

Abstract
Development of clinically relevant tumor model systems for glioblastoma multiforme (GBM) is important for advancement of basic and translational biology. One model that has gained wide acceptance in the neuro-oncology community is the primary xenograft model. This model entails the engraftment of patient tumor specimens into the flank of nude mice and subsequent serial passage of these tumors in the flank of mice. These tumors then can be used to establish short-term explant cultures or intracranial xenografts. The focus of this manuscript is to review the procedures associated with the establishment, maintenance and utilization of a primary GBM xenograft panel.
Key terms: glioblastoma, xenograft, mouse models
INTRODUCTION
Pre-clinical novel therapeutic testing traditionally has been performed using tumor cell lines that have been selected for and maintained in cell culture for many years. As a result of prolonged tissue culture, the genetic and morphologic characteristics of these tumors often do not accurately reflect those typically found in primary human tumors. In glioblastoma multiforme (GBM), one of the most striking examples of this is the lack of tumorigenic cell lines that harbor amplified epidermal growth factor receptor (EGFR) despite this genetic lesion being found in approximately 40% of primary GBM tumor specimens (Frederick, 2000, Smith, 2001). Moreover, prolonged cell culture leads to progressive hypermethylation of the DNA O6-methylguanine methyltransferase (MGMT) promoter (Danam, 2001, Pegg, 1990, Yamada, 2001), and as a consequence, approximately 80% of glioma cell lines are MGMT hypermethylated, as compared to a 40% incidence of promoter hypermethylation in clinical samples. To address these shortcomings, we have developed a panel of primary GBM xenografts that were established by implanting patient tumor specimens directly into the flank of nude mice. Maintenance of these xenograft lines by serial transplantation in mice preserves key genetic features of the corresponding patient tumor, including EGFR amplification and MGMT promoter methylation status (Carlson, 2009, Pandita, 2003). When transplanted into the brain, these xenograft lines form tumors with histopathological features, such as invasion, commonly seen in primary GBM tumors (Giannini, 2005). The maintenance of these important molecular and histopathological features of primary GBM tumors makes this model highly useful for both basic and translational studies.
This unit outlines the procedures required to establish and maintain primary xenograft tumor lines with the intention of using these tumor models for basic or translational studies. The specific sections are as follows:
Basic Protocol 1: Implantation of patient tumor samples into mice
Basic Protocol 2: Serial passage of flank tumor xenografts
Basic Protocol 3: Cryo-preservation of xenograft tissue
Basic Protocol 4: Establishing short-term explant cultures
Basic Protocol 5: Intracranial and flank tumor implantation
NOTE: All protocols using live animals must first be reviewed and approved by an Institutional Animal Care and Use Committee (IACUC) or must conform to governmental regulations regarding the care and use of laboratory animals.
Basic Protocol 1: Implantation of patient tumor samples into nude mice
Processing of patient tumor samples for implantation into mice is the first step towards developing primary xenografts. For development of our model, we have exclusively relied on direct implantation of primary patient tumor samples into the flank of nude mice. Others have either used direct implantation into the brain or initial in vitro culturing prior to implantation. A complementary approach to xenografting would be to establish brain tumor initiating cell (BTIC) cultures from patient tumor samples, although those techniques are beyond the scope of this manuscript (Pollard, 2009). Prior to initiating a xenograft, the appropriate institutional approvals must be in place for use of human tissue and animals in research. From a laboratory safety point-of-view, tumors derived from patients with blood-borne viral infectious diseases, such as HIV or hepatitis, should not be used to establish tumor lines. Following implantation, tumors can take up to one year to develop, but on subsequent tumor passage, the growth rates of the tumors typically increase. Experience in multiple labs suggests that approximately a third of tumors implanted will establish viable xenografts. At Mayo Clinic, we have implanted 135 patient tumors using the method described below, and currently 45 of these implants have formed flank tumors that can be serially passaged. Of these tumors, we have evaluated the tumorigenicity of 35 tumor lines in the orthotopic location, and 33 of these tumors form intracranial tumors that cause neurologic decline and death in mice within 180 days from implantation.
NOTE: All of these GBM xenograft models are publicly available from the Sarkaria laboratory at the Mayo Clinic. After execution of an academic (or commercial) materials transfer agreement, viable tumor tissue, cryopreserved tumor tissue, or flash frozen or formalin-fixed paraffin-embedded (FFPE) tumor samples are available to investigators for a reasonable fee. Isolated RNA, DNA and protein lysates from flank tumor samples are available for many of the lines. For intracranial tumors, archived FFPE and frozen OCT blocks are available for most lines. In addition, gene expression profiling and copy number variation studies have been performed on most lines using tissue from intracranial tumor samples. These data are currently available upon request in the context of a collaboration. We are in the process of developing a web-site to allow for more ready access to this genomic data. All requests for tumor samples or genomic data can be directed to the corresponding author via email: sarkaria.jann@mayo.edu.
Materials
Tumor sample
NOTE: Ideally 1 cm3 of tumor tissue is used for implantation, although tumor samples as small as 0.125 cm3 have been used to implant a single mouse. Tissue obtained in the operating room using a Cavitron Ultrasonic Surgical Aspirator (CUSA) works well for tumor implantation. Sterile 50-ml conical tube (Falcon)
Hanks Balanced Salt Solution (HBSS, Irvine Scientific)
4–5 week old Female Athymic Nude mice (Harlan Sprague Dawley Athymic Nude-Foxn1nu mice this mouse colony originated from NCI Frederick)
Animal ear-tag punch (Fisher Scientific)
Centrifuge
1-cc syringe
16-G, 1½ inch needles
Isoflurane (Novaplus)
Bell jar desiccator for anesthesia
Fume hood
BD Bioscience Growth Factor Reduced BD Matrigel™ Matrix (subsequently referred to as Matrigel through the remainder of the manuscript)
10% povidone-iodine (Betadine)
Scalpels
Petri dish
Wet ice
Process patient tumor sample in preparation for injection
-
1)
Transport tumor sample from pathology to laboratory in HBSS at room temperature and record relevant patient information (i.e. age, sex, date of resection, medical record number).
While the time from resection to implantation is not critical, we implant all tumors within eight hours of resection. However, tumors remain viable for 24 to 48 hr, so in theory, they could be shipped at room temperature via overnight courier and implanted the next day.
-
2)
Upon receipt of the tumor sample, spin down the sample in a centrifuge at 320 RCF for three minutes. All subsequent steps are performed under aseptic conditions. Aspirate off the transport media, and move the specimen to a sterile Petri dish. Because GBM tumors are typically quite soft, mechanical disaggregation or other homogenization steps are not performed. Using a 1-cc syringe without a needle, pull up approximately 100 μL of the tumor into the syringe and then cap the syringe with a 16-G needle. Place the syringe with capped needle on wet ice. If injecting more than one mouse, prepare individual syringes for each mouse.
The tumor tissue typically is quite soft and does not require mechanical disaggregation. However, if necessary, the tumor sample can be broken up in a sterile Petri dish using a sterile scalpel blade.
-
3)
Thaw out enough Matrigel on ice for all the injections (100 μL per syringe).
In step 4, you will add Matrigel to the tumor tissue. Since Matrigel solidifies at room temperature, the syringes and tumor material that contacts the Matrigel must be kept as cold as possible to avoid solidification prior to injection.
Matrigel that has not been used can be re-frozen for use at a later time, provided that it is kept on ice and not contaminated with cells. Matrigel that has solidified is unusable.
-
4)
Draw up 100 μL of Matrigel through the needle and mix the tumor and the Matrigel by moving the plunger back and forth. Dispel the air from the syringe (total volume should be approximately 200 μL of a 1:1 solution), recap the needle and place on wet ice until ready to inject.
Anesthetize the mice
-
5)
Mice are anesthetized with isoflurane in a plastic desiccator. Place the desiccator into an externally vented fume hood. If a hood is not available, the biosafety department should test that laboratory personnel are not being exposed to excessive isoflurane fumes using this method.
-
6)
Place a paper towel in the bottom of the desiccator and add 1–2 mL of anesthetic to the towel.
Add additional anesthetic as needed to maintain the required effect.
-
7)
Place an individual mouse in the desiccator.
-
8)
Once the mouse is unconscious and not moving, remove it from the desiccator and mark the ear using an ear punch or other method of animal identification. Because the procedure is quite quick, we typically do not confirm depth of anesthesia and we do not warm the animals during anesthesia. We do observe the mice and if they are unconscious for more than 5 min, we will typically group them with other mice to maintain their body temperature.
Inject tumor sample into mouse
-
9)
Swab the back of the mouse with Betadine or rubbing alcohol over the injection site. Although the operator should wear sterile gown, gloves and mask (typical garb for handling nude mice), an aseptic field with a sterile drape is not necessary.
