. Before use, the slide surface was deactivated by a 30 min incubation in 10% ammonium hydroxide solution (Fluka, Deisenhofen, Germany), followed by three washing steps in water for 10 min each.
Generation of DNA targets
All PCR products were amplified in a thermal cycler (TC9700 or TC2400, Perkin Elmer) under the conditions described below. Reaction mixtures of 50 µl contained 100 µM of each dNTP (Roche Molecular Biochemicals, Mannheim, Germany), 20 pmol of each primer, 100 ng of a genomic DNA preparation, 1× PCR buffer and 1–2 U AmpliTaq (Perkin Elmer).
Double-stranded PCR products were generally amplified under the following conditions: initial denaturation (5 min at 94°C) followed by 35 cycles of denaturation (1 min at 94°C), annealing (1 min at the respective primer annealing temperature) and extension (1 min at 72°C). A final extension step was carried out for 10 min at 72°C. Double-stranded PCR products used as targets were denatured after the last cycle by heating at 95°C for 5 min followed by snap cooling on ice. Purification of PCR products was performed using the QIAquick PCR purification kit (Qiagen).
Asymmetric single-stranded PCR products used in this study were generated from different double-stranded PCR templates: a 426 bp template was amplified with primers 3153L (5′-TTC ACA AAG CGC CTT CCC CC-3′)/3579H (5′-TAT GGG GAG GGG GGT TCA TAG TAG-3′), three templates of 4 kb were amplified with primers L 3170 (5′-AGG CGC CTT CCC CCG TAA AT-3′)/R 7117 (5′-AAG GTG TAG CCT GAG AAT AGG GGA AA-3′), L 12210 (5′-AAA GCT CAC AAG AAC TGC TAA CTC ATG C-3′)/R 16052 (5′-ATG GGT GAG TCA ATA CTT GGG TGG-3′) and L 8316 (5′-TTA ACC TTT TAA GTT AAA GA-3′)/R 12788 (5′-GAT ATA ATT CCT ACG CCC TCT CAG CC-3′), respectively. The asymmetric PCR conditions were as follows: after an initial denaturation step (5 min at 94°C), 40 cycles of denaturation (30 s at 94°C), annealing (30 s at 56°C) and extension (30 s at 72°C) were carried out.
To generate single-stranded targets from double-stranded PCR products, PTO-modified primers were used for PCR amplification followed by 5′–3′ exonuclease digestion of the unmodified strand. Primers for amplification of a 426 bp product were 3153L/3579H-PTO; primers for product A were V-15421-L (5′-AAT CAA AGA CGC CCT CGG CT-3′) and R 3343-PTO (5′-TGG GTA CAA TGA GGA GTA GGA G-3′); for product B, 3153L and R 7117-PTO; for product C1, L-6325-L (5′-CCT CCG TAG ACC TAA CCA TCT TCT CCT-3′) and R-8810-PTO (5′-GTT GGT GTA AAT GAG TGA GGC A-3′); for product C2, N-8821-L (5′-TCT ATA AAC CTA GCC ATG GC-3′) and R-12017-PTO (5′-TGA GTG AGC CCC ATT GTG TTG TG-3′); for product D, T-11741-L (5′-TGC CTA GCA AAC TCA AAC TA-3′) and R-16052-PTO (5′-ATG GGT GAG TCA ATA CTT GGG TGG-3′). PCR amplification of products A, B, C1, C2 and D was performed using the Expand Long Template PCR System from Roche Molecular Biochemicals. Purified PCR product (2 µg) was incubated with 1× T7 Gene 6 Exo buffer and 10 U of T7 Gene 6 Exonuclease (Amersham Pharmacia Biotech, Freiburg, Germany) for 30 min at 37°C followed by 15 min at 85°C. The digested product was used directly in a primer elongation reaction.
