

Abstract
Background and Purpose
Phosphoinositide 3-kinase (PI3K) gamma is linked to inflammation and oxidative stress. This study was conducted to investigate the role of the PI3Kgamma in the blood-brain barrier (BBB) dysfunction and brain damage induced by focal cerebral ischemia/reperfusion.
Methods
Wild-type and PI3Kgamma knockout mice were subjected to middle cerebral artery occlusion (60 min) followed by reperfusion. Evans blue leakage, brain edema, infarct volumes and neurological deficits were examined. Oxidative stress, neutrophil infiltration, and matrix metallopeptidase-9 (MMP-9) were assessed. Activation of NF-kB and expression of proinflammatory and pro-oxidative genes were studied.
Results
PI3Kgamma deficiency significantly reduced BBB permeability and brain edema formation, which were time-dependently correlated with preventing the degradation of the tight junction protein, claudin-5, and the basal lamina protein, collagen IV, and the phosphorylation of myosin light chain (MLC) in brain microvessels. PI3Kgamma deficiency suppressed ischemia/reperfusion-induced NF-kB p65 (Ser536) phosphorylation and the expression of the pro-oxidant enzyme NADPH oxidase (Nox1, Nox2, and Nox4) and pro-inflammatory adhesion molecules (E- and P-selectin, ICAM-1) at different time points. These molecular changes were associated with significant inhibition of oxidative stress (superoxide production and malondialdehyde content), neutrophil infiltration, and MMP-9 expression/activity in PI3Kgamma knockout mice. Eventually, PI3Kgamma deficiency significantly reduced infarct volumes and neurological scores at 24 hours after ischemia/reperfusion.
Conclusions
Our results provide the first direct demonstration that PI3Kgamma plays a significant role in ischemia/reperfusion-induced BBB disruption and brain damage. Future studies need to explore PI3Kγ as a potential target for stroke therapy.
Keywords: blood-brain barrier, PI3 kinase-gamma, ischemic stroke, oxidative stress, metallopeptidases
INTRODUCTION
Effective stroke therapies require early restoration of cerebral blood flow to ischemic brain tissue. However, early reperfusion can cause the blood-brain barrier (BBB) disruption, leading to cerebral edema, brain hemorrhage, and neuronal death.1 These complications of early reperfusion, which result from excess production of reactive oxygen species (ROS) and overexpression/activation of metalloproteinases (MMPs), especially MMP-9, significantly limit the benefits of stroke therapies.1, 2 The blood–brain barrier (BBB) is primarily formed by specialized brain endothelial cells that are interconnected by well-developed tight junctions and provides a dynamic interface between the blood and the brain.3 BBB disruption is a critical event in the pathogenesis of acute ischemic stroke; however, the molecular mechanisms involved are not completely understood.4 Experimental evidence and clinical data suggest that ROS and MMP-9 disrupt the BBB by degrading the tight junctions (e.g. claudins and occludin) and basal lamina proteins (e.g. collagen IV, laminin, fibronectin).4–6 Although a plethora of drugs targeting ROS and MMP-9 are effective in the treatment of acute stroke in animal models, their translation into clinical practice has completely failed.1, 2 Thus, there is urgent need to identify molecular signaling pathways that can be targeted with innovative therapies.7
Phosphoinositide 3-kinases (PI3Ks) are a family of enzymes characterized by protein and lipid kinase activity. Although these kinases have been linked to Akt/PKB activation and neuroprotection during cerebral ischemia,8 disappointedly, there is no direct and conclusive evidence of a causal link between the PI3Ks and the development of disease. Previous experimental studies that investigated the roles of PI3Ks in stroke are limited in number, and almost all of the studies used pan-PI3K inhibitors (e.g. wortmannin, Ly294002). These pan-PI3K inhibitors also inhibit a number of other kinases, such as MAP kinase, mTOR, and DNA-dependent protein kinases.9 To date, the roles of specific PI3K isoforms in the disease process of stroke has never been investigated.
Recent studies, using genetically modified mice and isoform-selective PI3K inhibitors, have revealed that different PI3K isoforms often exert distinct cellular functions downstream of specific receptors in various cell types. 10 The class Ib PI3K (p11oγ), primarily expressed in leukocytes and also in endothelial cells, regulates different cellular functions relevant to inflammation, oxidative stress, and tissue damage. 10–12 Increasing evidence reveals that PI3Kγ is critically involved in a number of inflammatory and autoimmune diseases. Genetic deletion and pharmacological inhibition of PI3Kγ have been shown to protect from atherosclerosis, myocardial ischemia/reperfusion injury, and rheumatoid arthritis in mice.13–15 Currently, pharmacological targeting of PI3Kγ represents one of the most promising approaches for developing novel safe treatments for inflammatory diseases. 9,10
Our recent study has shown that PI3Kγ is constitutively expressed in normal brain microvessels and significantly upregulated in postischemic brain primarily in activated microglia following cerebral ischemia in mice.16 Genetic deletion of PI3Kγ not only inhibits neutrophil infiltration but also blocks microglia activation and expansion in vivo. 16 This study was designed to investigate the role of PI3Kγ in BBB dysfunction and brain damage elicited by focal cerebral I/R and explore potential mechanisms involved.
Materials and Methods
Mice
Male C57BL/6J mice (wild type [WT]) and PI3K-p110γ knockout (PI3Kγ KO) mice (8–10 weeks old, 22–25 g) were used. PI3Kγ KO mice (backcrossed onto a C57BL/6 background for >10 generations) exhibit normal size, weight, viability, fertility, blood cell count, and chemistries as described (Suppl. Table S1).17 All animal protocols were approved by the Institutional Animal Care and Use Committee.