-
10)
Inject all 200 μL of the tumor/Matrigel mixture into the flank of the mouse.
The injection site is typically on the posterior/lateral aspect of the lower rib cage. Insert the needle through the skin into the subcutaneous space to inject. You should lift up the skin with your needle prior to injecting to insure that you are not in the muscle. The needle should be inserted approximately 5 mm beyond the end of the needle bevel.
-
11)
While removing the syringe, pinch the injection site for 15 to 30 sec so that the tumor/Matrigel mixture does not leak out of the injection site.
-
12)
Place the animal back in their cage and repeat the process until all animals are injected. Label the cage with the appropriate xenograft number and record the appropriate information in a laboratory book or computer file.
-
13)
Observe mice weekly for presence of tumor growth. Tumors may take up to one year to grow following implantation of a primary tumor. Most implantations will form palpable tumor within 6 months, but some may take longer. There are no other indicators that we have identified that can be used to predict which tumor samples will form usable xenografts.
Basic protocol 2: Serial passage of flank tumor xenografts
Serial passage of tumors in the flank of nude mice provides a ready supply of tumor cells for use in both in vitro and in vivo studies. While tumor propagation via prolonged cell culture promotes loss of EGFR amplification and gain of MGMT promoter methylation, these and other genetic features are stably maintained even with multiple serial tumor passage in the xenograft model (Carlson, 2009, Giannini, 2005). In this section, methods are described for propagation of flank tumors through serial passaging in mice. While this method is quite reliable for serial passage, approximately 1% of the time a tumor will not grow after passage. Thus, xenografts are typically passaged into three recipient mice for the first passage, and then a tumor line is subsequently maintained in three mice at any one time (see previous section). Typical growth time of an established line is one to two months, depending on the tumor line. The passage of tumors can be staggered to provide a near continuous supply of tumors for use in laboratory studies. With each tumor passage, tissue samples are routinely archived in liquid nitrogen and/or paraffin, and, at least in early generations, cryo-preserved as well to facilitate restoration of early passage tumors as discussed below.
Materials
Mouse bearing tumor (Basic Protocol 1)
CO2 source
Forceps
Betadine
Scalpels
100-mm Sterile Culture Plates
Tissue Path Disposable Base Molds (Fisher Scientific)
2 ounce Specimen Containers (Kendall)
OCT Media (Sakura Tissue-Tek)
10% Buffered Formalin (Fisher Scientific)
1.8-mL Cryotube (Nunc or Corning)
1-cc syringe
Hypodermic 16-G needles
Matrigel
Isoflurane (Novaplus)
Animal ear-tag punch (Fisher Scientific)
1.5-mL micro-centrifuge tube
Dry ice
Wet ice
Tumor resection and processing
The description below is for passaging a single tumor measuring 1 to 1.5 cm in greatest dimension into one to three recipient mice.
Euthanize the tumor-bearing mouse by CO2.
Swab the skin around the tumor with Betadine to minimize the risk of bacterial or fungal infection. As described above, sterile technique is used, but sterile draping is not particularly required.
Dissect out the tumor using a sterile scalpel and separate it from the skin.
Place tumor into a sterile 100-mm culture dish.
Tissue is routinely archived for future studies from each tumor generation using the following techniques, as outlined in steps 6 through 9 below. This archived tissue can be very useful for molecular and histopathological studies. If no tissue is to be archived, proceed to step 10 below.
Using sterile scalpels cut a cross-section of the tumor (~1–2 mm thick slice) for OCT embedding. Add a small amount of OCT to the base mold, place the tissue section on the OCT bed. Using forceps, press the tissue into the OCT, such that the tumor is lying flat in the base mold, and then encase the rest of the tissue with OCT to ensure that the tissue is not degraded over time in the −80°C freezer. Place OCT sample between two blocks of dry ice for freezing and then transfer to a −80°C freezer.
-
Cut another cross-section of the tumor (~ 1–2 mm thick) and place it in a specimen jar filled with formalin. Optimal fixation can be achieved with overnight fixation prior to paraffin embedding.
Fixation in formalin for more than two weeks prior to paraffin embedding is sub-optimal, since the tissue becomes brittle, sections poorly, and can affect antigen recognition for IHC staining.
Place another portion of tumor in a labeled 1.8-mL Cryotube for fresh frozen tissue. Immediately place this sample on dry ice or into liquid nitrogen, and subsequently transfer the specimen into a −80°C freezer.
Additional tumor can be processed for cryo-preservation as described in the subsequent section.
After the appropriate tissues are archived, thaw Matrigel on wet ice and mince the remaining tissue into small pieces using scalpels in a sterile 100-mm culture dish.
-
Use a 1-cc syringe to break up the tumor into even smaller pieces by repeatedly drawing up and expelling the tissue. Finally, draw up 100 μL of tumor into the syringe, chill on ice, and then draw up an equal volume of Matrigel. Mix the tumor and Matrigel in the syringe and then return the syringe to the ice.
Using a syringe without a needle to break up the tissue reduces the risk of tissue shearing.
If there is limited tumor available or it is difficult to draw up in the syringe, the tumor can be moved, after dicing, to a 1.5-mL micro-centrifuge tube, and the syringe can be used as a “pseudo” pestle to further disaggregate the tumor.
Anesthetize the mice and inject tumor cell mixture subcutaneously as described in the previous section.
Basic protocol 3: Cryopreservation of xenograft tissue
Cryopreservation of xenograft tissues is a key methodology used to re-establish heterotopic xenografts from archived tissue. Because extended tumor passage in vivo likely leads to genetic and epigenetic drift of the tumors away from the original patient tumor characteristics, early passage xenograft material is cryo-preserved for each xenograft line. For xenograft lines that are maintained continuously in vivo for experimental studies, the xenograft lines are passaged only 15–20 times before being restored using early passage material. Similarly, lines that are used infrequently can be preserved in liquid nitrogen and then restored upon demand for a specific experiment. Standard procedure with each new xenograft line is to cryo-preserve tissue from five passages of the given heterotopic xenograft, which should provide adequate tissues to restore the xenograft line 50 times. If this resource nears depletion for any given line, then a restored early passage xenograft will be expanded and cryo-preserved. In this way, cycles of cryo-preservation and restoration can be used to maintain xenograft lines as early passage tumors indefinitely.
Materials
Mouse bearing tumor measuring 1 to 1.5 cm in greatest dimension (for cryo-preservation)
Recipient mouse (for restoration of cryo-preserved tumor tissue)
1.8-mL Cryotube (Corning or Nunc) Scalpels Sterile Culture Plates
Betadine
1-cc syringe
16-G hypodermic needle
-
Freezing Media:
DMEM (Mediatech)
Penicillin/Streptomycin (Cellgro; 5000 I.U./mL Pen, 5000 μg/mL strep (P/S))
DMSO (Fisher Scientific)
Fetal Bovine Serum (FBS) Premium (Atlanta Biologicals)
Cryo 1°C Freezing Container (Nalgene)
Reduced Matrigel (BD Biosciences)
Isoflurane (Novaplus)
Animal ear-tag punch (Fisher Scientific)
150-mL sterile filter (Nalgene)
Sterile phosphate-buffered saline (PBS) or HBSS (Cellgro)
Wet ice
Dry ice
Cryo-preservation of a flank tumor sample
Preparation
-
1)
Identify an appropriate mouse tumor for use, preferably measuring 1 to 1.5 cm in greatest dimension. Label the cryotubes with the lineage information for the tissue that is being preserved prior to euthanizing the mouse.
-
2)
Make up freezing media by adding 50 mL of FBS and 15 mL DMSO to 85 mL of Complete DMEM, then add penicillin and streptomycin to a final concentration of 1% each. Sterile filter the resulting freezing media. The freezing media can be stored at 4 C for up to 3 months.
-
3)
Prior to euthanizing the animal, label all specimen containers with tumor lineage and passage information.
This will be important for future studies to record the data in a file where the passaging, archiving and tumor preservation can be tracked.
Tumor Resection and Cryo-preservation
-
4)
Euthanize the tumor-bearing mouse by CO2 and swab the tumor area with Betadine.
-
5)
Cut out the tumor using a sterile scalpel, separate the tumor from the skin, and process as described in the previous section by mincing the tumor with sterile scalpels.
-
6)
Use a 1 cc syringe to break up the tumor into smaller chunks, pull up about 0.5 cc of tumor into the syringe, and then pull up about 0.5 cc of freezing media.
-
7)
Place tumor and freezing media into a pre-labeled cryotube (from step 3). Set tissue aside at room temperature for at least 30 min but no more than 60 min.