Primer elongation reaction on microarrays
A reaction mixture of 30 µl final volume containing 3 U of a thermostable Taq polymerase (RedTaq, Sigma), the appropriate buffer (1×), 80 µM of each dATP, dCTP, dTTP (Roche Molecular Biochemicals), 40 µM Cy3-dUTP (Amersham) and the respective DNA target was applied to the slide. The area containing the oligonucleotide array was either sealed with FrameSeal slide chambers (BIOzym, Oldendorf, Germany) if a thermal cycler from BIOzym (PTC 200 with in situ block) was used, or covered with Ampli Cover Discs (Perkin Elmer) clamped over the slide with the help of an Assembly Tool (Perkin Elmer) for the use of the Gene Amp In Situ PCR System 1000 (Perkin Elmer). The temperature profile of the primer elongation reactions was as follows: 3 min at 90°C followed by 15 cycles of denaturation (30 s at 90°C), annealing (30 s at 56°C) and extension (15 s at 72°C).
Washing and scanning of microarrays
Immediately following the elongation reactions, slides were washed three times in Petri dishes, with MilliQ water, for 10 min each. After centrifugation (1000 g, 3 min), slides were ready for scanning. Laser scanners from Beecher Instruments (Silver Springs, MD) and the Affymetrix 428 scanner (Affymetrix, Santa Clara, CA) were used for the detection of incorporated Cy3-dUTP.
Data analysis
Sixteen-bit TIFF images of 10 µm resolution were imported into custom-made software (Y.Chen, National Human Genome Research Institute, NIH, Bethesda, MD; personal communication) that runs as an extension on IPLab Spectrum Software. After subtraction of local background intensity, mean intensities of individual spots were used to calculate match to mismatch signal intensity ratios of spot pairs corresponding to different alleles.
RESULTS AND DISCUSSION
Principle of allele-specific primer elongation reaction on microarrays
The novel SNP detection protocol described here relies on the power of a Taq polymerase enzyme to discriminate single base pair mismatches in an elongation reaction of immobilized oligonucleotide primers on microarrays (Fig. 1). As with allele-specific PCR, the strong dependence of Taq polymerase function on matched base pairs is utilized. For each SNP to be detected, two oligonucleotide primers were used that were immobilized on the microarray at their 5′ N-terminus. Primers used in early experiments consisted of a 5′ T spacer of 15 nt followed by a stretch of specific sequence of 25 nt and a variable base at the free 3′-end corresponding to the allelic difference. In subsequent experiments (see below) we used oligonucleotide primers that did not contain an oligoT spacer, and consisted of a specific sequence of only 50 nt. We found that these produced superior results. The allele-specific elongation reaction took place in the presence of all four dNTPs (one of which was Cy3-dUTP) along a stretch of single-stranded DNA target sequence containing a segment complementary to the specific sequence of the immobilized oligonucleotide primers. Compared to Cy5-dUTP, Cy3-dUTP gave stronger fluorescent signals and was therefore used in all experiments. In contrast to the single nucleotide primer extension or minisequencing reaction on microarrays (22), this technique results in an extension product for which the size is dependent on the length of the target sequence between the variant base and the 5′-end, as illustrated in Figure 1. To enhance the yield of extended products, repeated reactions were performed consisting of 15 cycles of denaturation, annealing and extension.
Figure 1.

Principle of allele-specific primer elongation on microarrays. Single-stranded DNA targets serve as templates in a Taq polymerase-catalyzed elongation reaction of immobilized oligonucleotide primers. The match and mismatch primers differ at their free 3′-end by a variable base, which is discriminated by the enzyme. Elongation and thereby incorporation of Cy3-dUTP takes place in a template-dependent manner.
Detection of single base mismatches in synthetic oligonucleotide targets
Initially, synthetic 50mer oligonucleotides were used as targets to determine the power of the Taq polymerase-catalyzed elongation reaction for discriminating single base pair mismatches.
Four different oligonucleotides corresponding to a mtDNA sequence of 50 bp but differing in their central base were used as targets. Four oligonucleotide primers of 26 bp were immobilized in duplicate as probes on the microarray. Their sequences were complementary to all four target oligonucleotides except for the free 3′ base, which carried an A, T, G or C (Fig. 2). Each 50mer target was used in a single primer elongation reaction on the microarray and signal intensities at the position of each probe were measured to calculate ratios of mismatch to match signal intensities.
Figure 2.