Middle Cerebral Artery Occlusion and Reperfusion
Focal cerebral ischemia was induced by transient occlusion of the left middle cerebral artery (tMCAO) with a 6-0 silicone-coated nylon monofilament (Doccol Corp.), as previously described.18 Subgroups of WT mice and PI3Kγ KO mice were randomly selected to receive MCAO or sham procedures. In ischemic groups, animals were subjected to 60 min tMCAO followed by reperfusion for indicated times. In the sham controls, the arteries were visualized but not disturbed. Rectal temperature was maintained at 37± 0.5°C throughout the procedure from the start of the surgery until the animals recovered from anesthesia with a feedback-regulated heating pad. Systemic parameters including pH and blood gases were all within normal range and there were no differences between wildtype and KO mice. Regional cerebral blood flow (CBF) was monitored by laser Doppler flowmetry (MSP300XP; ADInstruments Inc) as previously described.19 Only animals that exhibited a reduction in CBF>85% during MCAO and a CBF recovery by >80% after 15 min of reperfusion were included in the study. The number of animals used for each experimental group are provided in the figure legends and summarized in the Suppl. Table S2.
Determination of Evans Blue Leakage, Edema, and Water Content
Evans blue leakage was assessed as previously described.20 2% Evans blue (Sigma-Aldrich, 6 mL/kg) in 0.9% saline was injected into the right femoral vein, and animals were killed 3 hours after the injection. Each hemisphere was homogenized in 2 mL of N,N-dimethylformamide (Sigma-Aldrich), incubated for 18 hours at 55°C, and centrifuged. The absorption of the supernatant was measured at 620 nm by spectrophotometry. Brain tissue water content was measured by the wet and dry weight method.21 It was calculated as =100×(wet weight − dry weight)/wet weight. Edema volume was calculated as = [(ipsilateral hemisphere’s volume/contralateral hemisphere’s volume) − 1] × 100.21
Infarct volumes and neurological deficits
Mice were killed 24h after tMCAO, and the brains were removed and 2-mm-thick coronal sections were immersed in 2% 2,3,5-triphenyltetrazolium chloride (Sigma) at 37°C for 30 minutes.18 The area of infarction in each slice was determined by a computerized image analysis system (NIH Image), and the average infarct volume was calculated by multiplying the distance between sections. All measurements were performed by two “blinded” researchers and the mean of their results wan calculated. The calculated infarct volumes were corrected for brain swelling as previously described.21
24 h after tMCAO the modified Bederson score 22 was used to determine global neurological function according to the following scoring system: 0, no deficit; 1, forelimb flexion; 2, decreased resistance to lateral push; 3, unidirectional circling; 4, longitudinal spinning; 5, no movement. Motor function and coordination were evaluated by the grip test.23 For this test, the mouse was placed midway on a string between two supports and rated as follows: 0, falls off; 1, hangs onto string by one or both forepaws; 2, as for 1, and attempts to climb onto string; 3, hangs onto string by one or both forepaws plus one or both hindpaws; 4, hangs onto string by fore- and hindpaws plus tail wrapped around string; 5, escape (to the supports). Neurological scores were always assessed by a blinded independent investigator.
Immunohistochemistry
Immunostaining was performed as described previously.24, 25 The primary antibodies were: goat anti-MMP-9 (1:500; R&D Systems), rabbit anti-mouse PMN antibody (1:500; Accurate Chemical) to label neutrophils, Iba-1 (1:1000; Wako) to label microglia, GFAP (1:500, Dako) to label astrocytes, NeuN (1:1000; Millipore) to label neuronal cells, CD31 (1:100; BD Pharmingen) to label endothelial cells, rabbit anti-claudin-5 (1:200; Zymed), rabbit anti-collagen-IV (1:500; Abcam), and phospho-myosin light chain 2 (Ser19) mouse mAb (1:500; Cell Signaling). For double-labeling immunofluorescence, after primary antibody incubation, sections were incubated with Alexa fluorochrome-conjugated secondary antibodies (1:1000 in PBS; Invitrogen). At lease five animals were used per group per time point. The number of PMN-positive and MMP-9-positive cells was counted in predefined areas of the ischemic hemisphere at five coronal levels (rostrocaudal levels, +1.6, +1.0, +0.4, −0.2, −0.8 mm from bregma).24 Cell counts were performed by two “blinded” researchers under light microscopy (x200) and the mean of their results was calculated and adjusted to express as mean cell number/mm2.
Determination of ROS production
Mice were killed at 4 or 24h after tMCAO. ROS production in the brain was assessed using in vivo dihydroethidium (DHE) staining as previously described 25 with minor modifications. DHE, a cell-permeable oxidative-sensitive fluorescent dye, is oxidized to ethidium by superoxide but not by other ROS. 26, 27 Ethidium intercalates into DNA and emits a red fluorescence mainly from the nuclei.26 DHE (Sigma-Aldrich) was prepared as a 1.0 mg/ml solution in 1% DMSO. DHE solution (1.0 mg/kg) was injected into the femoral vein of anesthetized mice and allowed to circulate for 4 h. Mice were killed and perfusion-fixed with 4% PFA at 4h and 24h after reperfusion. Cryostat sections (20 um thickness) were prepared and photographed with a Nikon fluorescent microscope with excitation at 510–550 nm and emission >580 nm to detect oxidized DHE. DHE fluorescence intensity was analyzed by two “blinded” researchers using Nikon NIS-Elements software (Basic Research 3.0) and the mean of their results wan calculated. Fluorescence intensity was measured in predefined areas of the ischemic cortex and striatum at five coronal levels as described above. The sum of the fluorescence intensity for each region was divided by the total number of pixels analyzed and expressed as fluorescence intensity relative to the level in contralateral controls.27
Quantitative Real-time PCR
Mice were killed at multiple time points (1, 3, 6, 12, 24, 72h) after tMCAO, and the brains were removed. Total RNA was extracted from the ipsilateral cortices (between coronal levels +2 mm to −3 mm relative to bregma) by using the TRIzol extraction kit (Life Technologies). Samples from sham-operated animals served as basal controls. Expression levels of NADPH oxidase subunits (Nox1, Nox2, and Nox4) and adhesion molecules (E-Selectin, P-selectin, ICAM-1) were determined by quantitative real-time RT-PCR with a Bio-Rad thermocycler and an SYBR green kit (Bio-Rad) according to the recommendations of the manufacturer. Primer sequences are given in Suppl. Table S3. All primers were purchased from Invitrogen. All samples were run in triplicate. Relative levels of mRNA were normalized to the mouse HPRT housekeeping gene, and the results are expressed as fold changes versus the sham controls.