-
8)
Place cryotubes into the freezing container and place the freezing container into a −80°C freezer overnight. Transfer the cryo-preserved tissue from the −80°C freezer into a liquid nitrogen storage tank for long-term storage.
The freezing container controls the rate of cooling of the tumor samples.
-
9)
Record the pertinent tumor information on the tissue preserved and the archival location in a file exclusively for tracking xenograft information (e.g., an Excel spreadsheet).
Restoration of a xenograft from cryo-preserved material
Preparation
-
10)
Locate the tissue in the liquid nitrogen tank and update the tissue log to record its use.
-
11)
Pull the sample from the liquid nitrogen tank and place it on wet ice. Loosen the top of the cap slightly to allow any liquid nitrogen that entered the tube during the archiving process to escape or the tube may explode.
-
12)
Thaw Matrigel aliquot on wet ice.
Flank injection from cryo-preserved tissues
-
13)
Spin down the sample in a centrifuge at 320 RCF for 3 min, aspirate off the freezing media from the specimen, and re-suspend the entire contents of the tube in 200 μL of sterile PBS.
-
14)
Using a 1-cc syringe, pull up 200 μL of the tumor into a 1-cc syringe. Insert the syringe into a capped 16-G needle and place it back on wet ice.
-
15)
Draw up 200 μL of Matrigel into each syringe/needle and mix with the tumor by rapidly pulling the plunger back and forth. After dispelling the air from the tip of the needle, the total volume should be about 400 μL of a 1:1 solution.
-
16)
Inject tumor sample into the mouse as described in previous sections.
-
17)
Observe the animals weekly for visible tumor growth. Using our cryo-preservation technique, approximately 90–95% of cryo-preserved tumor samples are successfully restored. A restored tumor may take 2 to 3 months to start growing.
Basic Protocol 4: Establishing short-term explant cultures from xenograft lines
Short-term explant cultures can be readily derived from most established xenograft lines, and these cultures can be used for in vitro studies or in preparation for establishing intracranial tumors. Similar techniques are used for establishing both ‘standard cell cultures’ using serum-containing media or BTIC cultures using serum-free stem cell media. Although beyond the scope of the current manuscript, either culture model can be used for assessing in vitro drug sensitivity using cell proliferation or colony formation assays, performing mechanistic studies regarding drug efficacy, or for studying basic cancer biology in highly relevant tumor models.
When needed in preparation for tumor implantation, these short-term cultures can be transduced with lentiviral vectors for gene knock-down or over-expression studies or for in vivo imaging studies (Sarkaria, 2007). The ability to develop short-term cell cultures from xenografts provides tremendous versatility in the use of this xenograft model and can facilitate numerous experimental approaches.
Materials
Matrigel
Sterile Filtered complete DMEM (2.5% FCS and 1% Pen/Strep)
Sterile Filtered complete DMEM (10% FCS and 1% Pen/Strep)
100-mm culture dish (Sarstedt)
150-mm Tissue Culture plate (Corning)
Scalpels
1-cc syringe Wet ice
Short term explant cultures
Preparation
-
1)
Thaw Matrigel aliquots on wet ice.
-
2)
Make up a stock solution for coating plates by adding 400 μL of Matrigel to 3.6 mL of DMEM (2.5% FCS and 1% pen/strep) for each 150-mm plate to be coated. Coat plates with Matrigel by adding about 4 mL to each plate and then tipping the plates in order to coat the entire culture surface.
-
3)
Allow the plates to sit at room temperature on a level surface for about 30 min prior to use to allow the Matrigel to properly adhere to the plates.
Short term explant culturing
-
4)
Euthanize a mouse with a 1 to 1.5 cm tumor by CO2.
-
5)
Swab the tumor area with Betadine, remove the tumor, and mince as described above in an uncoated Petri dish.
-
6)
Add approximately 3 mL of complete DMEM (2.5% FCS and 1% P/S) to the plate and continue to disrupt the tumor chunks using a 1-cc syringe.
Tip the culture dish on its side and break up the tumor into smaller chunks by pulling it into the syringe and expelling it back into the plate many times. This will allow for better disruption of the tumor and a more even distribution of the cells on the tissue culture plates.
-
7)
Pull up 1-cc of tumor cells/media and place into each Matrigel-coated plate. If there are still cells/media left over, distribute evenly to each plate. Add an additional 10–25 mL of DMEM (2.5% FCS, 1% P/S) to the plate.
Additional media can be added after tumor disaggregation depending on the number of plates being seeded. Cells seem to grow best using Corning tissue culture plates.
Use of low percentage fetal bovine serum favors growth of glioma cells over murine fibroblasts. No other media supplements are required.
-
8)
Maintain cells in an incubator at 37°C and 5% CO2.
-
9)
Check the plates daily until the cells have adhered to the plates.
Depending on the tumor line, this may take between one to seven days. Take care to not disturb the cells until they have fully adhered to avoid cell loss.
-
10)
Once cells adhere to the plate, remove the debris from the plate by vigorously shaking the plate and then aspirating the media, debris, and non-adherent cells.
Do not beat the flasks against your hand as this may dislodge the cells. Similarly, do not wash the flasks with additional media, as this also may dislodge cells. A sterile Pasteur pipette can be used to remove any stubborn debris or tissue chunks.
-
11)
Replace media with DMEM containing 10% FBS and 1% P/S media once the cells have adhered to the plate.
-
12)
Monitor the cells until they are at about 80–90% confluent and change media as necessary.
Make sure that the cells are well fed. If the media turns yellow, they may not recover from the stress or they may change their response, making it difficult to reproduce experimental results.
The ability to passage cultured cells varies significantly for each tumor line. Some tumor lines require passaging at 1:2 or 1:3 dilutions, while others can be cut more aggressively. The dilution ratio must be determined empirically. For those lines obtained from Mayo Clinic, this culture dilution conditions can be provided if necessary.
The ability of the cultures to form xenografts tends to decrease with serial in vitro passaging, so only use cells that have been maintained in culture for less than 30 days.
-
13)
When re-plating the cells for in vitro assays, it is not necessary to use Matrigel-coated plates.
Short term explant cultures can be used for in vitro studies for evaluation of drug sensitivity or resistance and for understanding cellular mechanisms of sensitivity or resistance. As one example, temozolomide (TMZ) sensitivity was evaluated in short-term explant cultures derived from GBM12 and GBM43 flank tumor xenografts (Kitange, 2009). As seen in Figure 1A, GBM43 was significantly more resistant to TMZ as compared to GBM12. O6-methylguanine methyltransferase (MGMT) is a key protein that directly removes the cytotoxic lesion induced by TMZ. In both in vitro (Fig. 1B) and in vivo (not shown here) studies, we demonstrated that TMZ treatment was associated with a marked induction of MGMT expression in the TMZ resistant GBM43 line but not in the sensitive GBM12 tumor line. Aside from associative studies, the use of shRNA knockdown or gene overexpression in these tumor lines can be a powerful technique that can be used to demonstrate mechanistically the influence of a particular gene target on drug sensitivity or resistance. As mentioned above, we only use explant cultures for 30 days following removal from a flank tumor, but during that time, they can be used in an analogous manner to established cell cultures for a variety of translational and basic molecular investigations.
Figure 1. Analysis of TMZ resistance in short-term cell cultures.


Short-term explant cell cultures from GBM12 and GBM43 were treated with TMZ. (A) The effects on cell proliferation were evaluated 96 hours after treatment with graded concentrations of TMZ using a methylene blue staining assay. Mean ±SD for relative optical density (OD) vs. TMZ concentrations are plotted from 3 independent experiments. (B) Following treatment with 100 μM TMZ, cells were harvested at the indicated time points and processed for western blotting for MGMT and then βactin. Data reproduced with permission from (Kitange, 2009).
Basic Protocol 5: Intracranial and flank tumor implantation
Large numbers of animals can be implanted with tumor in the flank or intracranially in a single session. Several different methods can be used for generating a uniform population of cells for implantation including direct isolation of a single cell suspension from flank tumors or short-term explant cultures of cells. While direct isolation of a single cell suspension eliminates the use of cell culture and may have some theoretical advantages, we have used short-term explant cultures for establishing our large-scale tumor studies. Short-term explant cultures can provide a relatively pure population of tumor cells with reproducible intracranial growth characteristics. Moreover, when implanting a large number of mice, this technique allows one to trypsinize cells throughout the day, so that isolated tumor cells are not kept on ice for longer than three to four hours. This ensures optimal viability of cells throughout the implantation procedure. While the methods described above for direct flank to flank tumor injection for serial passaging can be scaled up to inject large numbers of animals with flank tumors, we have found that injection of flank tumors using short-term cultured cells provides more reproducible and uniform tumor growth than direct tumor passaging (unpublished data), which can be important for therapy evaluation experiments. Below, the techniques are described for both flank and intracranial tumor implantation from short-term explant cultures.