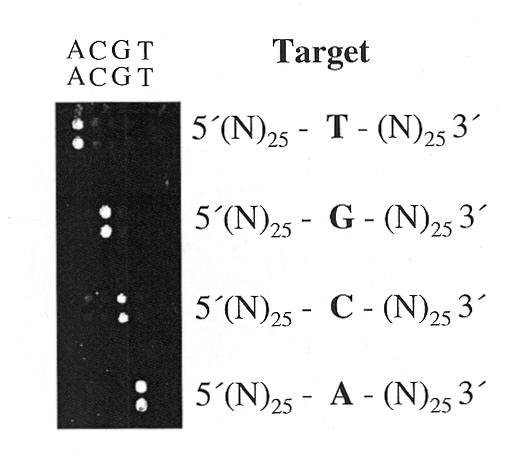
Mismatch detection using synthetic oligonucleotides as targets. Four primer elongation reactions were performed with a 50mer oligonucleotide carrying a central single nucleotide exchange, in each reaction. Oligonucleotide primers spotted as duplicates on the microarray, consisted of a 5′(T)15 spacer sequence linked to a 25 bp specific sequence complementary to the 25 nt at the 3′-end of the targets and carried one of the four bases at their free 3′-ends. Positions of the primers with the respective variable base are indicated above.
Mismatch to match ratios were in the range of 1:10 to 1:50 with the highest discrimination detected for A (primer):G (target) and T:G, and the lowest for C:T mismatches. These results are comparable to the mismatch discrimination of Taq polymerases in allele-specific PCR amplification. A comprehensive study reported the highest specificity of this enzyme in allele-specific PCR for A (primer):G (template), G:A, G:G, C:C and A:A mismatches, and lowest discrimination power for C:T mismatches (29).
False positive signals under different experimental conditions
Primer elongation reactions were always performed both with and without a DNA target added to the reaction, in order to further monitor background activity. Even without target DNA, strong signals were obtained in a few cases. From these results, we concluded that inter- or intramolecular DNA double strands can lead to an elongation reaction. A close inspection of the sequences of immobilized DNA probes revealed that primer–dimer formation of adjacent molecules within one spot is the most plausible explanation. Of the six most intense false positive signals in our experiments, the sequence of five of them indicates possible primer–dimer formations as shown in Figure 3. This mechanism leading to false positive signals was reported earlier in a study by Nikiforov et al. (30).
Figure 3.

False positive signals under different experimental conditions. Primer elongation reactions were performed under standard conditions (see Materials and Methods) without a target on differently coated slides. (A) 1%, (B) 2%, (C) 3% aminopropyl-trimethoxysilane and (D) aminosilane slides from Perkin Elmer. Prominent false positive signals are denoted by arrows. Possible primer–dimer structures of five of these six sequences are shown below. The false positive signal at position 2 cannot be explained by the formation of primer–dimers or hairpin loops. Varying parameters of the cycling protocol influence the intensity of false positive signals: (E) standard conditions, (F) increased cycle number (25 instead of 15), (G) decreased annealing temperature (48 instead of 56°C), (H) prolonged annealing time (60 instead of 30 s) and (I) prolonged elongation time (30 instead of 15 s).
The proportion of these kinds of false positive signals could be altered by varying experimental conditions. To test the influence of the surface chemistry on the occurrence of false positive signals, 96 allele-specific primers were immobilized on different aminosilane-coated slides. Silanization was performed using 1, 2 and 3% aminopropyl-trimethoxysilane, respectively, in addition to commercially available aminosilane slides (see Materials and Methods). The results of primer elongation reactions on these four kinds of slides without any target can be seen in Figure 3A–D. The intensity and number of false positive signals increased on self-silanized slides with the aminopropyl-trimethoxysilane concentration. The highest rate of false positive signals was detected on commercially available slides. Control experiments using an asymmetric PCR product as target (see also Fig. 4C) revealed that under the same experimental conditions, the best discrimination rates were obtained using slides coated with 1% 3-aminopropyl-trimethoxysilane. Moreover, certain parameters of the cycling protocol influenced the rate of false positive signals. With 96 allele-specific primers immobilized on 1% 3-aminopropyl-trimethoxysilane slides, the intensity of false positive signals differed with increased numbers of cycles (25 instead of 15, Fig. 3F), decreased annealing temperature (48 instead of 56°C, Fig. 3G), prolonged annealing time (60 instead of 30 s, Fig. 3H) and a prolonged elongation time (30 instead of 15 s, Fig. 3I). As depicted in Figure 3E, minimal intensities of false positive signals were obtained under standard conditions, which also revealed the best discrimination power using the asymmetric PCR product as a control. Generally, oligonucleotide primers that cause false positive signals even under optimized conditions could be replaced by primers detecting the complementary DNA sequence.