Western blot
Mice were killed at indicated times after tMCAO. Brains were stored at −80°C until analysis. Western blot analysis was performed as described previously.28 Briefly, whole-cell protein was prepared from the ischemic cortices (between coronal levels +2 mm to −3 mm relative to bregma). Samples from sham-operated animals served as basal controls. Brain tissues were homogenized in 10× vol of cold protein extraction buffer (complete tablet; Roche), centrifuged at 13,000 rpm for 20 min at 4 °C, and the supernatant was used for analysis. After adding the 2× volume of sample buffer (Invitrogen) to the supernatant, equal amounts were loaded per lane. Antibodies used were as follows: rabbit polyclonal antibodies to total NF-kB p65 and phospho-p65 at serine 536 (1:1000; Cell Signaling), and rabbit anti-phospho-MLC (ser19) (1:1000; Cell Signaling), and rabbit anti-mouse MPO monoclonal antibody (1:1000, Upstate). Protein samples (30 μg protein each lane) were separated by SDS-PAGE and proteins transferred to polyvinylidene difluoride membrane. Membraneswere blocked [10% nonfat milk in 0.01% PBS-Tween20 (PBS-T)] and incubated in primary antibody (diluted in 1% bovine serum albumin in PBS-T) overnight at 4°C. Membranes were then incubated with horseradish peroxidase-conjugated secondary antibody (in 5% milk in PBS-T). Immunopositive bands of horseradish peroxidase (HRP)-conjugated secondary antibodies were detected with an ECL system (GE Healthcare) and exposure to ECL Hyperfilm.
Gelatin Zymography
MMP-9 or MMP-2 activity in brain homogenates were determined by gelatin zymography as previously described.29 Briefly, brain were removed quickly and dissected on ice, and the ipsilateral and contralateral hemisphere (between coronal levels +2 mm to −4 mm relative to bregma) were homogenized with lysis buffer (50 mmol/L Tris-HCl (pH 7.4), 150 mmol/L NaCl, 5 mmol/L CaCl2, 0.05% Brij-35, and 1% Triton X-100). Supernatants obtained from homogenized brain tissues were incubated with gelatin-Sepharose 4B (GE Healthcare). Equal amounts of protein (20 μg) was loaded per lane and separated on 10% Tris-glycine gels with 0.1% gelatin as substrate. A mixture of MMP-9 and MMP-2 (CC073, Chemicon International) was used as a gelatinase standard. Images of the gels were scanned with a densitometer (GS-700, Bio-Rad Laboratories), and quantification was analyzed with Multi-Analyst 1.0 software (Bio-Rad Laboratories).
ELISA Assays
ELISA assays of brain cortice homogenates were performed to detect total MMP-9 protein using Quantikine mouse MMP-9 (total) immunoassay kit (R&D Systems), myeloperoxidase (MPO) using the mouse ELISA kit (HK210, Hycult Biotechnology), and malondialdehyde (MDA) using NWLSS™ MDA assay kit (Northwest Life Science Specialties, LLC, WA, USA) according to the instructions by the manufacturers. MDA is, one of the end products of lipid peroxidation and a commonly used marker of oxidant stress. All samples were run in triplicate.
Statistical Analysis
Data were expressed as mean ± s.d. Statistical analysis was performed using ANOVA and Student’s t-test. Differences with p < 0.05 were considered statistically significant.
Results
PI3Kγ deficiency reduces Evans blue leakage and brain edema
In WT mice, Evans blue leakage was markedly increased within the first 4 h of reperfusion, followed by an obvious decrease before 24 h of reperfusion (data not shown), and a second rise was observed between 24 and 72 h (maximally at 48 h) (Figure 1A). These findings are consistent with the time course described in a rat stroke model,30 and suggest that there is a biphasic opening of the BBB after I/R in our mouse model. Evans blue leakage was largely reduced in PI3Kγ KO mice (Figure 1A). Brain water content and brain swelling were significantly reduced in KO mice compared with WT mice (Figure 1B). There were no significant differences between WT and KO mice, with respect to vascular anatomy (Suppl. Figure 1) and cerebral blood flow (CBF) (Suppl. Figure 2) and systemic parameters (Suppl. Table S1).
Figure 1. PI3Kγ deficiency reduces BBB leakage, edema formation, infarct volume, and improves neurological outcome.