Flank tumor implantation
Materials
Short term explant cultured tumor cells (from Basic Protocol 4)
Matrigel
Trypsin-EDTA (Cellgro; 0.05% trypsin/0.53 mM EDTA in HBSS)
Complete DMEM (10% FCS and 1% P/S)
15-mL and 50-mL Conical Tubes (Falcon)
Sterile phosphate-buffered saline (PBS) 18-G × 1½ inch hypodermic needles
1-cc syringes
Isoflurane (Novaplus)
Centrifuge
Hemacytometer
Trypan Blue
Wet ice
Preparation
-
1)
Determine the number of flasks required for the study based on the number of cells that are required per animal and the number of animals in the study. Make sure enough cells are available to inject all the animals (most of the Mayo xenograft lines yield anywhere from 15 to 25 millions cells per flask at 80% confluence).
Each animal is implanted with 5×106 or 1×107 cells using short-term explant cultures established as described in the previous section.
-
2)
Aspirate off the media and trypsinize the cells with an ample amount of trypsin. A 150-mm flask will require 10 to 12 mL of trypsin. Flasks are incubated at room temperature for 3 to 10 min. Once cells begin rounding up and can be dislodged from the plate after striking against your palm, proceed to the next step.
-
3)
Add an equal volume of serum-containing media (10 to 12 ml) to inactivate the trypsin, transfer the cells and media to a conical tube, and spin down the cells at 320 RCF for 3 min. Resuspend the cells in 10 to 20 ml of PBS. Remove a small aliquot of cells for counting and spin the remaining cells at 320 RCF for 3 min. Count the cells using a hematocytometer and calculate the volume of PBS required for a cell concentration of 5×106 to 1×107 cells per 100 microL.
-
4)
Aspirate off the PBS from the cells, making sure not to disturb the cell pellet, and resuspend cells in the appropriate volume of PBS. Place cells on wet ice and pre-chill the syringes.
-
5)
Using a 1-cc syringe and an 18-G needle, draw up 100 μL of cells and 100 μL of Matrigel. Mix the Matrigel and cells in the syringe and remove the air from the syringe.
Using Matrigel should provide close to a 99% take rate.
Keep your syringes on wet ice to prevent the Matrigel from solidifying prior to injection.
-
6)
Anesthetize the mice with isoflurane and inject tumor cells subcutaneously as described in Section 1.
-
7)
Observe the mice two to three times per week for tumor development and growth. Depending on the tumor line, detectable tumors that are growing may take 1 to 12 weeks to appear.
Intracranial tumor implantation
Materials
Short term explant cultured tumor cells (from Basic Protocol 4)
Trypsin-EDTA (Cellgro; 0.05% trypsin/0.53 mM EDTA in HBSS)
DMEM (10% FBS, 1% P/S)
Centrifuge
Sterile PBS
1.5-mL sterile microcentrifuge tubes
Wet ice
Ketamine (100 mg/ml)
Xylazine (20 mg/ml)
Childrens liquid Tylenol (32 mg/ml)
0.5-cc tuberculin syringes
Dremel drill with a #7 or #8 bit
Betadine
Scalpels
Alcohol
10-μL Hamilton syringe with a 26-G needle
Spore-klenz or similar disinfectant
Stereotactic frame (ASI Instruments) with a neonatal rat adaptor (Stoelting)
50 mL sterile conical (Falcon)
4–0 vicryl with rb-1 needle (Ethicon J30 4H)
Artificial tears (Petrolatum opthalmic ointment, Puralub Vet Ointment, Dechra)
Triple antibiotic (Bacitracin, Neomycin, Polymyxin B sulfate, G&W Laboratories)
Heating pad (optional)
Radiofrequency identification (RFID) chips and reader (optional; Datamars Companion Animal ID; www.datamars.com).
Preparation of cells
-
1)
Prepare cells for injection as described above for flank tumor injection, but resuspend the cells in sterile PBS at 100,000 cells/μL in a 1.5-mL sterile microcentrifuge tube.
Some cell lines can be difficult to draw into the syringe, so test this prior to anesthetizing the animals. If the cells are difficult to draw up, then discard the cells and trypsinize a back-up flask(s) of cells.
-
2)
Place the tube containing the cell suspension on wet ice.
Cells are viable for up to four hours when kept on wet ice.
Anesthesia and skull preparation for injection
-
3)
Provide Children’s Tylenol in the animal drinking water (final concentration 1.33 mg/mL water) starting 24 hr prior to the procedure, and maintain the Tylenol in the water for at least 48 hr after the procedure.
Monitor the water for signs of bacterial or fungal growth associated with the flavoring of the Tylenol and change water if necessary.
-
4)
Prepare the anesthetic mixture by adding 2 mL Ketamine and 1 mL of Xylazine to 17 mL of normal saline. For a 20-g mouse, use approximately 200 μL of the ketamine/xylazine mixture injected intraperitoneal (IP) with a 0.5-cc syringe for a dose of 100 mg/kg ketamine and 10 mg/kg xylazine.
This dose usually provides 30 min of anesthesia. The amount of anesthetic will vary based upon the strain and size of the animals being used. For example, SCID mice require less anesthetic for effective anesthesia. Adequate anesthesia is assessed by toe pinch of the hind leg. If the mouse withdraws from this pinch, then either wait longer for the anesthetic to take effect or consider providing an additional dose of anesthetic.
-
5)
Use a disinfectant regularly throughout the injection process (e.g. Spore-Klenz) to ensure that your hands remain as sterile as possible to reduce the risk of infection in the mice.
-
6)
Swab the head with Betadine, and lubricate the eyes with artificial tears. Make a 1-cm midline incision extending from just behind the eyes to level of the ears using a sterile scalpel while applying pressure to the skin so that the skin separates as you are making the incision.
-
7)
Using your fingers, push apart the skin to expose the skull. From the bregma, identify a point 1 mm anterior and 2 mm lateral, and drill through the skull using a #7 or #8 bit in a Dremel drill.
The bregma is the junction of the sagittal and coronal sutures as shown in Figure 2.
There is a tactile sensation of a slight “popping” when the bit penetrates the skull. Keep a firm grip on the drill and the animal to prevent the drill bit from skipping across the skull and to prevent the mouse from moving as the drill bit establishes purchase on the skull.
A Dremel drill can be purchased from any local hardware store.
Figure 2. Skull anatomy of a mouse.
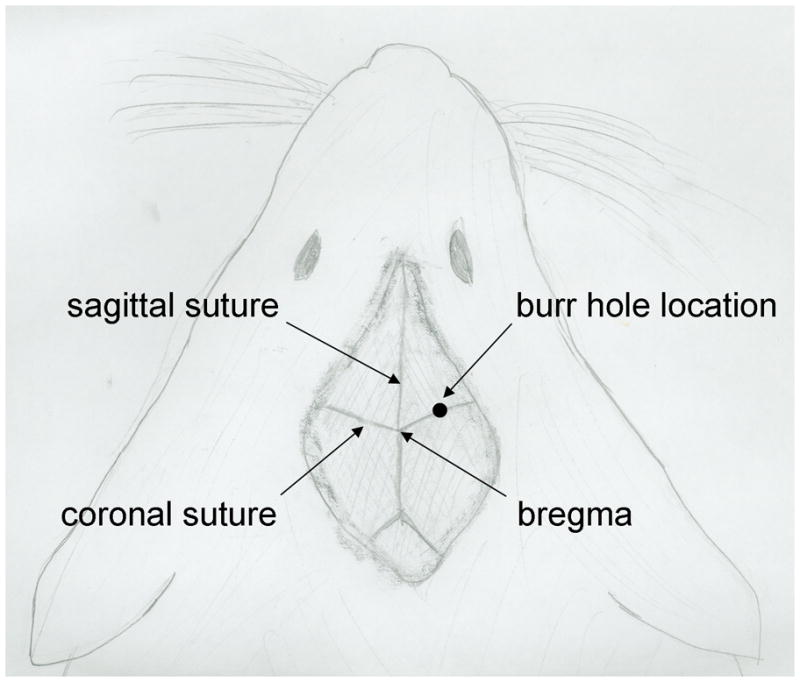
The diagram shows the typical exposure of the skull bones following a mid-line incision. The relationship between the sagittal and coronal sutures are shown as well as the location of the bregma. The burr hole location typically used is 1 mm anterior and 2 mm lateral to the bregma.
Stereotactic injection
-
8)
Clean the Hamilton syringe/needle assembly thoroughly by drawing up alcohol into the syringe and dipping the plunger into the alcohol. Rinse the syringe and needle by drawing PBS into the syringe multiple times. Sterilize the injection jig by wiping it down with Spore-Klenz and drape it with a sterile towel.
