

Abstract
We report the preparation and physical and biological characterization of human serum albumin-based micelles of approximately 30 nanometers diameter for the delivery of amphipathic drugs, represented by doxorubicin. The micelles were surface conjugated with cyclic RGD peptides to guide selective delivery to cells expressing the alpha v/beta 3 integrin. Multiple poly(ethylene glycol)s (PEGs) with molecular weight of 3400 daltons were used to form a hydrophilic outer layer, with the inner core formed by albumin conjugated with doxorubicin via disulfide bonds. Additional doxorubicin was physically adsorbed into this core to attain a high drug loading capacity, where each albumin was associated with about 50 doxorubicin molecules. The formed micelles were stable in serum but continuously released doxorubicin when incubated with free thiols at concentrations mimicking the intracellular environment. When incubated with human melanoma cells (M21+) that express the alpha v/beta 3 integrin, higher uptake and longer retention of doxorubicin was observed with the RGD-targeted micelles, than in the case of untargeted control micelles, or free doxorubicin. Consequently the RGD-targeted micelles manifested cytotoxicity at lower doses of drug than control micelles or free drug.
Keywords: Human serum albumin, doxorubicin, PEGylation, RGD, targeted delivery, micelle, nanoparticle
INTRODUCTION
A major disadvantage of hydrophobic drugs is low water solubility and associated poor pharmacokinetic profiles, which in turn can contribute to adverse effects and low therapeutic efficacy. A variety systems, including polymers, micelles, liposomes, nanoparticles, and microspheres, have been investigated to improve the parental delivery of hydrophobic agents. Albumin is a major blood protein, and due to its abundance, low immunogenicity, good biocompatibility, and its ability to be transcytosed across vascular endothelia, it has been utilized in delivery systems for moieties such as anticancer drugs 1, 2, photosensitizers 3, antifungal drugs, and diagnostic imaging agents 4, 5. In one approach the albumin carrier is used as a single molecule, where the drugs are covalently conjugated to the free thiol in the cysteine-34 position 6 or to introduced free thiols 7. This type of conjugation strategy maintains the physiochemical properties of albumin while significantly improving the tumor uptake of conjugated drugs. In this strategy, however, only one or a few therapeutic molecules are carried by each albumin molecule, leading to low delivery efficiency. Albumin can also be formulated into microspheres or nanoparticles through precipitation in organic solvent followed by cross-linking 8–10. The formed particles can encapsulate tens to thousands of drug molecules per particle, achieving high drug-loading efficiency. However, with sizes in the range of several hundred nanometers to several micrometers, these particles are prone to uptake by reticuloendothelial system in liver and spleen. Another approach has been reported that can formulate hydrophobic drugs into albumin nanoparticles in the size range between 100 and 200 nm 11,12. Thus Abraxane, a paclitaxol-albumin nanoparticle with average size of 130 nm, has been extensively investigated in animal and clinical studies 13.
Polymeric micelles with sizes around 20–100 nm provide effective carriers for delivery of hydrophobic or amphipathic compounds 14, 15. In aqueous media, amphiphilic block copolymers spontaneously assemble into micelles with a distinct core-shell structure. The hydrophilic portion of the copolymer, usually PEG with molecular weights ranging from 2 to 15 kD, forms a coronal layer; the hydrophobic portion, typically consisting of poly(propylene oxide), poly(D,L-lactic acid), poly(e-caprolactone), poly (glutamic acid), or poly(L-aspartic acid), to name a few, forms a core that acts as a reservoir for drugs such as doxorubicin, paclitaxel, docetaxel, camptothecin, and others 16–19. Polymeric micelles often have high drug payloads in the form of either chemically conjugated or physically entrapped drug. They typically have high stability in the circulation owing to low critical micelle concentration, and long circulating lifetime due to a size above renal filtration threshold and due to PEG’s ability to evade recognition by the reticuloendothelial system (RES). These properties aid in the preferential accumulation of micelles in tumor tissue through the enhanced permeability and retention (EPR) effect 20–22, resulting in reduced toxicity to healthy tissues and improved antitumor efficacy. However, there remain concerns about the possible toxicities of polymeric micelles, including general cytotoxicity, hematoxicity, immunogenicity, carcinogenicity and other aspects 23, making high biocompatibility a prerequisite for synthetic copolymer micelles.
With these considerations in mind, we report the first example of targeted albumin-based nanoscale micelles for delivery of amphipathic drugs. We hypothesize that increasing the hydrophobicity of PEGylated albumin by addition of drug, and diminishment of primary amine groups on the protein surface, can drive the aggregation and formation of polymeric micelles. In addition, functionalization of the distal ends of the PEG chains can be used to incorporate ligands into the micelle outer layer to guide active targeting. We use doxorubicin, an anthracycline agent widely used for the treatment of leukemia, lymphoma and solid tumors, as a model compound 24, 25. Doxorubicin is conjugated to the amine group on albumin using a bi-functional linker that provides a disulfide bond between the protein and the drug, while additional doxorubicin molecules become physically entrapped in the micelle. A cyclic RGD peptide was conjugated to the micelle PEG chains since it is a ligand with high binding affinity to αvβ3 integrin, a membrane protein highly expressed in angiogenic endothelia and in some cancer cells 26, 27. This targeted albumin micelle delivery system is described herein with focus on its synthesis, physiochemical characterization, and uptake and pharmacological efficacy in cultured cells.