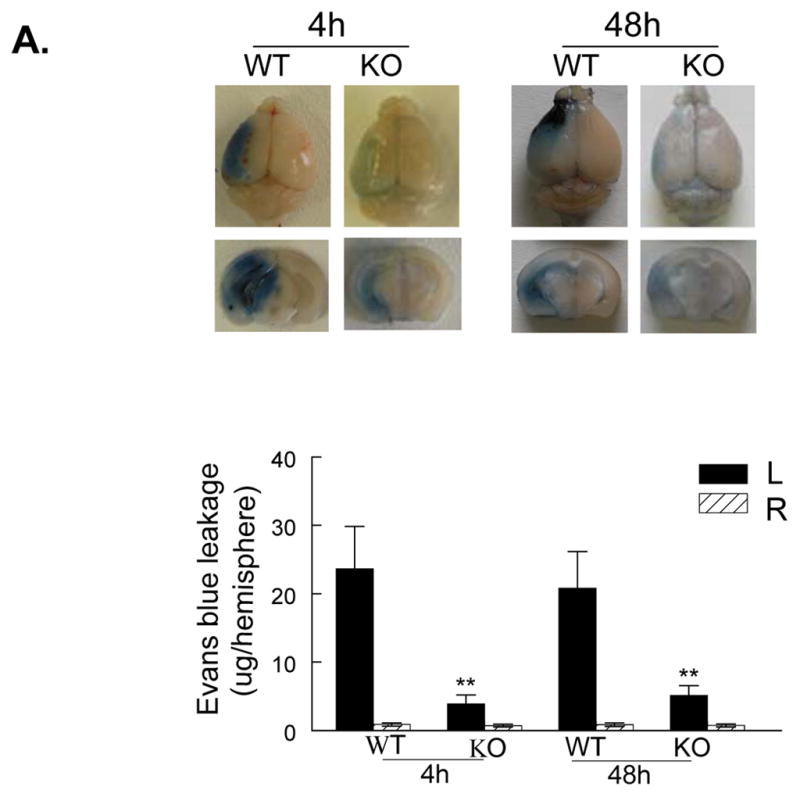


A, Upper panel shows representative pictures of Evans blue leakage in the brains and coronal sections (bregma +0.70 mm) in wildtype (WT) and PI3Kγ knockout (KO) mice at 4 and 48 h after tMCAO. Lower panel shows the quantitative analysis of Evans blue leakage. L represents the ischemic (left) side, and R represents the contralateral (right) side. n= 5 mice per time point per group, **P<0.01 versus WT controls. B, Brain tissue water content and edema volume in WT and KO mice at 24 and 48 h after tMCAO. n=8 mice per group, *P<0.05 versus WT controls. C, Infarct volumes. Representative photographs of TTC staining (upper panel) and infarct volumes (lower panel) at 24h after tMCAO in WT and KO mice (n=12/group). D, Neurological outcomes. Neurological Bederson score (upper panel) and grip test score (lower panel) as assessed at day 1 after tMCAO in WT and KO mice (n=12/group). *P<0.05 versus WT mice.
PI3Kγ deficiency reduces brain infarction and neurological deficits
24 hours after tMCAO, the infarct volume that developed in PI3Kγ KO mice was significantly smaller than inWT mice (23.5±7.2 mm3 vs 52.8±9.3 mm3, P<0.05, Figure 1C). The reduction in infarct size was functionally relevant, becausethe Bederson score assessing global neurological function and the grip test score assessing motor function were significantly better in PI3Kγ KO mice than in WT mice at 24 h aftertMCAO (Figure 1D).
PI3Kγ deficiency reduces the damage to BBB structural components
Immunostaining was performed to demonstrate I/R-induced time-dependent changes in the tight junction protein, claudin-5, and the basal lamina protein, collagen IV, because both proteins are critical for maintaining BBB integrity.3, 4 For claudin-5 staining, in WT mice claudin-5 levels in the ischemic cortical regions were markedly decreased at 4 h, followed by partial recovery at 24 h, and then reduced more markedly at 48 h after reperfusion (Figure 2A). In KO mice, the damage to claudin-5 was profoundly attenuated at all these time points (Figure 2A). Double immunostaining with anti-claudin-5 and anti-CD31 antibodies showed strong co-localization in the non-ischemic (contralateral) hemisphere, indicating the endothelial origin of the claudin-5 in microvessels (Suppl. Figure 3A). For collagen IV staining, a similar pattern in the extensive and dense networks of the collagen IV was noted in WT and KO non-ischemic (contralateral) hemisphere (Figure 2B). In WT mice, Collagen IV levels were decreased slightly at 4 h, but decreased obviously at 24 and 48 h after reperfusion (Figure 2B). In KO mice, the damage to collagen IV was profoundly prevented at all these time points (Fig. 2B). These findings were confirmed by Western blotting analysis of claudin-5 and collagen IV in brain homogenates (Suppl. Figure 4).
Figure 2. PI3Kγ deficiency reduces cerebrovascular claudin-5 and collagen IV degradation and myosin light chain (MLC) phosphorylation.