-
9)
Using a neonatal rat adaptor in association with the stereotactic device, place the mouse in the jig by their front teeth (Figure 3).
Using the ear pins is not necessary.
Placing the mouse on the jig prior to loading the needle will help to minimize the risk of a needle stick.
-
10)
Mix the cells by flicking the tube with your finger several times and draw up 3 μL of cells in a 10-μL Hamilton syringe with a 26-G needle. Make sure that you visualize the cells being drawn up into the syringe. There is usually a small air bubble that is noticeable in the syringe and is a good indicator that the cells are loaded in the syringe.
Confirming that cells have been pulled up into the syringe is critical, since failure to inject the appropriate number of cells will have an obvious impact on the formation of intracranial tumors and can lead to significant variability in the time to reach a moribund state.
-
11)
Insert the needle and syringe into the holder and place the needle just within the drill hole. Using the stereotactic controls, drive the needle 3 mm into the brain (for large mice) or 2 mm (for particularly small mice).
-
12)
Inject 1 μL of cells per min over 3 min. This can be done either manually or with a syringe pump. Allow the needle to remain in the brain for an additional minute (total injection time is 4 min). There is no need to use cement or other material to seal the drill hole in the skull.
-
13)
Remove and clean the needle and syringe with alcohol followed by PBS.
-
14)
Remove the mouse from the jig, and suture the wound with 4-0 vicryl with rb-1 needle (Ethicon J304H).
Use two to three sutures depending on the size of the wound for the head.
If applicable, use one suture for closing the wound from inserting the RFID chips (see
step 15).
Apply triple antibiotic liberally to the incision and stitches to prevent infection.
-
15)
Insert the RFID chips (if appropriate) while the mouse is under anesthetic. This can be done either before or after stereotactic injection. The RFID chip typically comes pre-loaded in 13-G trocar. Insert the trocar under the skin in the subcutaneous space and depress the plunger to push the chip out under the skin.
Turn the chip approximately perpendicular to the surgical wound to ensure that the chip does not get extruded from the injection site.
-
16)
Place a paper towel in a clean cage on the bedding chips on the opposite side from the water bottle.
Hypothermia can be a major problem for mice as they recover from anesthesia. Therefore, lay 5 mice per cage next to and on top of each other to ensure adequate body temperature is maintained. Alternatively, place a portion of the cage over a heating pad. If you use this approach, make sure the heating pad is not too hot otherwise the mice will die from overheating.
Make sure the animals are not getting water dripped on them as this also can lead to hypothermia.
-
17)
Observe the animals to make sure that they wake up from the anesthetic. Once mice have fully recovered from anesthetic (walking around and normally active), their cages can be moved to the housing unit.
-
18)
Mice with intracranial tumors must be observed daily, as neurologic decline can occur rapidly, at which point mice should be euthanized. Typical signs of neurologic decline include a hunched posture with an arched back, circling, walking on their tip-toes, balance issues, eyes that are not opened, weight loss, hyperactivity, and seizures.
Figure 3. Stereotactic injection set-up.



A–B)The stereotactic injection jig with a mouse in place is shown. C) By using multiple injection jigs in an assembly line fashion, 2 or 3 technicians can implant 100 mice in 4 hours. Photographs courtesy of Cory Petell.
Evaluation of standard or novel therapeutic strategies in orthotopic GBM xenografts can provide important translational insights into the potential for efficacy and mechanisms of anti-tumor effect. In one study, response to therapy with the EGFR inhibitor erlotinib was evaluated in 12 GBM xenograft lines in an orthotopic therapy evaluation model (Sarkaria, 2007). While previous studies had suggested that wild-type PTEN status and EGFR amplification were required for sensitivity to erlotinib therapy, our data demonstrated that only 2 of 5 tumor lines with this genetic phenotype were responsive to erlotinib therapy (Figure 4A) and none of the 7 tumors without this phenotype responded to therapy. Using tumors from mice treated with placebo or erlotinib for 5 days, immunohistochemistry for the MIB-1 proliferation marker demonstrated a significant reduction in tumor proliferation in GBM39 tumors (Figure 4B). To further understand how erlotinib therapy influences tumor growth, short-term explant cultures were transduced with a lentivirus expressing firefly luciferase and subsequently established orthotopic xenografts were imaged by bioluminescent imaging. These studies demonstrated that after approximately 2 weeks of erlotinib therapy the tumor burden was significantly reduced with erlotinib treatment, and that once treatment was discontinued, the tumors regrew rapidly (Figure 4C-D). Similar studies have been performed by our group or the evaluation of numerous standard and novel therapeutic strategies including responses to radiation, temozolomide, everolimus and viral therapy (Allen, 2008, Carlson, 2009, Clarke, 2009, Dinca, 2008, Dinca, 2007, Kitange, 2009, Kitange, 2009, Liu, 2007, Sarkaria, 2006, Yang, 2008).
Figure 4. Evaluation of erlotinib efficacy in orthotopic xenografts.
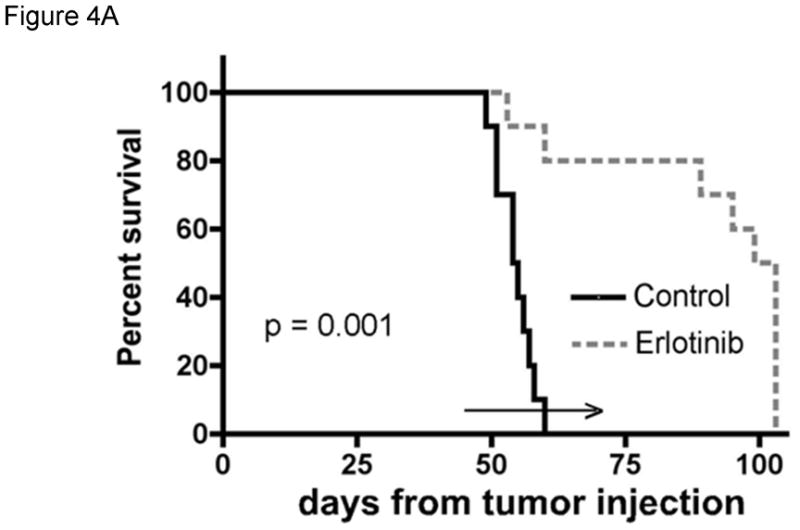
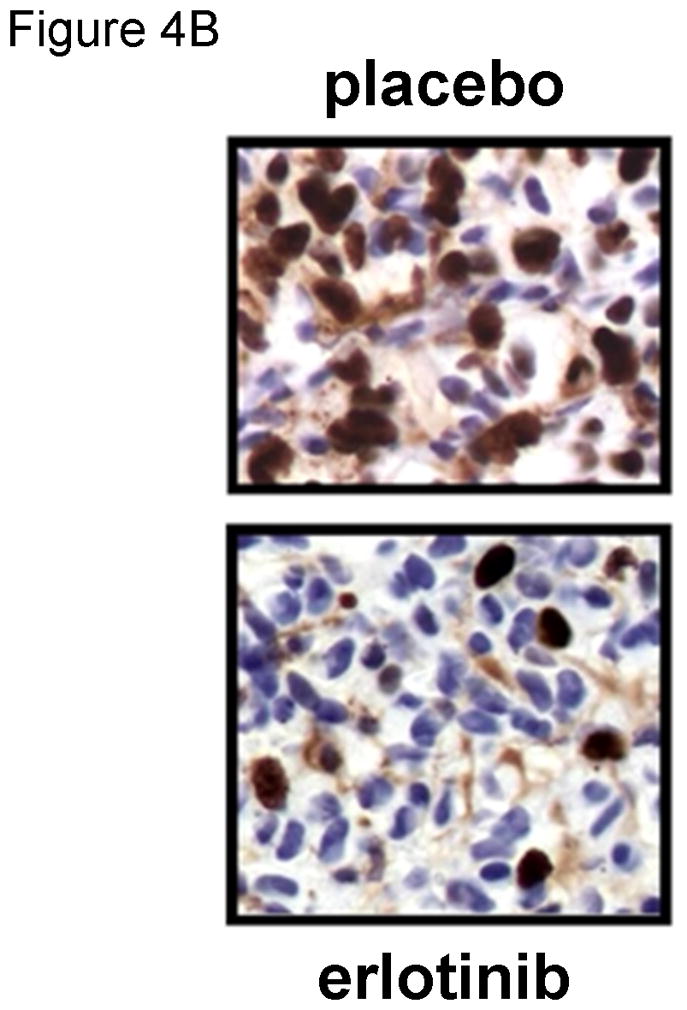


The efficacy of erlotinib was evaluated in mice bearing orthotopic GBM39 tumors transduced with a luciferase-expressing lentivirus prior to implantation. A) Kaplan-Meier survival curves for mice treated with daily oral erlotinib or placebo as shown by the horizontal arrow. B) Moribund mice were processed for MIB-1 IHC to assess changes in tumor cell proliferation in vivo. C). Luminescence intensity overlays showing serial results for a single placebo treated mouse and a single erlotinib treated mouse, with images recorded at days 42, 53, and 60 days for each, and additionally at days 67, 74, and 81 for the erlotinib-treated mouse. D) Luminescence readings were converted to normalized values by dividing each mouse’s luminescence readings with its corresponding maximal pre-treatment luminescence reading recorded at day 42. Mean normalized bioluminescence and corresponding standard error values for placebo and erlotinib groups have been plotted for each imaging session. The duration of treatment is indicated by the horizontal arrow. Data reproduced with permission from (Sarkaria, 2007).