MATERIALS AND METHODS
Materials
Human serum albumin, Ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EDTA), doxorubicin hydrochloride, 2-mercaptoethanol, ninhydrin, hydrindantin, and 2-methoxyethanol were purchased from Sigma-Aldrich (St. Louis, MO, USA). AlexaFluor 488 C5 maleimide was from Invitrogen (Carlsbad, CA, USA). Hydroxylamine hydrochloride, sulfosuccinimidyl 6-(3′-[2-pyridyldithio]-propionamido) hexanoate (Sulfo-LC-SPDP) and 2-iminothiolane were from Pierce (Rockford, IL, USA). Bifunctional poly(ethylene Glycol) (PEG), Malhex-NH-PEG-O-C3H6-CONHS, was purchased from Rapp Polymere (Tubingen, German). Cyclo[RGDfK(Ac-SCH2CO)] peptide was from Peptide International (Louisville, KY, USA).
Preparation of albumin conjugates with Cyclic RGD PEG or 2-mercaptoethanol PEG
Human serum albumin (30 mg, 4.44×10−7 mol) was reacted with AlexaFluor 488 C5 maleimide (1 mg, 1.39×10−6 mol) for 4 hours at room temperature in phosphate saline buffer (PBS, pH 7.4) containing 1 mM EDTA. This was followed by dialysis in a MWCO 3500 cassette (Pierce, Rockford, IL) overnight at room temperature. Amine groups from lysine residues on albumin surface were then reacted with Mal-hex-NH-PEG5000-C3H6-CONHS (60 mg, 1.2×10−5 mol) in PBS with 1 mM EDTA (pH 7.4) for 4 hours. The unreacted PEG derivative was removed by dialysis in a MWCO 100,000 chamber (Havard Apparatus, Holliston, MA, USA). The PEGylated albumin (equivalent to 15 mg of albumin) was reacted with cyclo[RGDfK(Ac-SCH2CO)] (10 mg, 1.47×10−5 mol) in PBS with 1mM EDTA and 50 mM hydroxylamine for 24 hours at room temperature. The product, denoted as RPA, was purified by extensive dialysis in a MWCO 100,000 chamber for 24 hours. MPA was prepared by conjugating PEGylated albumin (equivalent to 2 mg of albumin) with 100 microgram of 2-mercaptoethanol in PBS with 1 mM EDTA for 24 hours and dialysis in a MWCO 100,000 chamber overnight. The number of PEG molecules conjugated was estimated by determining the loss of surface amine groups using a ninhydrin test.
Preparation of RGD-PEG-albumin-doxorubicin conjugates (RPA-DOX)
RPA (equivalent to 2 mg albumin) was reacted with sulfo-LC-SPDP (0.5 mg, 9.5×10−7mol) in PBS with 1 mM EDTA for 4 hours at room temperature, followed by dialysis in a MWCO 100,000 chamber overnight. Doxorubicin hydrochloride (1.5 mg, 2.6×10−6mol) was reacted with 2-Iminothiolane (0.36 mg, 2.6×10−6 mol) for 2 hours at PBS with 1mM EDTA to create thiolated (−SH) doxorubicin (ESI-MS: [M+1]+: 647.47 (observed), 645.79 (calculated)). An excess of the resulting doxorubicin-thiol was then reacted with the product from the RPA and sulfo-LC-SPDP reaction; the final product RGD-PEG-Albumin-doxorubicin conjugates (denoted as RPA-DOX) was thus prepared. RPA-DOX was further purified by dialysis in PBS with 1 mM EDTA for 24 hours, and stored in a refrigerator at 4 °C. The number of doxorubicin molecules conjugated via the SPDP linker was estimated using the ninhydrin reaction. Please note that, for the sake of simplicity, here and in the sections that follow we refer to these materials as albumin-doxorubicin conjugates although it is actually a thiolated form of doxorubicin that is conjugated. Likewise the drug that is released from the conjugate is termed doxorubicin although it is actually the thiolated form.
Preparation of 2-mercaptoethanol-PEG-albumin-doxorubicin conjugates (MPA-DOX)
The preparation of MPA-DOX was exactly parallel to that of RPA-DOX excepting that MPA lacking RGD groups was utilized.
FPLC-QELS and Zeta Potential
Samples (100 microliter) containing albumin conjugates at 0.1 to 0.5 mg protein per ml were loaded on a fast protein liquid chromatography (FPLC) system (GE Healthcare) equipped with a Superdex-200 size exclusion column and quasi-elastic light scattering (QELS) (Wyatt, Santa Barbara, CA, USA). The data was collected and analyzed using Astra software (Wyatt, Santa Barbara, CA, USA). The zeta potentials of the RPA-Dox and MPA-Dox were measured using a Zetasizer (Malvern Instruments, Westbury, MA).
TEM
The RPA-Dox and MPA-Dox were diluted and dropped on 200 mesh carbon coated copper grids (Ted Pella, Redding CA), and allowed to attach for two minutes. Uranyl acetate aqueous solution (2–5%) was then added on the grid for 2 minutes to counterstain the nanoparticles. The TEM images were then acquired with magnification of 17,000 times using a JEOL 100CX II transmission electron microscope.
Conjugate serum stability
RPA-DOX and MPA-DOX containing 20 micromolar doxorubicin were incubated in PBS containing 10% serum, using PBS as control. After incubation at 37 °C for 0.3, 2, 4 and 8 hours, a fraction was collected and ultrafiltered at 3400 rpm for 3 min using a YM-50 filter (Millipore, Bedford, MA, USA). Both the eluted phase and the residual volume on top of the filter were collected. The volumes were measured (Vup denoted residual volume and Vdown denotes volume of filtrate buffer) and the absorbance at 481 nm was determined (Aup denotes absorbance of residual and Adown denotes absorbance of filtered volume). The percentage of released doxorubicin was calculated by the equation:
Percentage of released doxorubicin (%) = ((Adown × (Vup + Vdown))/(Aup × Vup + Adown × Vdown) − 6%)× 100. The 6% is an estimate of the nonspecific leaking of RPA-DOX or MPA-DOX themselves. The serum stability curve was produced by plotting the percentage of released doxorubicin versus time.