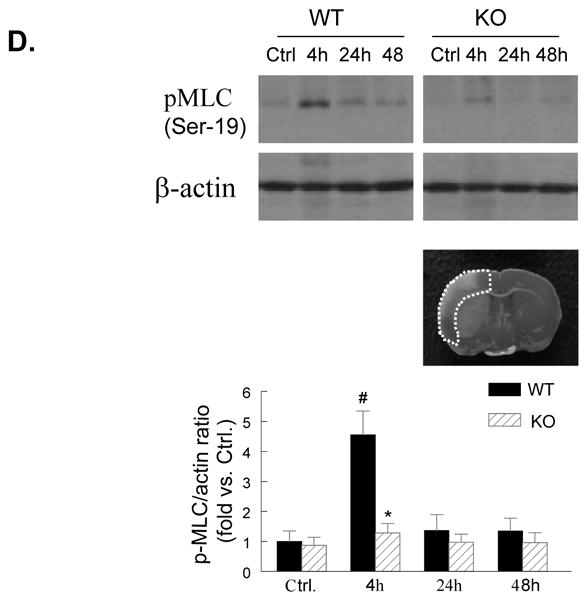
A through C, Immunostaining of claudin-5 (A), collagen IV (B), and phospho-MLC (Ser19) (C) in WT and PI3Kγ KO mice at 4, 24, and 48 h after tMCAO. The region of interest is shown in adjoining coronal section. Ctrl and Ipsi. represents contralateral and ipsilateral hemispheres, respectively. n=5 per time point per group. Scale bar: 50 μm. D, Western blot analysis of the phospho-MLC(ser19) (18 kDa) in the ischemic cortex (region of interest shown in adjoining coronal section). Samples from sham-operated animals served as controls. Quantified band intensities are presented as fold-changes of control (Ctrl). n= 4 per time point per group. *P<0.05 vs WT, #P<0.05 vs Ctrl.
PI3Kγ deficiency reduces microvascular endothelial MLC phosphorylation
The phosphorylation of the myosin light chain (MLC) in brain microvessels has been linked to the acute (4h) damage of the tight junction protein claudin-5 after cerebral I/R.25 In the non-ischemic (contralateral) hemisphere, the phosphorylation of the MLC (Ser19) in microvessels was minimal (Figure 2C), consistent with the previous data.25 In WT mice, I/R induced a marked phosphorylation of the MLC (Ser19) in the ischemic cortex at 4 h, but the MLC phosphorylation levels were largely decreased at 24 h and thereafter (Figure 2C). In KO mice, the I/R-induced MLC phosphorylation in microvessels was markedly inhibited (Figure 2C). This finding was confirmed by Western blotting analysis of the phosphor-MLC (Ser19) in brain homogenates (Figure 2D). Double immunostaining with anti-MLC (Ser19) and anti-CD31 antibodies showed strong co-localization in the ischemic cortex of WT mice at 4 h, indicating the endothelial origin of the phosphorylated MLC in microvessels (Suppl. Figure 3B).
PI3Kγ deficiency attenuates NF-kB activation and inflammatory gene expression
Transcription factor NF-κB is known to be activated in cerebral ischemia.31 We used PI3Kγ-deficient mice to determine whether PI3Kγ is required for the activation of NF-kB elicited by tMCAO (Figure 3A). Western blot analysis showed that the phosphorylation of NF-kB p65 (ser536) was dramatically induced at 4 h and more prominently at 24 h after tMCAO in WT mice, this result is consistent with previous findings.28 In PI3Kγ KO mice, the phosphorylation of NF-kB p65 (ser536) was profoundly attenuated.
Figure 3. PI3Kγ deficiency reduces NF-kB p65 phosphorylation and proinflammatory gene expression.



A, Western blot analysis of the phospho-p65 (ser536) and total p65 in the ischemic cortex at 4 and 24 h after tMCAO. The region of interest is shown in adjoining coronal section. Samples from sham-operated animals served as controls. Similar results were obtained in three separate experiments. B & C, Real-time RT-PCR analysis of mRNA expression of the NADPH oxidase subunits (Nox-1, Nox-2, Nox-4) (B) and the leukocyte-endothelial adhesion molecules (E-selectin, P-selectin, ICAM-1) (C) in the ischemic cortex (region of interest shown in adjoining coronal section) at indicated times after tMCAO. n= 4 per time point per group. *P<0.05 vs WT, #P<0.05 vs sham control.
NF-kB plays a critical role in regulating the expression of many different genes involved in oxidative stress and proinflammatory response.31 Next, we examined the effect of PI3Kγ deficiency on mRNA expression of the NADPH oxidase subunits (Nox1, Nox2, Nox4) at multiple time points after tMCAO (Figure 3B). In WT mice, the expression of Nox-1 and Nox-2 was elevated significantly within early hours (1–6 h) and continued to increase or maintain high level up to 72h after tMCAO, this result is consistent with previous findings.24, 32, 33 However, a significant increase of Nox-4 mRNA expression was detected at relatively late time points (12–72 h), this result is consistent with a recent study. 34 In PI3Kγ KO mice, the expression of Nox-1, Nox-2, and Nox-4 was significantly attenuated (Figure 3B).
Leukocyte adhesion and transmigration across the blood-brain barrier is regulated by multiple proinflammatory adhesion molecules, including selectins and ICAM-1. Thus, we examined the effect of PI3Kγ deficiency on the expression of proinflammatory adhesion molecules (E-selectin, P-selectin, ICAM-1) (Figure 3C). In WT mice, I/R induced a significant increase in the expression of E- selectin and P-selectin observed from 3–24 h (peak at 12 h), while ICAM-1 increase observed from 3–72 h (peak at 24 h). In PI3Kγ KO mice, the expression of these adhesion molecules was significantly attenuated (Figure 3C).
PI3Kγ deficiency attenuates ROS production and oxidative stress
Using the superoxide-sensitive dye dihydroethidium (DHE) staining technique, we assessed the effect of PI3Kγ deficiency on ROS production after tMCAO. In WT mice, the ROS signal in the ischemic cortex and striatum was markedly increased at 4 and 24 h (greatest at 4h) after reperfusion (Figure 4A, 4B), this result is consistent with previous findings.27 In PI3Kγ KO mice, ROSproduction was profoundly attenuated at both time points (Figure 4A, 4B).