COMMENTARY
Background Information
The Mayo Clinic GBM xenograft panel is comprised of 45 flank tumor xenografts and has been widely used for basic and translational studies. Of the 35 lines tested in an orthotopic location, 33 tumor lines form intracranial tumors within 180 days with histopathological features highly similar to those seen in patient tumor samples (unpublished data) (Giannini, 2005). As discussed above, flank tumors can be used for direct tumor passage, or they can be grown in short-term explant cultures and these cultures can be used for in vitro studies or subsequent tumor implantation into recipient mice. Tumor lines from the Mayo Clinic GBM xenograft panel have been extensively distributed to over 45 different institutions worldwide and any given tumor line is available for a nominal fee following execution of a materials transfer agreement. Tumor lines are shipped to recipient institutions either as fresh flank tumor specimens suspended in HBSS, or as short-term explant cultures shipped in tissue culture media. Archived flash frozen flank tumor specimens and formalin-fixed paraffin embedded intracranial tumor specimens also are available from all tumor lines for basic and translational studies upon request. Thus, the Mayo Clinic xenograft panel is a readily available resource of primary GBM xenograft models for basic and translational studies. Although we do not yet have a website developed, contact the corresponding author for more information or to request tumor tissues.
A subset of the Mayo Clinic GBM xenograft lines have been extensively characterized for both molecular features and response to various treatments. Common molecular features observed in each of the tumor lines are summarized in Table 1. Of these lines, 10 of 20 lines have non-amplified EGFR, 6 of 20 have amplified full-length EGFR, and 3 of 20 have the EGFR-viii activating truncation mutation. Similarly, 7 of 20 have p53 mutation and 8 of 20 have either PTEN mutation or deletion. Interestingly, 18 of 20 tumors have p16 gene deletion, which is much higher than the approximately 50% rate of p16 deletion observed in patient tumor samples (2008). Given that only a third of patient tumor samples engrafted form robust xenograft lines, these data suggest that p16 deletion may be a molecular lesion that promotes xenograft development. In addition to molecular characterization, multiple xenograft lines have been evaluated for response to erlotinib, everolimus, radiation, and temozolomide (Carlson, 2009, Sarkaria, 2007, Yang, 2008), and the results from the studies with radiation and temozolomide are summarized in Table 2. MGMT promoter methylation is an important driver of temozolomide sensitivity, and the status of MGMT methylation also is noted in this table. Collectively, these molecular and phenotypic results can be useful for planning both basic and translational studies.
Table 1.
Molecular features of Mayo GBM panel
| GBM | EGFR amp | Pten status | p53 mutation | p16 deletion |
|---|---|---|---|---|
| 5 | No | Wt | None | No |
| 6 | Yes viii | Wt | 273: arg>cys | Yes |
| 8 | Yes wt | HD | None | Yes |
| 10 | No | Wt | None | Yes |
| 12 | Yes wt | Wt | 5, 3 Osplice | Yes |
| 14 | No | 2bp del, exon 1 | None | Yes |
| 15 | Yes wt | Wt | None | Yes |
| 16 | No | HD | None | Yes |
| 22 | No | Wt | 273: arg>cys | Yes |
| 26 | Yes wt | HD | None | Yes |
| 28 | No | 132: gly>asp | 246: met>thr | No |
| 34 | Yes wt | Wt | None | Yes |
| 36 | No | HD | 132:lys>met | Yes |
| 38 | Yes wt | Wt | 110: arg>cys | Yes |
| 39 | Yes viii | Wt | None | Yes |
| 43 | No | Wt | 270: phe>cys | Yes |
| 44 | No | Wt | None | Yes |
| 46 | Yes vii | Wt | N/A | Yes |
| 58 | No | HD | None | Yes |
| 59 | Yes viii | HD | None | Yes |
Table 2.
Response to radiation (RT) and temozolomide (TMZ) of Mayo Primary GBM panel
| Survival Ratio (p-value) | ||||
|---|---|---|---|---|
| GBM# | MS-PCR | TMZ | RT | TMZ/RT |
| GBM5 | M | 5.84 (<0.0001) | 2.53 (0.0003) | 6.35 (<0.0001) |
| GBM6 | U | 1.39 (0.0005) | 1.02 (0.23) | 2.39 (0.04) |
| GBM8 | M | 2.11 (<0.0001) | 1.83 (0.001) | 3.23 (0.07) |
| GBM10 | U | 1.34 (0.10) | 1.61 (0.003) | 1.85 (0.0003) |
| GBM12 | M | 3.53 (<0.0001) | 2.47 (<0.0001) | 5.70 (<0.0001) |
| GBM14 | M | 5.64 (<0.0001) | 1.68 (<0.0001) | 5.70 (0.0003) |
| GBM15 | M | 4.68 (0.0001) | 4.63 (<0.0001) | 4.67 (0.0001) |
| GBM16 | M | 6.85 (0.02) | 1.35 (0.47) | 7.22 (0.10) |
| GBM22 | M | 3.59 (0.0009) | 1.00 (0.29) | 7.57 (0.0009) |
| GBM26 | U | 1.21 (0.003) | 1.58 (0.0001) | 1.58 (0.0001) |
| GBM28 | U | 1.70 (0.003) | 2.06 (0.0001) | 2.50 (<0.0001) |
| GBM34 | U | 4.31 (<0.0001) | 1.84 (0.01) | 4.60 (0.0001) |
| GBM36 | M | 1.82 (0.14) | 1.55 (0.05) | 2.45 (0.02) |
| GBM38 | U | 1.45 (0.0001) | 1.04 (0.21) | 1.36 (0.003) |
| GBM39 | M | 3.87 (<0.0001) | 1.63 (<0.0001) | 6.52 (<0.0001) |
| GBM43 | U | 1.79 (0.003) | 1.71 (0.04) | 2.29 (0.002) |
| GBM44 | U | 1.69 (0.05) | 2.97 (0.008) | 3.23 (<0.0001) |
| GBM46 | M | 1.13 (0.22) | 2.31 (0.0007) | 2.54 (<0.0001) |
| GBM58 | U | 1.11 (0.25) | 1.09 (0.35) | 1.21 (0.14) |
| GBM59 | M | 1.96 (<0.0001) | 1.61 (0.0004) | 5.52 (<0.0001) |
MS-PCR – methylation specific PCR; M – methylated, U – unmethylated Survival ratio is the ratio of median survival for treatment vs. placebo. p-value refers to the comparison of survival for the indicated treatment relative to placebo treatment. Reproduced from (Carlson, 2009). Mice were treated with TMZ at 66 mg/kg/day for 5 days and/or RT at 2 Gy twice per day × 5 days. Dosing was initiated 2 weeks prior to mice anticipated to become moribund for a given tumor line.
Prolonged serial xenograft passage can select for more aggressive growth characteristics over time. Within the Mayo Clinic GBM xenograft panel, we initially maintained all tumor lines by continuous serial passage, with some tumor lines being passaged for up to 50 generations. During the course of tumor passage, flank tumor samples were used repeatedly to establish orthotopic xenografts for basic or translational studies: a total of 114 intracranial experiments were performed using 19 different tumor lines. To evaluate whether prolonged tumor passage affected intracranial growth, an association between tumor generation and median survival for placebo-treated mice for each experiment was considered. Analyzing each tumor line individually, there was an inverse correlation between median survival and tumor generation in 14 of 19 lines (74%), and on univariate analysis in which all tumor lines are pooled, this inverse correlation was highly significant (r=−0.40, p<0.001; Figure 5). In a multivariate analysis accounting for both generation and tumor line, there was no significant generation by tumor line interaction (p=0.247), while when adjusting for tumor line, there remained a significant interaction between survival time and generation (p<0.001). These data suggest that the xenograft lines become more aggressive with extended flank tumor passaging. On the basis of these data and a general concern that prolonged tumor passage may lead to genetic drift, all GBM xenograft lines are now maintained as relatively early passage generations (passage 5 to 20) using cryo-preserved tumor material to restore early passage tumors as required.