Release of doxorubicin by sulfhydryls
RPA-Dox and MPA-Dox containing 20 micromolar doxorubicin were incubated at 37 °C in PBS (pH 7.4) containing 10 millimolar glutathione and 100 micromolar cysteine, or in unmodified PBS as control. Samples were collected after 15, 120, 240 minutes and 8, 24 and 48 hours of incubation and ultrafiltered at 3400 rpm for 3 min using a YM-50 filter (Millipore, Bedford, MA, USA). The percentage of released doxorubicin was calculated and the time course of doxorubicin release was then generated by plotting percent of released doxorubicin versus time.
Cell uptake
M21+ human melanoma cells that express the alpha v/beta 3 integrin were seeded on 24 well plates at 4 × 104 cells per well and cultured overnight. The cells were either preincubated with 100 micromolar cyclic (RGDfV) for half an hour or directly incubated with doxorubicin, RPA-DOX, or MPA-DOX at concentrations ranging from 100 nanomolar to 2 micromolar in Opti-MEM medium. After 4 hours incubation, the cells were harvested by trypsinization. The fluorescence Intensity at 488/575 nm of doxorubicin 28 per cell was determined using a Cyan flow cytometer (Beckman Coulter, Miami, FL).
Confocal microscopy
M21+ cells were cultured in complete DMEM medium in a glass bottom culture dish (MatTek, Ashland, MA) overnight. The medium was replaced with Opti-MEM medium containing 2 micromolar free doxorubicin, RPA-DOX or MPA-DOX and incubated for 4 hours at 37 °C. Additional samples were continued in culture in drug-free complete DMEM medium for 12 hours. The doxorubicin distribution was imaged using a Zeiss confocal microscopy with a 63x objective lens and a 488 nm laser light source. The images were analyzed using Zeiss LSM software.
Cytotoxicity
M21+ cells were seeded into 96-well plates at a density of 5000 cells per well. After overnight incubation, the medium was aspirated and fresh DMEM medium containing different concentrations of free doxorubicin, RPA-Dox or MPA-Dox was added; the concentrations of RPA-Dox and MPA-Dox are expressed in doxorubicin equivalents. The cells were continuously cultured at 37 °C, 5% CO2 for 3 days. The cytotoxicity was then assessed by a modified MTT assay. Briefly, the medium was replaced by a mixture of 200 microlitre of fresh DMEM medium and 20 microlitre of MTT solution (5 mg/ml MTT in PBS), followed by 2 hours incubation. The culture medium was then removed by aspiration. Then 100 microlitre DMSO was added into each well, stirred and absorbance at 540 nm of the cell suspension was determined using a FLUOstar Omega Microplate Reader (BMG Labtech, Offenburg, Germany). The cytotoxicity was evaluated through comparing absorbance of treated cells against the untreated controls. The results of cytotoxicity assay were used to construct dose response curves and IC50 doses were determined 29. The toxicities of RPA and MPA, without doxorubicin, were determined in a parallel assay. In a separate experiment the toxicity of doxorubicin itself versus its thiolated derivative was also compared using the MTT assay.
RESULTS
Synthesis and characterization
The preparation of albumin-doxorubicin micelles is outlined in Figure 1. First, human serum albumin (HSA) was labeled with the green fluorescent dye Alexa Fluor 488 at the free thiol group at Cys34. Subsequently, several Mal-PEG-NHS moieties with molecular weight of 3400 were conjugated to primary amines on the HSA surface to form PEGylated albumin. The unreacted PEG derivative was removed by dialysis in a MWCO 100,000 bag. Excess cyclo[RGDfK(AcSCH2CO)] was then incubated with the PEGylated albumin in the presence of hydroxylamine to form RGD-PEG-HSA (RPA). Alternatively, 2-mercaptoethanol was used to react with PEGylated albumin to form 2-mercaptoethanol-PEG-HSA (MPA) as a control. The amount of PEG per albumin was determined using a ninhydrin test 30 to compare the primary amine density before and after the PEGylation reaction. RPA has an average of 17.5 PEGs per albumin molecule while MPA has 15.1 PEGs. Additional primary amine groups on the surface of RPA and MPA were reacted with sulfo-LC-SPDP followed by conjugation with the thiolated doxorubicin derivative to form PEGylated albumin doxorubicin complexes termed as RPA-DOX or MPA-DOX. The reaction between thiol and SPDP created a disulfide linker that can be potentially cleaved by endogenous thiols to liberate the thiolated doxorubicin.
Figure 1.

The preparation of RPA-Dox.
The physical aspects of the conjugates were monitored, as summarized in Figure 2, supplementary Figure S.1, Figure 3 and Tables 1 and 2. Significant size and apparent molecular weight increases were observed after the addition of the PEG derivative followed by cyclic RGD or 2-mercaptoethanol. Both RPA and MPA exist primarily as monomers and trimers with narrow molecular weight distributions, indicated by low polydispersity indices. However, FPLC-QELS analysis showed that the RPA-DOX has a hydrodynamic radius of about 15 nm and molecular weight more than 1 million daltons, with a single distribution and a low polydispersity index. The RPA-DOX was visualized by transmission electromicroscopy (TEM), revealing a diameter averaging about 30 nanometers, which is consistent with its hydrodynamic size (Figure 3). Compared with RPA, RPA-DOX has much larger particle size as well as apparent molecular weight. Thus the RPA-DOX must contain several albumin molecules that have co-aggregated. We postulate that RPA-DOX monomers oligomerize to form nanoscale micelles. This may be due to the amphiphilicity of PEGylated RPA-DOX, where the hydrophilicity can be attributed to PEG, and the hydrophobicity is from albumin that has lost a significant fraction of its primary amines, with hydrophobicity further increased by the conjugation of doxorubicin molecules. The zetasizer revealed that the formed micelles had a slightly negative surface potential (Table 2).