Figure 4.
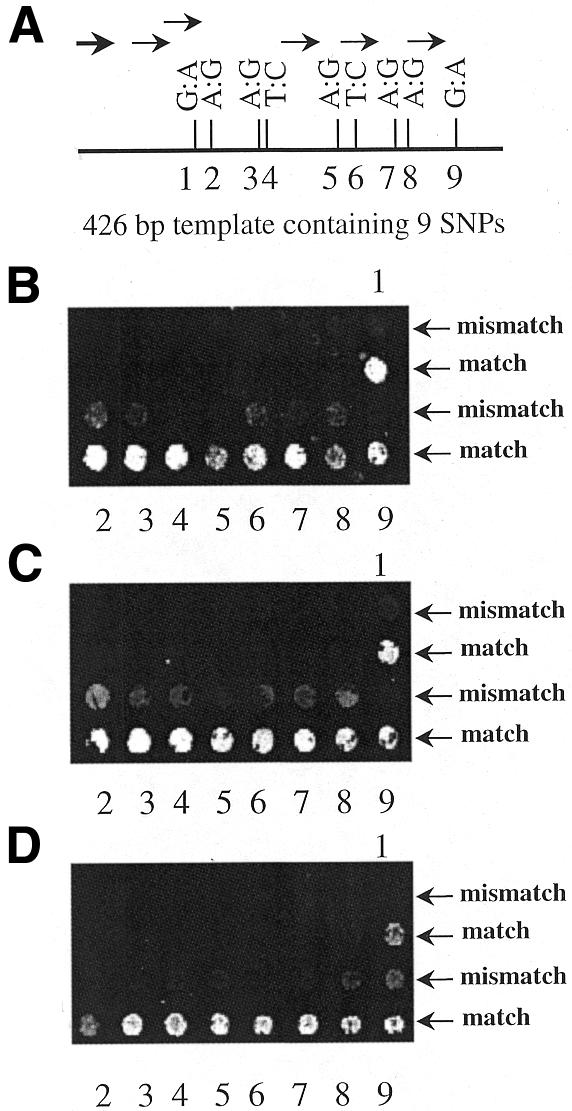
Detection of nine SNPs in a 426 bp sequence using differently generated single-stranded DNA targets. (A) Illustration of the relative position and kind of SNPs within the template with primers indicated by an arrow. (B) Result of a primer elongation reaction with asymmetric PCR products as targets, which were generated in a reaction with a single PCR primer as indicated by a bold arrow in (A). (C) SNP detection using asymmetric PCR products generated with all six primers in a multiplex reaction. (D) An exonuclease-treated PTO-modified PCR product spanning the complete 426 bp sequence was used in a primer elongation reaction.
Mismatch detection of single-stranded asymmetric PCR products
Since dsDNA sequences were found to yield very weak signals, we generated ssDNA targets using two different approaches. First, asymmetric PCR was employed using primers upstream of one or more SNP sequences. Figure 4A illustrates an experiment in which one primer upstream of nine SNPs (indicated by a bold arrow) contained in a 426 bp sequence was used in an asymmetric PCR reaction. The primer elongation with these single-stranded products was performed on microarrays containing two oligonucleotide primers with the respective 3′ base exchanges for each of the nine SNPs. The detected signals are shown in Figure 4B. All nine SNPs could be typed, with match to mismatch ratios ranging from 1.67 to 6.28 and averaging 3.60. The distance between the PCR primer used in the asymmetric PCR reaction and the position of the SNP in the template sequence determines the length and thus the incorporation of fluorescent dUTP of the elongated product. The optimal PCR primer to SNP distance was found to be 25 bp or longer (data not shown).