Figure 4. PI3Kγ deficiency reduces ROS and MDA production.


A, (Left panel) Representative DHE fluorescence images showing the superoxide signal taken from the ischemic cortex and striatum 4h after tMCAO. The region of interest is shown in adjoining coronal section indicated by asterisks. B, Semiquantitative analysis of ROS production in cortex and striatum as described in Methods at 4 and 24 h after tMCAO. Data represent mean ± S.E. n = 5 per time point per group, *P<0.05, **P<0.01 versus WT. C, ELISA assay for MDA protein in the ischemic cortex (region of interest shown in adjoining coronal section) at 4 and 24 h after tMCAO in WT and KO mice. MDA level is also expressed as ratio of ipsilateral/contralateral (ipsi/ctrl). n=5 per time point per group, **P<0.01 versus WT.
Malondialdehyde (MDA) is an end product of lipid peroxidation and an indicator of oxidative stress. In WT ischemic hemispheres, the MDA levels did not altered during the first 4 h compared with sham controls, but were significantly increased 24 h after reperfusion (Figure 4C), this result is consistent with a previous study.33 In KO mice, MDA levels in postischemic hemispheres were not significantly increased after I/R (Figure 4C).
PI3Kγ deficiency reduces neutrophil infiltration and neutrophil-associated MMP-9
The cellular sources of MMP-9 in the postischemic brain include mainly infiltrated neutrophils (PMNs) but also cerebral microvascular endothelial cells after acute ischemic stroke in animals and humans.5, 6, 25 Immunostaining showed that PMNs and MMP-9 were not detected either in the contralateral hemisphere or in the sham-operated mice (Figure 5A, 5B). In WT mice, only a few PMNs and MMP-9-positive (MMP-9+) cells were noted in the ischemic hemisphere at 4h (not shown), but the number of positively stained cells was markedly increased between 24 and 72 h after tMCAO (Figure 5A, 5B). In PI3Kγ KO mice, the number of PMNs and MMP-9+ cells in the ischemic hemisphere was largely reduced (Figure 5A, 5B). It is worth to note that PMNs and MMP-9+ cells exhibited similar immunostaining patterns at different time points. Moreover, ELISA assay of myeloperoxidase (MPO), a marker of neutrophil infiltration, confirmed that neutrophil infiltration was inhibited in PI3Kγ KO mice compared with WT control (Figure 5C). Using double immunostaining, we determined the cellular sources of MMP-9 in postischemic brain. Our data showed strong co-localization of MMP-9 signal with the neutrophil-specificmarker (PMN), but not with neuronal (neuN), microglial/macrophage(Iba1), or astrocytic (GFAP) markers (Figure 5D), indicating that infiltrated neutrophils were the major cellular source of MMP-9 in our model.
Figure 5. PI3Kγ deficiency reduces neutrophil infiltration and neutrophil-associated MMP-9.


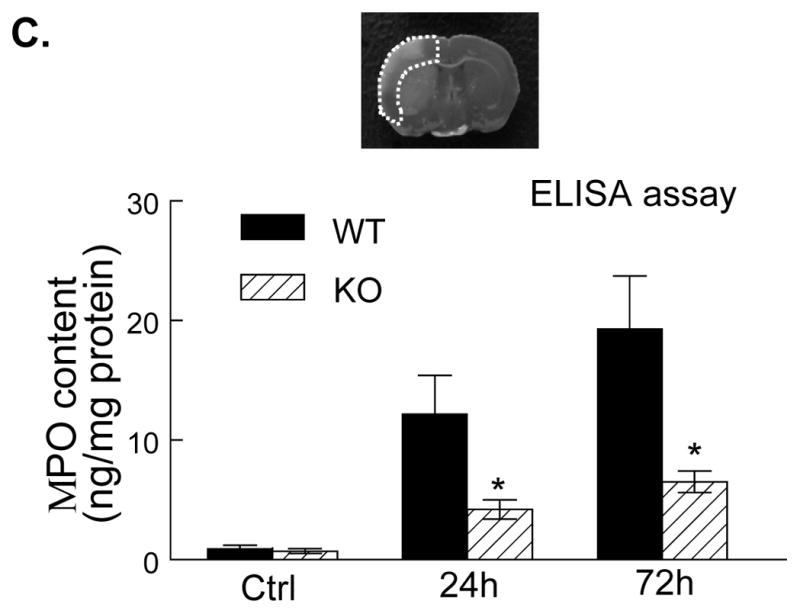

A&B, Immunostaining for neutrophils (A) and MMP-9 (B) in the ischemic cortex in WT and PI3Kγ KO mice at the indicated times after tMCAO. The number of the cells positively stained for neutrophils and MMP-9 was calculated in the 3 predefined regions of interest shown in adjoining coronal section. Bar = 50μm. n = 5 per time point per group, *P<0.05 versus WT control. C, Neutrophil infiltration was quantified by MPO assay. ELISA assay of MPO protein in ischemic cortex (region of interest shown in adjoining coronal section) 24 h after tMCAO. n=5 for each group. *P<0.05 versus WT control. In A–C, Samples from sham-operated animals served as controls (Ctrl). D, Double immunostaining showing the co-localization of MMP-9 with the specific neutrophil marker (PMN), but not with microglial (Iba1), astrocytic (GFAP), or neutronal (neuN) markers in the ischemic cortex at 24 h after tMCAO. Bar = 50μm.