Figure 2.

The reaction progress of RPA-Dox. The intermediates and final products were analyzed by FPLC-QELS. Dotted line: molar mass. Solid line: Rayleigh ratio.
Figure 3.

TEM image of RPA-Dox (170,000x). The sample is counterstained by uranyl acetate. The average particle size is around 30 nm.
Table 1.
Physiochemical properties of RPA-DOX, MPA-DOX and intermediates.
| Rh(z) (nm) | Mw | Mn | PDI | ||
|---|---|---|---|---|---|
| HSA | Peak 1 | 3.2 | 2.16×105 | 2.15×105 | 1.01 |
| Peak 2 | 3.8 | 1.30×105 | 1.29×105 | 1.01 | |
| Peak3 | 3.2 | 6.68×104 | 6.68×104 | 1.00 | |
|
| |||||
| MPA | Peak 1 | 13.3 | 3.81×105 | 3.25×105 | 1.17 |
| Peak 2 | 6.9 | 1.21×105 | 1.18×105 | 1.03 | |
|
| |||||
| RPA | Peak 1 | 14.1 | 3.77×105 | 3.36×105 | 1.12 |
| Peak 2 | 9.3 | 1.27×105 | 1.25×105 | 1.02 | |
|
| |||||
| MPA-DOX | Peak 1 | 16.8 | 1.35×106 | 1.07×106 | 1.26 |
|
| |||||
| RPA-DOX | Peak 1 | 15.2 | 1.27×106 | 1.15×106 | 1.10 |
Note: the peaks were numerated and displayed at Figure 2 and Figure S.1.
Table 2.
Physiochemical properties of RPA-Dox and MPA-Dox
| PEG per albumina | Total Doxorubicn per albuminb | Drug Loadig ratio (%) | Conjugated doxorubicin per albumina | Aosorbed doxorubicin per albumin | Hydrodynamic size (Rh (z), nm)c | Mwc | Poly dispersityc | Zeta Potential (mV)d | |
|---|---|---|---|---|---|---|---|---|---|
| RPA-Dox | 17.5 | 50.6 | 56.2 | 24.6 | 26 | 15.2 | 1.27×106 | 1.10 | −2.6 |
| MPA-Dox | 15.1 | 56.5 | 62.3 | 20.7 | 35.8 | 16.8 | 1.35×106 | 1.26 | −1.9 |
determined by ninhydrin test;
determined by spectrum method;
determined by FPLC-QELS;
Measured by zeta sizer.
As determined by a ninhydrin test, RPA-DOX had approximately 24.6 chemically conjugated doxorubicins per albumin. However, the total amount of doxorubicin associated with the albumin micelle was greater, as determined by measuring absorbance at 481 nm, where doxorubicin (as well as its thiolated derivative) has an extinction coefficient of 10410 M−1cm−1 at 25°C 31. This revealed that an average 50.6 doxorubicin molecules were associated with each albumin molecule. This suggests that extra doxorubicin molecules were physically accommodated in the hydrophobic domain of the formed micelle, probably through hydrophobic interaction and van de Waals forces. This substantially increased the drug loading of the albumin micelles (Table 2).
The driving force of micelle formation was further evaluated by synthesizing a series of RPA derivatives, where each RPA was conjugated to sulfo-LC-SPDP and then to doxorubicin derivatives at ratios from 1:5:5 to 1:30:90. The FPLC analysis (supplementary Figure S.2) showed that higher molecular weight complexes started to form when each RPA was conjugated to doxorubicin derivatives at a molar ratio of 1:5. However, in this condition UV absorbance showed that only a part of RPA-DOX was formed into micelles with most of it staying at the same form and distribution as drug-free RPA. With the doxorubicin amount increased from 5 per RPA to 30 per RPA, additional RPA was incorporated into micelles. When doxorubicin was reacted to RPA at a molar ratio of 1:90, with excess doxorubicin derivatives, almost all RPA was formed into micelles with a single peak distribution. This result clearly shows that higher amounts of doxorubicin can function as a driving force for micelle formation. The doxorubicin conjugates on RPA were determined by observing the release of pyridine-2-thione λmax = 343 nm) from the reaction intermediate, and the final amount of doxorubicin associated with RPA was determined by the absorbance of doxorubicin at 481 nm, Table S.1. It shows that RPA5 and RPA30 lost significant amounts of loaded doxorubicin during dialysis while RPA90 retained approximately 44 doxorubicin molecules per RPA. This result shows that using extra amounts of doxorubicin in the formulation process can result in more doxorubicin loaded on RPA. Taken together, it suggests that formulating RPA with a high amount of doxorubicin will drive the formation of micelles that stably hold associated doxorubicin molecules.
The stability of the formed micelles was tested (Figure 4). After five weeks storage in PBS buffer with 1 mM EDTA at 4 °C, only trace amounts of doxorubicin were detected after ultrafiltration of the material. This result shows that the doxorubicin is stably held inside the micelle. The stored batch of RPA-DOX was also analyzed by FPLC-QELS. This showed that RPA-DOX kept its single peak distribution profile with a low polydispersity index and maintained its size close to freshly prepared material. This suggests that RPA-DOX does not aggregate into larger particles under this storage condition. The critical micelle concentration (CMC) was not determined. However, when diluted to concentrations lower than 0.2 micromolar albumin equivalent, the micelles maintained their structure when visualized by TEM. No doxorubicin leaked out into solution during ultracentrifugal filtration with a MWCO 50 kDa filter. This indicates that the albumin micelle has a similar low CMC value as other polymeric micelles, typically on the order of 10−6–10−7 M.