We then generated asymmetric PCR products using shorter PCR primer to SNP distances. Six single primers were designed and combined in a multiplex PCR reaction. The distances from these primers to downstream SNP loci ranged from 35 to 49 bp. Results are shown in Figure 4C. Match to mismatch ratios in this experiment were not superior to the previous experiment with only one PCR primer, and ranged from 1.6 to 3.8 with an average of 2.76.
In order to detect 48 SNPs contained in three templates, each ∼4 kb in size, in a single primer elongation reaction, we combined targets generated in three multiplex asymmetric PCR reactions with 13, 15 and 20 primers, respectively. As seen in Figure 5, all but two SNPs could be typed with match to mismatch ratios between 1.6 and 30.7. The detection of two SNPs failed due to ratios of 0.9 and 1.2, respectively. Sequencing of the three templates revealed that this failure was not due to the presence of both alleles as could be expected in cases of heteroplasmy.
Figure 5.

Primer elongation reaction on a microarray carrying paired oligonucleotide primers corresponding to 48 different mitochondrial SNPs. Asymmetric PCR products generated in three multiplexed reactions with 13, 15 and 20 primers were used as targets. Match (marked in gray) and mismatch bases of the immobilized primers as well as match to mismatch ratios of the fluorescent signals are listed in the table according to the position on the microarray.
Mismatch detection of single-stranded targets generated by exonuclease digestion of PTO-modified PCR products
Another method we applied to generate ssDNA targets consisted of an initial PCR amplification with two primers, one of which was PTO-modified at the first three 5′ nucleotides. The advantage of this approach is the possibility of monitoring multiplex PCR reactions by the presence of distinct dsDNA bands in an agarose gel. In the case of asymmetric PCR reactions, the resulting smear of ssDNA probes of different lengths makes an analysis of multiplex reactions difficult. PTO groups provide protection against exonuclease activity, and subsequent treatment with a 5′–3′ exonuclease leads to selective digestion of the unmodified strand. The same principle was used in the ‘Genetic Bit Analysis’ method for typing SNPs to generate single-stranded products with high efficiency (30). Nine SNPs previously detected with asymmetric PCR targets were typed using a ssDNA target generated by exonuclease digestion of a 426 bp PTO-modified PCR product (Fig. 4D). Match to mismatch ratios ranged from 1.89 to 8.21 with an average of 4.25, indicating improved discrimination compared with asymmetric PCR products.
We next wanted to find out if larger PTO-modified PCR products containing multiple SNPs could be employed. Such targets would reduce the number of primers needed in the preceding amplification of DNA regions. All microarray-based methods reported so far require two primers per SNP, which is a major drawback of large-scale SNP typing. We PCR-amplified a 4 kb fragment of mtDNA (product B), which contains numerous SNPs, and designed oligonucleotide primers for 16 of these. In a primer elongation reaction all 16 SNPs could be typed with match to mismatch ratios between 1.44 and 5.90 and an average of 2.95 (Fig. 6B). We found that the position of a single SNP within the 4 kb product did not influence the specificity of the reaction. The application of large single-stranded targets is facilitated by repeated denaturation steps in our protocol, which lead to abrogation of secondary structures, thereby increasing the availability of DNA stretches for binding to oligonucleotide primers. Since the mitochondrial genome consists of only 16.5 kb of sequence, we designed four additional primer pairs to generate PCR products (A, C1, C2 and D) which, together with product B cover the mitochondrial genome (Fig. 6). Allele-specific primer pairs representing 46 SNPs were immobilized on a chip and each of the four exonuclease-digested PCR products was used in a primer elongation reaction. As can be seen in Figure 6, 44 out of 46 SNPs could be typed with match to mismatch ratios ranging from 1.4 to 6.4, averaging 2.99. The detection of two SNPs failed due to the absence of a fluorescence signal in one case (SNP 29) and no significant discrimination (ratio of 1.0) in another case (SNP 26). A primer elongation reaction with a larger target of 5.7 kb including the sequences of products C1 and C2 failed repeatedly, which indicates that the size limit of these single-stranded targets ranges between 4.4 (largest product used) and 5.7 kb.