Moreover, we examined the endothelial expression of MMP-9 after tMCAO. In WT and KO contralateral (non-ischemic) hemisphere, endothelial MMP-9 was not detected. In WT mice, the immunoreactivity for MMP-9 in the endothelium of microvessels in ischemic hemisphere was minimal at 4 h, but markedly increased at 24 h, declined to a low level at 48 h, and disappeared at 72 h after tMCAO (Suppl. Figure 5). MMP-9+neutrophils were shown abundantly around microvessels (i.e. perivascular MMP-9) at 72 h in WT ischemic hemisphere (Suppl. Figure 5). In KO mice, the expression of MMP-9 in the endothelium of microvessels was minimal at all these time points (Suppl. Figure 5).
PI3Kγ deficiency reduces MMP-9 activity and protein content
Using gelatin zymography, we assessed the effect of PI3Kγ deficiency on MMP activation at 24 h after reperfusion. Two clear bands could be detected for MMP-9 corresponding to pro-MMP-9 at 92 kDa and cleaved form at 82 kDa (Figure 6A). Total MMP-9 levels (including pro-MMP-9 and cleaved form) and cleaved MMP-9 were significantly increased in homogenates of the ischemic hemispheres in WT mice, but this increase was largely inhibited in KO mice (Figure 6A, 6B). In contrast, no significant change for MMP-2 (63 kDa) was noted (Figure 6A, 6B), consistent with the previous finding in rat.35
Figure 6. PI3Kγ deficiency reduces MMP-9 activity and protein content.


A, Gelatin zymogram. Note that MMP-9 (92 and 82 kDa) and MMP-2 (63 kDa) bands were detected in the homogenates of ischemic (left, L) and contralateral (right, R) cortex 24 h after tMCAO. The region of interest is shown in adjoining coronal section. B, Densitometric analysis of the bands shown in A. Data represent mean ± s.d. from 3 separate experiments. *P<0.05 versus WT (saline-treated) control. C. ELISA assay of MMP-9 protein in ischemic (left) and contralateral (right) cortex (region of interest shown in adjoining coronal section) 24 h after tMCAO. n=5 for each group. **P<0.01 versus WT control.
Consistent with the above findings, ELISA assay showed that total MMP-9 protein content was increased in homogenates of the WT ischemic hemispheres at 24h time point, but this increase was largely attenuated in PI3Kγ KO mice (Figure 6C).
Discussion
The present study provides the first direct demonstration that PI3Kγ plays a central role in I/R-induced BBB disruption and brain damage. Among the class I PI3Ks, PI3Kγ is a key regulator of inflammatory and oxidative responses and has been implicated in a number of inflammatory diseases, such as atherosclerosis and myocardial ischemia/reperfusion injury. However, the role of PI3Kγ in the pathogenesis of stroke has not been investigated. Here, we demonstrate for the first time that genetic deletion of PI3Kγ prevented the BBB disruption and offered protection against brain damage in acute ischemic stroke. We identify PI3Kγ as a crucial player of cerebral inflammation and oxidative stress, possibly through modulating NF-kB activation, proinflammatory gene expression, neutrophil infiltration, and MMP-9 expression/activation in the postischemic brain.
In this study, we demonstrated that absence of PI3Kγ provided a profound protection against the damage to BBB structural integrity, eventually attenuating BBB dysfunction and brain damage elicited by focal cerebral I/R. We observed that I/R caused a biphasic opening of the BBB corresponding to the early (4 h) and late (48 h) increase in BBB permeability. This observation is consistent with the time course of the BBB opening previously described in a rat stroke model.30, 36 Despite extensive investigation, the molecular mechanisms underlying the I/R-induced biphasic BBB opening have not been well-defined. Among various components of the BBB, the tight junction protein, claudin-5, and the basal lamina protein, collagen IV, are two of the most widely studied components of the BBB, both are critical for maintaining the BBB structural integrity and permeability.2–4 The degradation of the cerebrovascular claudin-5, as well as the basal lamina collagen IV, has been highly correlated with the dynamic process of BBB disruption after cerebral ischemia. 25, 35, 37, 38 The results of this study suggest that the early BBB opening correlated with an acute disruption of the tight junction protein, claudin-5, and also with an acute phosphorylation of the myosin light chain (MLC) in brain microvessels, while the late BBB opening correlated with a sustained disruption of claudin-5, and a marked loss of the basal lamina protein, collagen IV. Thus, PI3Kγ signaling is important for maintaining the structural integrity of the BBB in acute stroke.
Oxidative stress is probably one of the most important mechanisms involved in BBB disruption and neuronal death in acute ischemic stroke. 39 PI3Kγ is known to play an essential role in ROS production in peripheral tissues and many cell types, including leukocytes and endothelial cells through activation of NADPH oxidase.40,41 However, in vivo the exact role of PI3Kγ in ROS production in the brain under physiological and pathophysiological conditions are unknown. The findings of the present study indicate that PI3Kγ is a critical regulator of oxidative stress in the postischemic brain after cerebral I/R. Consistent with the study by Cho et al,27 we observed that ROS production was induced in the ischemic cortex and striatum most robustly at 4 h, but was relatively low at 24 h after reperfusion in WT mice, while ROS production was markedly inhibited in PI3Kγ KO mice. It is unlikely that the attenuation of ROS production observed in PI3Kγ KO mice is a consequence rather than a cause of the reduced brain damage, because in rodent models of cerebral ischemia, the tissue damage is not fully developed at the time that ROS production was greatest (0–4 h).42
There is ample evidence that NF-κB is activated in cerebral ischemia and promotes ischemic brain injury through regulating expression of many different genes involved in oxidative stress and inflammation.28, 31, 43,44 Among the five NF-kB subunits, p65/RelA and p50 are known to be responsible for detrimental effect in cerebral ischemia, and the phosphorylation of NFkB p65 (Ser536) is required for activation and nuclear translocation of NF-KB.31 Consistent with previous studies,28, 43, 44 the present data showed that the phosphorylation of NFkB p65 (Ser536) was robustly induced in the postischemic brain at 4 and 24 h after cerebral I/R in WT mice, while the induction of p65 phosphorylation was markedly inhibited in PI3Kγ KO mice. Moreover, the results of the present study showed that I/R-induced upregualtion of NADPH oxidase subunits (Nox1, Nox2, and Nox4) was largely inhibited in PI3Kγ KO mice at all time points (1–72 h) after tMCAO. In this regard, we assume that regulation of NF-kB and NADPH oxidase may represent a possible mechanism whereby PI3Kγ promotes oxidative stress elicited by focal cerebral ischemia. Our findings may have potential implications for preventing ROS production at its source by blocking PI3Kγ signaling upstream of NADPH oxidase.