Figure 4.

Stability of RPA-Dox. RPA-Dox sample were analyzed by FPLC-QELS in 3 days (A) and 5 weeks (B) postsynthesis.
Doxorubicin release
RPA-DOX was tested by incubation in PBS or PBS with 10% serum at 37 °C for up to 8 hours (Figure 5A). Less than 5% of doxorubicin was released, suggesting that PBS or serum had little influence on the stability of RPA-DOX. RPA-DOX and MPA-DOX show similar doxorubicin release kinetics when incubated in PBS containing 10 millimolar L-glutathione and 100 micromolar cysteine, the typical free thiol concentrations of intracellular cytosol 32–34 (Figure 5B). The doxorubicin released from nanoparticles can pass through a MWCO 50,000 filter, while RPA-DOX and MPA-DOX show minimal filtration, thus allowing the liberated doxorubicin to be separated and quantified. Figure 5B shows that it took 8 hours to release approximately 15 % of doxorubicin from the micelle and 48 hours to release 40%. Considering the micelle would enter target cells through endocytosis, the breakdown of the albumin by proteinases in endosomal- and lysosomal compartments might be expected to accelerate the doxorubicin release. As a control, when incubated in PBS for 48 hours, approximately 5% of doxorubicin was released.
Figure 5.
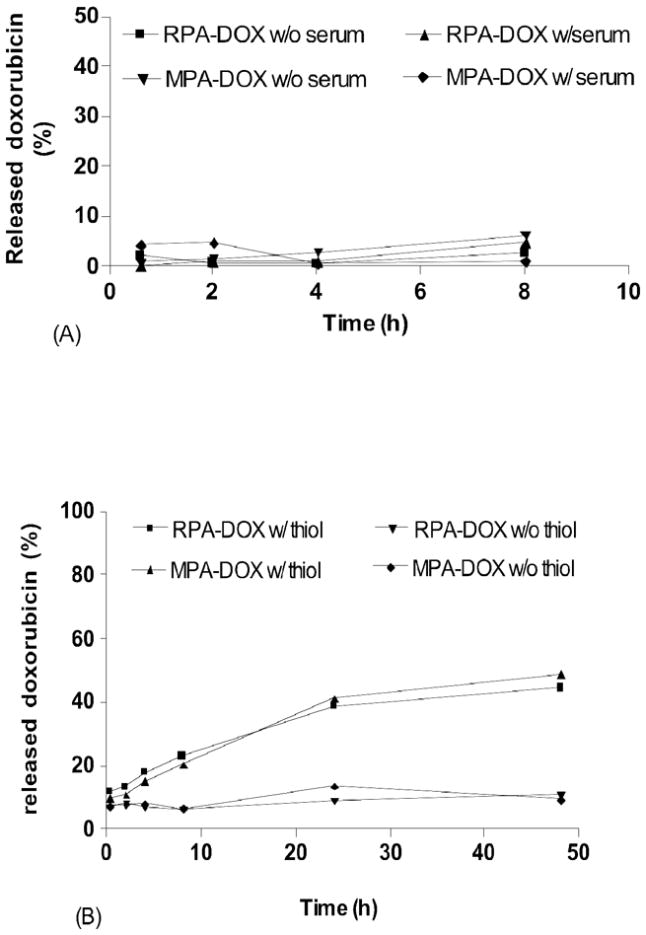
Doxorubicin release kinetics. (A) RPA-DOX and MPA-DOX were incubated with PBS only or PBS with 10% serum at 37 °C. (B) RPA-DOX and MPA-DOX were incubated at in PBS with thiols from 10 mM glutathione and 100 uM cysteine or PBS only at 37 °C.
Cell uptake
The uptake of doxorubicin was examined by flow cytometry, as seen in Figure 6. The fluorescence intensity of doxorubicin was excited by 488 nm wavelength light and detected at 575 nm. The mean fluorescence intensity per cell was approximately linear with doxorubicin concentration in all cases. At concentrations ranging between 200 and 1000 nanomolar, RPA-DOX displayed the highest doxorubicin uptake, followed by free doxorubicin and MPA-DOX. When the concentration of doxorubicin was 2 micromolar, free doxorubicin had the highest doxorubicin uptake, probably because it entered cells by passive diffusion while RPA-DOX and MPA-DOX entered cells through endocytosis that could be saturated or rate limited. With cells preincubated half an hour with 100 micromolar cyclic(RGDfV), a ligand for αvβ3 integrin, the uptake of RPA-DOX was suppressed to a level close to MPA-DOX. This result shows that conjugation of cyclic RGD on the surface of the albumin micelles supports receptor mediated endocytosis.
Figure 6.

Cellular uptake of doxorubicin formulation. In (A), M21 cells were incubated 4 hours in Opti-MEM medium containing medium only, 1 micromolar doxorubicin, RPA-DOX and MPA-DOX with 1 micromolar doxorubicin, and RPA-DOX with 1 micromolar doxorubicin with pre-incubation of 100 micromolar of cyclic(RGDfV), FL2 channel of flow cytometry was set to collect doxorubicin fluorescence. Figure (B) shows the dynamic cellular uptake of doxorubicin formulation. (mean +/− SD, from about 5000 cells).