Figure 6.
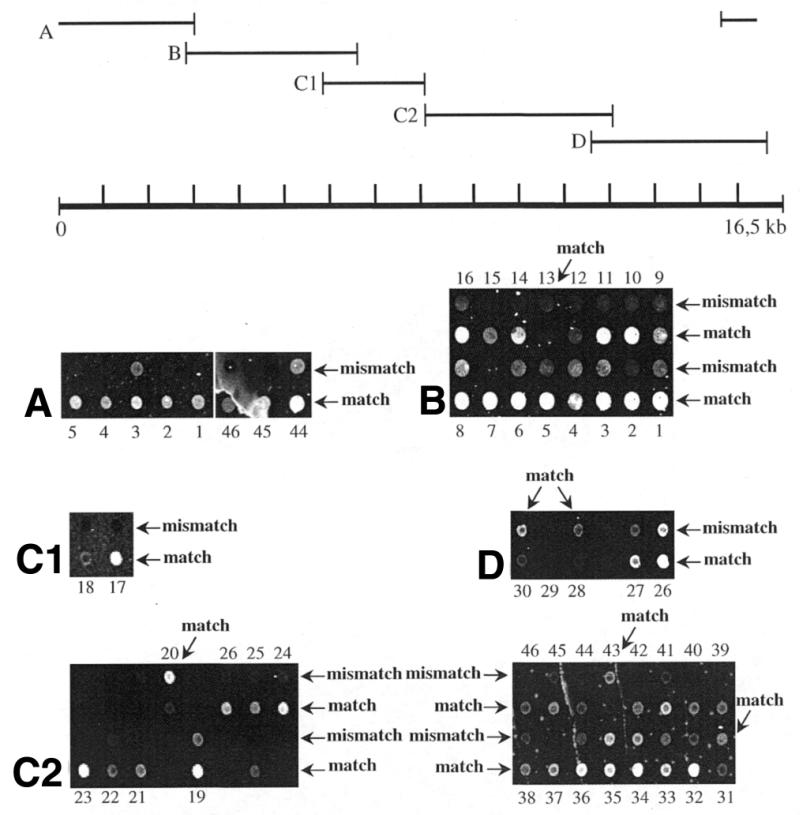
Detection of 46 mitochondrial SNPs contained in five large targets generated by exonuclease-treatment of the respective PTO-modified PCR products (A, B, C1, C2 and D), which span the mitochondrial genome. Particular SNPs are found in overlapping targets as depicted by identical numeration. Paired match and mismatch oligonucleotide primers are arranged in alternate rows with the exception of a few pairs, as indicated by oblique arrows.
CONCLUSION
In this study we describe a novel protocol for the detection of SNPs using oligonucleotide microarrays. The method relies on the allele-specific elongation of immobilized oligonucleotide primers during which a fluorescently labeled nucleotide (Cy3-dUTP) is incorporated and detected by confocal laser scanning. Cycled reactions consisting of a denaturation, annealing and elongation step were employed to increase the yield of elongated product. Using human mtDNA as a model system, we tested two different means of generating ssDNA targets used in the reactions: asymmetric PCR products and exonuclease-treated PTO-modified PCR products. Both proved to be suitable in this SNP detection system. Using asymmetric PCR products, we demonstrated that a single PCR primer upstream of nine SNPs in a 426 bp template is sufficient. Forty-six of 48 SNPs could be detected in this way using three multiplexed asymmetric PCR reactions. The disadvantage of asymmetric PCR reactions is the difficult judgement of multiplexed reactions since asymmetric PCR products appear as a smear of bands in agarose gels. The application of exonuclease-treated PTO-modified PCR products as targets is therefore advantageous. More importantly, however, we demonstrate that it is possible to type 44 out of 46 randomly distributed mitochondrial SNPs by using five targets between 2.5 and 4.4 kb, which together cover the entire mitochondrial genome. The size limit of such targets was found to lie between 4.4 and 5.7 kb. The approach described here simplifies PCR amplification of SNP loci, which is a major problem in transforming microarray-based SNP typing into a high-throughput method.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Kubilay Demir, Ralf Schulz and Bettina Lipkowitz for technical assistance. This work was supported by a grant (BEO 0311659) from the Bundesministerium für Bildung und Forschung (BMBF).