Figure 5. Changes in median survival with increasing generation.
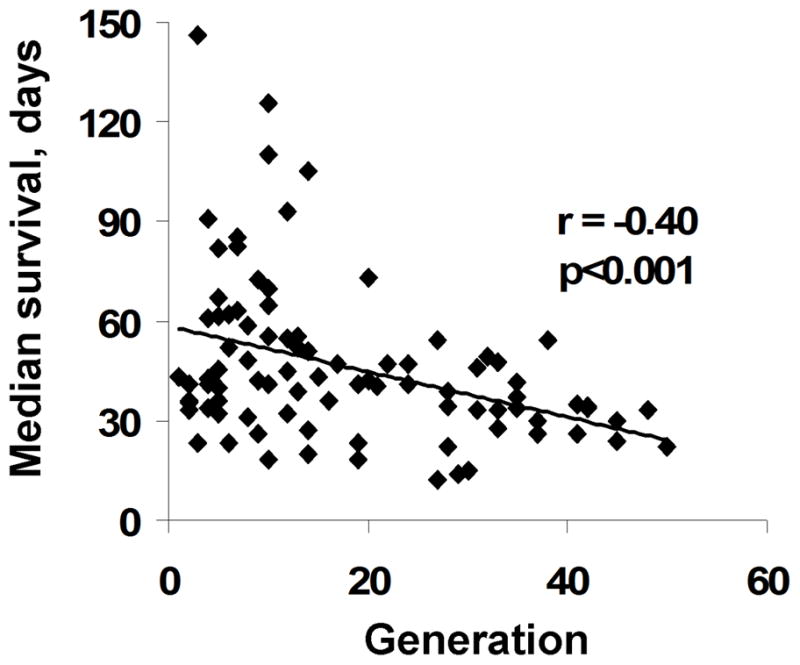
The median survival for placebo-treated mice entered into 114 intracranial survival studies involving 19 different xenograft lines is plotted relative to the generation from which any given study was derived.
The GBM xenograft model is one of four commonly used intracranial GBM tumor model systems for studying glioma therapeutics, with the other models being 1) established glioma tumor lines, 2) genetically-engineered mouse models (GEMMs), and 3) glioma stem cells (BTIC). Established glioma cell lines are the most pervasive models due to their convenience. These lines were initially established by culturing brain tumor specimens in serum-containing media on plastic, and most of these lines, such as U87 or U251 cell lines, grow extremely well in culture. They are highly characterized for numerous phenotypic and molecular features and have been used extensively in almost every aspect of glioma biology. However, the extended culturing of these glioma lines in tissue culture has led to significant genetic drift (Li, 2008). Many established tumor lines are not tumorigenic when implanted into the brain or they form tumors that are non-invasive and do not reflect the pathology of typical GBM tumors. Thus, established glioma models are highly convenient, typically have robust growth characteristics in vitro, but have the least similarity to primary human GBM tumors.
Multiple GEMM systems have been described that generate malignant gliomas with features highly similar to human brain tumors (reviewed in Huse, 2009). These models typically rely on deletion of a key tumor suppressor gene, often coupled with over-expression of an oncogene. These models are highly useful for evaluating mechanisms of gliomagenesis and for testing novel therapeutics in a tumor type with a select genetic defect. Because they are syngeneic, these models also are highly relevant for evaluating tumor microenvironment interactions and tumor-directed immunity. However, these models typically require serial imaging for detection of tumor-bearing mice and often less than 100% of mice develop tumors within the time-frame of the experiment. Also, by their nature, the genetic diversity that can be evaluated in these GEMM models is limited. Since there likely are multiple genetic factors that determine the efficacy of any one therapy, we believe these models provide a relatively limited understanding of potential molecular features that govern response to conventional or molecularly targeted therapeutics.
BTIC cultures, otherwise known as tumor stem cell cultures, are a highly complementary model system to GBM xenografts. While early experience suggested a 30–40% success rate in establishing robust BTIC cultures, recent publications suggest that modifications in the technique for establishing these cultures can yield high take rates with stem cell cultures derived from over 90% of tumor specimens (Fael Al-Mayhani, 2009, Pollard, 2009). This high take rate provides a broad genetic diversity of tumor models for evaluation of novel therapeutics. Moreover, while culture medium for BTIC is expensive, this model is more cost-efficient than maintaining a GBM xenograft panel by serial subcutaneous passaging in animals. However, in vitro expansion of BTIC cultures for implanting large numbers of mice for in vivo therapy evaluation studies can be challenging for some lines, and typically, the time to reach a moribund state for orthotopic xenografts established from BTIC cultures tends to be longer than with established xenograft lines. Thus, there are significant advantages for BTIC cultures, but some potential disadvantages specifically for testing therapeutic strategies in vivo.
Critical Parameters and Troubleshooting
Maintenance of a xenograft panel and large-scale in vivo experimentation requires careful attention to animal husbandry and detailed record keeping. While the regulatory requirement for use of anonymous tumor tissue is much less rigorous, access to the patient history and the primary patient tumor specimen from which a tumor was derived can be highly useful. For example, molecular analyses comparing patient tumors and the derivative xenograft lines are important for validating the clinical relevance of the derived tumor lines. Moreover, having the primary patient tissues and early archived xenograft samples can be useful in the event that xenograft lines become mislabeled. To allow definitive identification of any given tumor line, we have defined a molecular signature for each xenograft line using microsatellite analysis (unpublished data – signature of each line available upon request), and the signature of a specific tumor line can be compared to this baseline signature to identify mislabeled tumor lines.
A xenograft line is typically maintained in three mice at any one time. For those tumors with slow growth in the flank, staggering the passage of these tumors can provide a relatively steady supply of tumor material for in vitro and in vivo studies. Despite maintaining tumors in multiple mice, occasionally a tumor line will be lost if none of the three mice develop tumors upon passage or mice are lost due to non-tumor related issues, such as a leaking water bottle. In this case, we have used our cryo-preserved tumor samples to restore a xenograft line. Of note, tumors may take two to three months to begin growing when using cryo-preserved material.
The growth characteristic of each tumor line is relatively unique, and gaining experience with these tumor lines can improve the efficiency of using these lines. In our xenograft panel, the approximate time between tumor passage for flank tumor xenografts and the median time from intracranial tumor implantation to reaching a moribund state can vary between one and three months in flank tumors and 18 to 120 days for intracranial xenografts. Similarly, the relative ease of growing these tumor lines in cell culture and specific requirements for cell culture can vary. As with other primary tumor lines, the growth rate of cells in vitro generally increases with prolonged cell culture, which can be more convenient for in vitro studies, but also can lead to greater genetic divergence from the primary tumor. As noted above, available in vivo data suggest that xenograft lines become more aggressive with extended flank tumor passaging; again, a convenience for in vivo studies, but a phenotype which may be divergent from the primary GBM. Most tumor lines lose the ability to form intracranial tumors with extended passage in cell culture, and thus cells that have been cultured for longer than three weeks are not used to re-establish either intracranial or flank tumor xenografts.
Anticipated results
Primary xenografting is a resource-intensive exercise requiring a long-term commitment to the model. As discussed above, using our current techniques, approximately one-third of implanted primary patient specimens develop into usable, robust GBM xenograft lines. Following initial implantation of the patient specimen, a growing tumor is usually observed within six to nine months, but mice are observed for at least 12 months before euthanizing a mouse without a visible tumor. Tumor growth rate usually picks up in the second and third tumor generation, and becomes relatively stable by the fourth generation. Overall, a total of two years can be required to establish a usable xenograft line with robust growth characteristics.
Establishing short-term explant cultures can be done relatively routinely with a high success rate. Occasionally, a specific culture will grow poorly and such a culture should be discarded, and not used for intracranial implantation. These cultures grow slower than established tumor cell lines and may require a longer time to adhere to plates and resume growth following tissue culture passaging. Every attempt should be made to use the tumor cells with a minimum of in vitro passages, since tumor cell growth tends to slow with tissue culture passage. Use of robust cell cultures is especially important for orthotopic tumor implantation, and with careful attention to technique, the take-rate of intracranial tumor implantation should approach 99%.
Time considerations
The time required for implanting flank tumor samples is relatively short. An experienced technician can process and implant a primary patient tumor specimen into the flank of a nude mouse in less than one hour. A similar amount of time is required for passaging of a flank tumor from one mouse to another or to process a tumor for establishing a short-term explant culture. The rate of intracranial implantation can be highly variable depending on the level of experience of the technicians. In our laboratory, intracranial implantation is performed in a production-line fashion with two or three technicians using up to five stereotactic injection jigs, and with this technique, 25 to 30 mice can be implanted per hour. With a single operator using just one stereotactic injection jig, five to 10 mice can be implanted per hour. Once mice have been implanted with intracranial tumors, observing mice for neurologic signs associated with tumor burden takes less than one minute for each cage, so that therapy evaluations with survival as an endpoint can be performed relatively easily following the initial time investment to establish the intracranial tumors.