The subcellular distribution of doxorubicin was imaged by confocal microscopy (Figure 7). With free doxorubicin, after 4 hours incubation in Opti-MEM medium, the bulk of the fluorescence was located in the nucleus (Figure 7A). After a further 16 hours culture in drug-free medium, only limited doxorubicin fluorescence was retained inside nucleus, with the overall fluorescence significantly decreased. With RPA-DOX, after 4 hours incubation most of the doxorubicin fluorescence was in cytoplasmic vesicles in the area surrounding the nucleus (Figure 7B), indicating that RPA-DOX enters the cell via a pathway different from free doxorubicin. After 16 hours further culturing in drug-free medium, significant amounts of doxorubicin could be seen in the nucleus (Figure 7E), with overall fluorescence of doxorubicin remaining at a high level. This result indicates that doxorubicin delivered by RPA-DOX was retained in cells much longer than free doxorubicin and gradually infiltrated into the nucleus. The MPA-DOX showed similar subcellular distribution as RPA-DOX. However, the fluorescence intensity was much lower under the same imaging condition, indicating a lesser amount of doxorubicin was successfully delivered to cells by this nontargeted formulation.
Figure 7.

Confocal microscopy of doxorubicin uptake. Cells were incubated for 4 hours with 2 micromolar doxorubicin in free form (A), RPA-DOX (B) or MPA-DOX (C), or further cultured in drug-free medium for 12 hours, see (D), (E) and (F), respectively. Arrows point to the doxorubicin subcellular distributions.
Cytotoxicity
The cytotoxicity of the micelles to M21+ human melanoma cells was determined using an MTT assay and is shown in Figure 8. As shown, RPA-DOX has the highest potency, followed by free doxorubicin and then MPA-DOX. The IC50 doses for RPA-DOX, free doxorubicin and MPA-DOX were calculated to be approximately 200 nanomolar, 500 nanomolar, and 1200 nanomolar, respectively. The cytotoxicities of RPA and MPA were also evaluated by a similar method; after 72 hours incubation with M21+ cells, neither of them caused significant toxicity. As a free drug, the thiolated form of doxorubicin that is associated with the nanoscale micelles is slightly less toxic than doxorubicin itself (see supplementary Figure S.3). Thus the increased potency of RPA is not due to a greater effectiveness of the thiolated doxorubicin but rather to improved delivery. Since RPA-DOX contains both bound doxorubicin and conjugated doxorubicin, it is difficult to attribute the cytotoxic effect to one or the other of these components. Further, at present we do not know how quickly the conjugated drug is actually released within the cells. Although the data in Figure 5 suggest release over a period of hours in a reducing environment that mimics the cell interior, the actual release may be quicker due to contributions of endosomal proteases.
Figure 8.

Cytotoxicity of RPA-DOX, MPA-DOX and free doxorubicin on M21+ cells. The cytotoxicity of MPA and RPA equivalent to RPA-DOX and MPA-DOX containing 0.5 uM to 50 uM of doxorubicin was evaluated in a parallel assay, see inset.
DISCUSSION
In this report we describe targeted nanoscale micelles prepared from albumin and the amphipahtic anticancer drug doxorubicin. There has been substantial interest in conjugates of antineoplastic agents with proteins, including albumin and monoclonal antibodies, and several moieties have reached clinical trials or entered the clinic 35. However, in many cases only one or a few drug molecules are linked to the protein. In these cases better effects are achieved with extraordinarily potent agents such as the calicheamicins rather than more conventional drugs 36. Another approach has been to utilize micelles based on block copolymers to deliver antitumor agents 15, 37–39. This approach often has the advantage of allowing incorporation of a large number of drug molecules into micelles thus facilitating the utilization of well-known agents such as anthracyclines and taxanes. However, there are inevitably some concerns about possible toxicities of synthetic polymers 40, 41.
The albumin based nanoscale micelles described here may offer some advantages over other systems in that they provide a high degree of drug loading while being composed of non-toxic biocompatible materials. Our preparation process gives rise to multimeric aggregates of PEG-ylated albumin that contain about 50 doxorubicin molecules per albumin and that are relatively uniform in size with a mean diameter of about 30 nm. We describe this material as nanoscale micelles based on the fact that they are stable in aqueous suspension suggesting the presence of a hydrophilic outer region, but they non-covalently bind a substantial amount of doxorubicin suggesting a hydrophobic core. However, further work will be required to fully delineate the structure and assembly process for this material as well as the implications for pharmacological effects. Thus, at present we do not have detailed information on the structure of the RPA-Dox micelles or the placement of conjugated or bound doxorubicin moieties. Further, the relative contributions of the bound and conjugated drug to the overall pharmacological effect are not known.
The conjugation of RGD-terminated PEG to the albumin results in micelles capable of specifically targeting cells that express the αvβ3 integrin. Thus the RGD-containing RPA-DOX is taken up more efficiently than MPA-DOX, the control conjugate. Further, the uptake of RPA-DOX by αvβ3 positive cells is almost completely blocked by the presence of excess RGD illustrating that uptake is ligand-specific. The RPA-DOX micelles displayed excellent potency versus αvβ3 positive melanoma cells with an IC50 6-fold lower than that of the control conjugate. Once accumulated in cells, both the covalently linked and the non-covalently absorbed doxorubicin should be readily released from albumin carrier by a combination of reversal of disulfide bonds by intracellular thiols and by proteolytic processes in endosomes and lysosomes.
The strategy described here builds on our previous work using targeted albumin-PEG conjugates to deliver oligonucleotides 42. However in that case the conjugates remained as polydisperse but monomolecular entities. In the present case covalent linkage of doxorubicin to the PEG-albumin conjugates apparently drives their aggregation into nanoscale micelles. The ability of the micelles to accommodate additional non-covalently bound drug dramatically raises the drug-loading factor and may allow for very efficient delivery. However, the in vivo targeting and delivery capability of these constructs remains to be determined.
Supplementary Material
Acknowledgments
This work was supported by NIH grant P01GM059299 to R.L.J. Authors also thank Dr. Ashutosh Tripathy of the UNC Macromolecular Interactions Core Facility for his helpful advice on FPLC-QELS experiments.