References
- 1.Brookes A.J. (1999) The essence of SNPs. Gene, 234, 177–186. [DOI] [PubMed] [Google Scholar]
- 2.Zhao L.P., Aragaki,C., Hsu,L. and Quiaoit,F. (1998) Mapping of complex traits by single-nucleotide polymorphisms. Am. J. Hum. Genet., 63, 225–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cargill M., Altshuler,D., Ireland,J., Sklar,P., Ardlie,K., Patil,N., Lane,C.R., Lim,E.P., Kalyanaraman,N., Nemesh,J. et al. (1999) Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat. Genet., 22, 231–238. [DOI] [PubMed] [Google Scholar]
- 4.Halushka M.K., Fan,J.B., Bentley,K., Hsie,L., Shen,N., Weder,A., Cooper,R., Lipshutz,R. and Chakravarti,A. (1999) Patterns of single-nucleotide polymorphisms in candidate genes for blood-pressure homeostasis. Nat. Genet., 22, 239–247. [DOI] [PubMed] [Google Scholar]
- 5.Kruglyak L. (1999) Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat. Genet., 22, 139–144. [DOI] [PubMed] [Google Scholar]
- 6.McCarthy J.J. and Hilfiker,R. (2000) The use of single-nucleotide polymorphism maps in pharmacogenomics. Nat. Biotechnol., 18, 505–508. [DOI] [PubMed] [Google Scholar]
- 7.Housman D. and Ledley,F.D. (1998) Why pharmacogenomics? Why now? Nat. Biotechnol., 16 (Suppl.), 2–3. [DOI] [PubMed] [Google Scholar]
- 8.Nielsen R. (2000) Estimation of population parameters and recombination rates from single nucleotide polymorphisms. Genetics, 154, 931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hacia J.G., Fan,J.B., Ryder,O., Jin,L., Edgemon,K., Ghandour,G., Mayer,R.A., Sun,B., Hsie,L., Robbins,C.M. et al. (1999) Determination of ancestral alleles for human single-nucleotide polymorphisms using high-density oligonucleotide arrays., Nat. Genet., 22, 164–167. [DOI] [PubMed] [Google Scholar]
- 10.Kruglyak L. (1997) The use of a genetic map of biallelic markers in linkage studies. Nat. Genet., 17, 21–24. [DOI] [PubMed] [Google Scholar]
- 11.Guo Z., Guilfoyle,R.A., Thiel,A.J., Wang,R. and Smith,L.M. (1994) Direct fluorescence analysis of genetic polymorphisms by hybridization with oligonucleotide arrays on glass supports. Nucleic Acids Res., 22, 5456–5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hacia J.G., Brody,L.C., Chee,M.,S., Fodor,S.P.A. and Collins,F.S. (1996) Detection of heterozygous mutations in BRCA1 using high density oligonucleotide arrays and two-colour fluorescence analysis. Nat. Genet., 14, 441–447. [DOI] [PubMed] [Google Scholar]
- 13.Hacia J.G. (1999) Resequencing and mutational analysis using oligonucleotide microarrays. Nat. Genet., 21, 42–47. [DOI] [PubMed] [Google Scholar]
- 14.Winzeler E.A., Richards,D.R., Conway,A.R., Goldstein,A.L., Kalman,S., McCullough,M.J., McCusker,J.H., Stevens,D.A., Wodicka,L., Lockhart,D.J. et al. (1998) Direct allelic variation scanning of the yeast genome. Science, 281, 1194–1197. [DOI] [PubMed] [Google Scholar]
- 15.Wang D.G., Fan,J.B., Siao,C.J., Berno,A., Young,P., Sapolsky,R., Ghandour,G., Perkins,N., Winchester,E., Spencer,J. et al. (1998) Large-scale identification, mapping and genotyping of single-nucleotide polymorphisms in the human genome. Science, 280, 1077–1082. [DOI] [PubMed] [Google Scholar]