Acknowledgments
Grant Support: These studies supported by the Mayo Clinic and the National Cancer Institute (RO1 - CA127716 and the Mayo Brain Tumor SPORE - CA108961)
LITERATURE CITED
- 1.Allen C, Paraskevakou G, Iankov I, Giannini C, Schroeder M, Sarkaria J, Puri RK, Russell SJ, Galanis E. Interleukin-13 displaying retargeted oncolytic measles virus strains have significant activity against gliomas with improved specificity. Molecular Therapy: the Journal of the American Society of Gene Therapy. 2008;16:1556–1564. doi: 10.1038/mt.2008.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carlson BL, Grogan PT, Mladek AC, Schroeder MA, Kitange GJ, Decker PA, Giannini C, Wu W, Ballman KA, James CD, Sarkaria JN. Radiosensitizing Effects of Temozolomide Observed in vivo only in a Subset of O6-Methylguanine-DNA Methyltransferase Methylated Glioblastoma Multiforme Xenografts. Int J Radiat Oncol Biol Phys. 2009;75:212–219. doi: 10.1016/j.ijrobp.2009.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clarke MJ, Mulligan EA, Grogan PT, Mladek AC, Carlson BL, Schroeder MA, Curtin NJ, Lou Z, Decker PA, Wu W, Plummer ER, Sarkaria JN. Effective sensitization of temozolomide by ABT-888 is lost with development of temozolomide resistance in glioblastoma xenograft lines. Mol Cancer Ther. 2009;8:407–414. doi: 10.1158/1535-7163.MCT-08-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Danam RP, Howell SR, Remack JS, Brent TP. Heterogeneous methylation of the O(6)-methylguanine-DNA methyltransferase promoter in immortalized IMR90 cell lines. Int J Oncol. 2001;18:1187–1193. doi: 10.3892/ijo.18.6.1187. [DOI] [PubMed] [Google Scholar]
- 5.Dinca EB, Lu KV, Sarkaria JN, Pieper RO, Prados MD, Haas-Kogan DA, Vandenberg SR, Berger MS, James CD. p53 Small-molecule inhibitor enhances temozolomide cytotoxic activity against intracranial glioblastoma xenografts. Cancer Res. 2008;68:10034–10039. doi: 10.1158/0008-5472.CAN-08-1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dinca EB, Sarkaria JN, Schroeder MA, Carlson BL, Voicu R, Gupta N, Berger MS, James CD. Bioluminescence monitoring of intracranial glioblastoma xenograft: response to primary and salvage temozolomide therapy. J Neurosurg. 2007;107:610–616. doi: 10.3171/JNS-07/09/0610. [DOI] [PubMed] [Google Scholar]
- 7.Fael Al-Mayhani TM, Ball SLR, Zhao JW, Fawcett J, Ichimura K, Collins PV, Watts C. An efficient method for derivation and propagation of glioblastoma cell lines that conserves the molecular profile of their original tumours. J Neurosci Methods. 2009;176:192–199. doi: 10.1016/j.jneumeth.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 8.Frederick L, Wang XY, Eley G, James CD. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000;60:1383–1387. [PubMed] [Google Scholar]
- 9.Giannini C, Sarkaria J, Saito A, Uhm J, Galanis E, Carlson B, Schroeder M, James C. Patient Tumor EGFR and PDGFRA Gene Amplifications Retained in an Invasive Intracranial Xenograft Model of GBM. Neuro-Oncology. 2005;7:164–176. doi: 10.1215/S1152851704000821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huse JT, Holland EC. Genetically engineered mouse models of brain cancer and the promise of preclinical testing. Brain Pathol. 2009;19:132–143. doi: 10.1111/j.1750-3639.2008.00234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kitange G, Carlson B, Mladek A, Decker P, Schroeder M, Wu W, Grogan P, Giannini C, Ballman K, Buckner J, David James C, Sarkaria J. Evaluation of MGMT promoter methylation status and correlation with temozolomide response in orthotopic glioblastoma xenograft model. J Neurooncol. 2009;92:23–31. doi: 10.1007/s11060-008-9737-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kitange GJ, Carlson BL, Schroeder MA, Grogan PT, Lamont JD, Decker PA, Wu W, James CD, Sarkaria JN. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro-oncol. 2009;11:281–291. doi: 10.1215/15228517-2008-090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li A, Walling J, Kotliarov Y, Center A, Steed ME, Ahn SJ, Rosenblum M, Mikkelsen T, Zenklusen JC, Fine HA. Genomic changes and gene expression profiles reveal that established glioma cell lines are poorly representative of primary human gliomas. Molecular Cancer Research: MCR. 2008;6:21–30. doi: 10.1158/1541-7786.MCR-07-0280. [DOI] [PubMed] [Google Scholar]
- 14.Liu C, Sarkaria JN, Petell CA, Paraskevakou G, Zollman PJ, Schroeder M, Carlson B, Decker PA, Wu W, James CD, Russell SJ, Galanis E. Combination of measles virus virotherapy and radiation therapy has synergistic activity in the treatment of glioblastoma multiforme. Clin Cancer Res. 2007;13:7155–7165. doi: 10.1158/1078-0432.CCR-07-1306. [DOI] [PubMed] [Google Scholar]
- 15.Pandita A, Aldape KD, Zadeh G, Guha A, James CD. Contrasting in vivo and in vitro fates of glioblastoma cell subpopulations with amplified EGFR. Genes Chromosomes & Cancer. 2003;39:29–36. doi: 10.1002/gcc.10300. [DOI] [PubMed] [Google Scholar]
- 16.Pegg AE. Mammalian O6-alkylguanine-DNA alkyltransferase: regulation and importance in response to alkylating carcinogenic and therapeutic agents. Cancer Res. 1990;50:6119–6129. [PubMed] [Google Scholar]
- 17.Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, Bayani J, Head R, Lee M, Bernstein M, Squire JA, Smith A, Dirks P. Glioma Stem Cell Lines Expanded in Adherent Culture Have Tumor-Specific Phenotypes and Are Suitable for Chemical and Genetic Screens. Cell Stem Cell. 2009;4:568–580. doi: 10.1016/j.stem.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 18.Sarkaria JN, Carlson BL, Schroeder MA, Grogan P, Brown PD, Giannini C, Ballman KV, Kitange GJ, Guha A, Pandita A, James CD. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clin Cancer Res. 2006;12:2264–2271. doi: 10.1158/1078-0432.CCR-05-2510. [DOI] [PubMed] [Google Scholar]
- 19.Sarkaria JN, Yang L, Grogan PT, Kitange GJ, Carlson BL, Schroeder MA, Galanis E, Giannini C, Wu W, Dinca EB, James CD. Identification of molecular characteristics correlated with glioblastoma sensitivity to EGFR kinase inhibition through use of an intracranial xenograft test panel. Mol Cancer Ther. 2007;6:1167–1174. doi: 10.1158/1535-7163.MCT-06-0691. [DOI] [PubMed] [Google Scholar]
- 20.Smith JS, Tachibana I, Passe SM, Huntley BK, Borell TJ, Iturria N, O’Fallon JR, Schaefer PL, Scheithauer BW, James CD, Buckner JC, Jenkins RB. PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J Natl Cancer Inst. 2001;93:1246–1256. doi: 10.1093/jnci/93.16.1246. [DOI] [PubMed] [Google Scholar]
- 21.TCGA. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamada H, Vijayachandra K, Penner C, Glick A. Increased sensitivity of transforming growth factor (TGF) beta 1 null cells to alkylating agents reveals a novel link between TGFbeta signaling and O(6)-methylguanine methyltransferase promoter hypermethylation. J Biol Chem. 2001;276:19052–19058. doi: 10.1074/jbc.M100615200. [DOI] [PubMed] [Google Scholar]
- 23.Yang L, Clarke MJ, Carlson BL, Mladek AC, Schroeder MA, Decker P, Wu W, Kitange GJ, Grogan PT, Goble JM, Uhm J, Galanis E, Giannini C, Lane HA, James CD, Sarkaria JN. PTEN Loss Does Not Predict for Response to RAD001 (Everolimus) in a Glioblastoma Orthotopic Xenograft Test Panel. Clin Cancer Res. 2008;14:3993–4001. doi: 10.1158/1078-0432.CCR-07-4152. [DOI] [PMC free article] [PubMed] [Google Scholar]