Footnotes
SUPPORTING INFORMATION AVAILABLE
This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Kratz F, Beyer U. Serum proteins as drug carriers of anticancer agents: a review. Drug Deliv. 1998;4:281–299. doi: 10.3109/10717549809065759. [DOI] [PubMed] [Google Scholar]
- 2.Kratz F. Albumin as a drug carrier: design of prodrugs, drug conjugates and nanoparticles. J Control Release. 2008;132(3):171–183. doi: 10.1016/j.jconrel.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 3.Chen K, Preuss A, Hackbarth S, Wacker M, Langer K, Roder B. Novel photosensitizer-protein nanoparticles for photodynamic therapy: photophysical characterization and in vitro investigations. J Photochem Photobiol B. 2009;96(1):66–74. doi: 10.1016/j.jphotobiol.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 4.Bledin AG, Kim EE, Harle TS, Haynie TP, Chuang VP. Technetium-99m-labeled macroaggregated albumin arteriography for detection of abnormally positioned arterial catheters during infusion chemotherapy. Cancer. 1984;53(4):858–862. doi: 10.1002/1097-0142(19840215)53:4<858::aid-cncr2820530407>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 5.Chaumet-Riffaud P, Martinez-Duncker I, Marty AL, Richard C, Prigent A, Moati F, Sarda-Mantel L, Scherman D, Bessodes M, Mignet N. Synthesis and Application of Lactosylated, (99m)Tc Chelating Albumin for Measurement of Liver Function. Bioconjug Chem. 2010;21(4):589–596. doi: 10.1021/bc900275f. [DOI] [PubMed] [Google Scholar]
- 6.Kratz F, Muller-Driver R, Hofmann I, Drevs J, Unger C. A novel macromolecular prodrug concept exploiting endogenous serum albumin as a drug carrier for cancer chemotherapy. J Med Chem. 2000;43(7):1253–1256. doi: 10.1021/jm9905864. [DOI] [PubMed] [Google Scholar]
- 7.Di Stefano G, Lanza M, Kratz F, Merina L, Fiume L. A novel method for coupling doxorubicin to lactosaminated human albumin by an acid sensitive hydrazone bond: synthesis, characterization and preliminary biological properties of the conjugate. Eur J Pharm Sci. 2004;23(4–5):393–397. doi: 10.1016/j.ejps.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 8.Gupta PK, Hung CT. Albumin microspheres. I: Physico-chemical characteristics. J Microencapsul. 1989;6(4):427–462. doi: 10.3109/02652048909031165. [DOI] [PubMed] [Google Scholar]
- 9.Gupta PK, Hung CT. Albumin microspheres. II: Applications in drug delivery. J Microencapsul. 1989;6(4):463–472. doi: 10.3109/02652048909031166. [DOI] [PubMed] [Google Scholar]
- 10.Anhorn MG, Wagner S, Kreuter J, Langer K, von Briesen H. Specific targeting of HER2 overexpressing breast cancer cells with doxorubicin-loaded trastuzumab-modified human serum albumin nanoparticles. Bioconjug Chem. 2000;19(12):2321–2331. doi: 10.1021/bc8002452. [DOI] [PubMed] [Google Scholar]
- 11.Stinchcombe TE. Nanoparticle albumin-bound paclitaxel: a novel Cremphor-EL-free formulation of paclitaxel. Nanomed. 2007;2(4):415–423. doi: 10.2217/17435889.2.4.415. [DOI] [PubMed] [Google Scholar]
- 12.Desai N, Trieu V, Yao Z, Louie L, Ci S, Yang A, Tao C, De T, Beals B, Dykes D, Noker P, Yao R, Labao E, Hawkins M, Soon-Shiong P. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin Cancer Res. 2006;12(4):1317–1324. doi: 10.1158/1078-0432.CCR-05-1634. [DOI] [PubMed] [Google Scholar]
- 13.Gradishar WJ, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P, Hawkins M, O’Shaughnessy J. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol. 2005;23(31):7794–7803. doi: 10.1200/JCO.2005.04.937. [DOI] [PubMed] [Google Scholar]
- 14.Torchilin VP. Micellar nanocarriers: pharmaceutical perspectives. Pharm Res. 2007;24(1):1–16. doi: 10.1007/s11095-006-9132-0. [DOI] [PubMed] [Google Scholar]
- 15.Nishiyama N, Kataoka K. Current state, achievements, and future prospects of polymeric micelles as nanocarriers for drug and gene delivery. Pharmacol Ther. 2006;112(3):630–648. doi: 10.1016/j.pharmthera.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 16.Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW, Richie JP, Langer R. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci U S A. 2006;103(16):6315–6320. doi: 10.1073/pnas.0601755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamaguchi T, Kato K, Yasui H, Morizane C, Ikeda M, Ueno H, Muro K, Yamada Y, Okusaka T, Shirao K, Shimada Y, Nakahama H, Matsumura Y. A phase I and pharmacokinetic study of NK105, a paclitaxel-incorporating micellar nanoparticle formulation. Br J Cancer. 2007;97(2):170–176. doi: 10.1038/sj.bjc.6603855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uchino H, Matsumura Y, Negishi T, Koizumi F, Hayashi T, Honda T, Nishiyama N, Kataoka K, Naito S, Kakizoe T. Cisplatin-incorporating polymeric micelles (NC-6004) can reduce nephrotoxicity and neurotoxicity of cisplatin in rats. Br J Cancer. 2005;93(6):678–687. doi: 10.1038/sj.bjc.6602772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koizumi F, Kitagawa M, Negishi T, Onda T, Matsumoto S, Hamaguchi T, Matsumura Y. Novel SN-38-incorporating polymeric micelles, NK012, eradicate vascular endothelial growth factor-secreting bulky tumors. Cancer Res. 2006;66(20):10048–10056. doi: 10.1158/0008-5472.CAN-06-1605. [DOI] [PubMed] [Google Scholar]