- 16.Conner B.J., Reyes,A.A., Morin,C., Itakura,K., Teplitz,R.L. and Wallace,R.B. (1983) Detection of sickle cell β S-globin allele by hybridization with synthetic oligonucleotides. Proc. Natl Acad. Sci. USA, 80, 278–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Southern E.M., Maskos,U. and Elder,J.K. (1992) Analyzing and comparing nucleic acid sequences by hybridization to arrays of oligonucleotides: evaluation using experimental models. Genomics, 13, 1008–1017. [DOI] [PubMed] [Google Scholar]
- 18.Sosnowski R.G., Tu,E., Butler,W.F., O’Connel,J.P. and Heller,M.J. (1997) Rapid determination of single base mismatch mutations in DNA hybrids by direct electric field control. Proc. Natl Acad. Sci. USA, 94, 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilles P.N., Wu,D.J., Foster,C.B., Dillon,P.J. and Chanock,S.J. (1999) Single nucleotide polymorphic discrimination by an electronic dot blot assay on semiconductor microchips. Nat. Biotechnol., 17, 365–370. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen H.K., Fournier,O., Asseline,U., Dupret,D. and Nguyen,T.T. (1999) Smoothing of the thermal stability of DNA duplexes by using modified nucleosides and chaotropic agents. Nucleic Acids Res., 27, 1492–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gunderson K.L., Huang,X.C., Morris,M.S., Lipshutz,R.J., Lockhart,D.J. and Chee,M.S. (1998) Mutation detection by ligation to complete n-mer DNA arrays. Genome Res., 8, 1142–1153. [DOI] [PubMed] [Google Scholar]
- 22.Pastinen T., Kurg,A., Metspalu,A., Peltonen,L. and Syvänen,A.C. (1997) Minisequencing: a specific tool for DNA analysis and diagnostics on oligonucleotide arrays. Genome Res., 7, 606–614. [DOI] [PubMed] [Google Scholar]
- 23.Pastinen T., Raitio,M., Lindroos,K., Tainola,P., Peltonen,L. and Syvänen,A.C. (2000) A system for specific, high-throughput genotyping by allele-specific primer extension on microarrays. Genome Res., 10, 1031–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dubiley S., Kirillov,E. and Mirzabekov,A. (1999) Polymorphism analysis and gene detection by minisequencing on an array of gel-immobilized primers. Nucleic Acids Res., 27, e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hofmann S., Jaksch,M., Bezold,R., Mertens,S., Aholt,S., Paprotta,A. and Gerbitz,K.D. (1997) Population genetics and disease susceptibility: characterization of central European haplogroups by mtDNA gene mutations, correlation with D loop variants and association with disease. Hum. Mol. Genet., 6, 1835–1846. [DOI] [PubMed] [Google Scholar]
- 26.Wallace D.C. (1999) Mitochondrial diseases in man and mouse. Science, 283, 1482–1488. [DOI] [PubMed] [Google Scholar]
- 27.Liang M.H. and Wong,L.J. (1998) Yield of mtDNA mutation analysis in 2,000 patients. Am. J. Med. Genet., 77, 395–400. [PubMed] [Google Scholar]
- 28.Butler J.M. and Levin,B.C. (1998) Forensic applications of mitochondrial DNA. Trends Biotechnol., 16, 158–162. [DOI] [PubMed] [Google Scholar]
- 29.Huang M.M., Arnheim,N. and Goodman,M.F. (1992) Extension of base mispairs by Taq DNA polymerase: implications for single nucleotide discrimination in PCR. Nucleic Acids Res., 20, 4567–4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nikiforov T.T., Rendle,R.B., Goelet,P., Rogers,Y.H., Kotewicz,M.L., Anderson,S., Trainor,G.L. and Knapp,M.R. (1994) Genetic Bit Analysis: a solid phase method for typing single nucleotide polymorphisms. Nucleic Acids Res., 22, 4167–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]