- 20.Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, Jain RK, McDonald DM. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156(4):1363–1380. doi: 10.1016/S0002-9440(10)65006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 22.Matsumura Y, Fau-Maeda H, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 23.Ríhová B. Biocompatibility of biomaterials: hemocompatibility, immunocompatiblity and biocompatibility of solid polymeric materials and soluble targetable polymeric carriers. Advanced Drug Delivery Reviews. 1996;21(2):157–176. [Google Scholar]
- 24.Hortobagyi GN. Anthracyclines in the treatment of cancer. An overview. Drugs. 1997;54(Suppl 4):1–7. doi: 10.2165/00003495-199700544-00003. [DOI] [PubMed] [Google Scholar]
- 25.Arcamone F, Cassinelli G, Fantini G, Grein A, Orezzi P, Pol C, Spalla C. Adriamycin, 14-hydroxydaunomycin, a new antitumor antibiotic from S. peucetius var. caesius. Biotechnol Bioeng. 1969;11(6):1101–1110. doi: 10.1002/bit.260110607. [DOI] [PubMed] [Google Scholar]
- 26.Stupack DG, Cheresh DA. Integrins and angiogenesis. Curr Top Dev Biol. 2004;64:207–238. doi: 10.1016/S0070-2153(04)64009-9. [DOI] [PubMed] [Google Scholar]
- 27.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10(1):9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karukstis KK, Thompson EH, Whiles JA, Rosenfeld RJ. Deciphering the fluorescence signature of daunomycin and doxorubicin. Biophys Chem. 1998;73(3):249–263. doi: 10.1016/s0301-4622(98)00150-1. [DOI] [PubMed] [Google Scholar]
- 29.Malugin A, Kopeckova P, Kopecek J. Liberation of doxorubicin from HPMA copolymer conjugate is essential for the induction of cell cycle arrest and nuclear fragmentation in ovarian carcinoma cells. J Control Release. 2007;124(1–2):6–10. doi: 10.1016/j.jconrel.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaiser E, Colescott RL, Bossinger CD, Cook PI. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal Biochem. 1970;34(2):595–598. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- 31.Tian Y, Bromberg L, Lin SN, Hatton TA, Tam KC. Complexation and release of doxorubicin from its complexes with pluronic P85-b-poly(acrylic acid) block copolymers. J Control Release. 2007;121(3):137–145. doi: 10.1016/j.jconrel.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 32.de Graaf-Hess A, Trijbels F, Blom H. New method for determining cystine in leukocytes and fibroblasts. Clin Chem. 1995;45(12):2224–2228. [PubMed] [Google Scholar]
- 33.Andersson A, Lindgren A, Hultberg B. Effect of thiol oxidation and thiol export from erythrocytes on determination of redox status of homocysteine and other thiols in plasma from healthy subjects and patients with cerebral infarction. Clin Chem. 1995;41(3):361–366. [PubMed] [Google Scholar]
- 34.Deneke SM. Thiol-based antioxidants. Curr Top Cell Regul. 2000;36:151–180. doi: 10.1016/s0070-2137(01)80007-8. [DOI] [PubMed] [Google Scholar]
- 35.Kratz F, Abu Ajaj K, Warnecke A. Anticancer carrier-linked prodrugs in clinical trials. Expert Opin Investig Drugs. 2007;16(7):1037–1058. doi: 10.1517/13543784.16.7.1037. [DOI] [PubMed] [Google Scholar]
- 36.Ricart AD, Tolcher AW. Technology insight: cytotoxic drug immunoconjugates for cancer therapy. Nat Clin Pract Oncol. 2007;4(4):245–255. doi: 10.1038/ncponc0774. [DOI] [PubMed] [Google Scholar]
- 37.Vetvicka D, Hruby M, Hovorka O, Etrych T, Vetrik M, Kovar L, Kovar M, Ulbrich K, Rihova B. Biological evaluation of polymeric micelles with covalently bound doxorubicin. Bioconjug Chem. 2009;20(11):2090–2097. doi: 10.1021/bc900212k. [DOI] [PubMed] [Google Scholar]
- 38.Matsumura Y. Polymeric micellar delivery systems in oncology. Jpn J Clin Oncol. 2008;38(12):793–802. doi: 10.1093/jjco/hyn116. [DOI] [PubMed] [Google Scholar]
- 39.Upadhyay KK, Agrawal HG, Upadhyay C, Schatz C, Le Meins JF, Misra A, Lecommandoux S. Role of block copolymer nanoconstructs in cancer therapy. Crit Rev Ther Drug Carrier Syst. 2009;26(2):157–205. doi: 10.1615/critrevtherdrugcarriersyst.v26.i2.20. [DOI] [PubMed] [Google Scholar]
- 40.Rihova B. Immunocompatibility and biocompatibility of cell delivery systems. Adv Drug Deliv Rev. 2000;42(1–2):65–80. doi: 10.1016/s0169-409x(00)00054-5. [DOI] [PubMed] [Google Scholar]
- 41.Kabanov AV. Polymer genomics: an insight into pharmacology and toxicology of nanomedicines. Adv Drug Deliv Rev. 2006;58(15):1597–1621. doi: 10.1016/j.addr.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kang H, Alam MR, Dixit V, Fisher M, Juliano RL. Cellular delivery and biological activity of antisense oligonucleotides conjugated to a targeted protein carrier. Bioconjug Chem. 2008;19(11):2182–2188. doi: 10.1021/bc800270w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.