Although NADPH oxidase has been regarded as a major source of ROS in ischemic stroke,39 there is no conclusive evidence of a causal link between specific NADPH oxidase subunits and the development of disease. Contradictory results have been reported in the literature.32–34 Experimental data reported by Kahles et al 32 and Chen et al 33 consistently showed that genetic deletion of Nox2 prevented early BBB dysfunction and reduced infarct size in mice 24 h after tMCAO. Intriguingly, Kleinschnitz et al 34 showed that genetic deletion of Nox4 profoundly reduced oxidative stress, BBB dysfunction, and neuronal apoptosis 24 h after tMCAO, while deficiency of Nox1 or Nox2 had no impact on infarct size or functional outcome. Importantly, Kleinschnitz et al investigated the roles of Nox1, Nox2, and Nox4 under identical experimental conditions. The exact reasons for this discrepancy are unclear at present. 34
There is ample evidence, in human and animal stroke models, that increased expression and activation of MMP-9 plays a critical role in the BBB disruption (particularly in late BBB opening) after ischemic stroke. 2, 5, 6, 35 Clinical and experimental data have shown that increased MMP-9 in postischemic brain tissue is mainly a result of infiltrated leukocytes (particularly neutrophils) during acute ischemic stroke.6, 25, 45 Consistent with previous studies, the present data indicate that infiltrated neutrophils are the major source of MMP-9 in the ischemic hemisphere during the 24–72 h period after reperfusion in WT mice. In addition to infiltrated neutrophils, cerebral endothelial cells are considered as another important source of local MMP-9 in ischemic stroke.2, 5, 6 In this study, we observed that I/R induced MMP-9 expression in brain microvessels was minimally at 4h and prominent at 24h, declined at 48h, and disappeared at 72h after tMCAO.
NF-kB is known to regulate expression of many different proinflammatory genes.31 Absence of PI3Kγ blocked I/R-induced neutrophil adhesion and infiltration into postischemic brain, and this effect could be explained by our findings that I/R-induced expression of the adhesion molecules (E- and P-selectin, ICAM-1) was markedly inhibited in PI3Kγ KO mice. In vivo, PI3Kγ deficiency inhibited I/R-induced neutrophil adhesion and infiltration in the postischemic brain. In vitro, PI3Kγ deficiency blocked TNFα-induced ICAM-1 expression and neutrophil firm adhesion in cultured mouse brain microvascular endothelail cells (Suppl. Fig. 6).
Collectively, our findings reveal the molecular mechanisms by which blocking PI3Kγ inhibited neutrophil adhesion and infiltration and eventually reduced both neutrophil- and endothelial cell-associated MMP-9 in postischemic brain after tMCAO.
Study Limitation
There are several potential limitations to this study. First, although we demonstrated that PI3Kγ deficiency reduced ROS production possibly via NADPH oxidase-mediated mechanism, we cannot be sure which NADPH oxidase subunits and what mechanisms are exactly involved. Second, the precise cellular actions of PI3Kγ in stroke, especially regarding the contribution of the endothelial and microglial PI3Kγ to the development of ischemic stroke, need to be defined by future studies. Third, it is now apparent that no single animal model can precisely replicates human ischemic stroke, future studies in other stroke models (e.g. thromboembolic stroke model with or w/o tPA treatment) are warranted.
Conclusions
We have demonstrated that genetic deletion of PI3Kγ prevents ischemia/reperfusion associated BBB disruption and attenuates tissue damage in acute experimental stroke. These results have identified PI3Kγ as a crucial player in the pathophysiology of acute ischemic stroke. Future studies will be needed to identify the precise cellular actions of PI3Kγ in stroke and explore PI3Kγ as a potential target for stroke therapy.
Supplementary Material
Acknowledgments
The authors thank the animal core facility of the LSU Health Science Center-Shreveport for help with animal breeding and maintenance.
Sources of Funding
This work was supported by National Institutes of Health grants HL087990 (G.L.) and HL026441 (D.N.G.), and by a scientist development grant (0530166N, G.L.) from the American Heart Association.
Footnotes
Disclosures
None.
References
- 1.Jung JE, Kim GS, Chen H, Maier CM, Narasimhan P, Song YS, Niizuma K, Katsu M, Okami N, Yoshioka H, Sakata H, Goeders CE, Chan PH. Reperfusion and neurovascular dysfunction in stroke: from basic mechanisms to potential strategies for neuroprotection. Mol Neurobiol. 2010;41:172–179. doi: 10.1007/s12035-010-8102-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jin R, Yang G, Li G. Molecular insights and therapeutic targets for blood-brain barrier disruption in ischemic stroke: critical role of matrix metalloproteinases and tissue-type plasminogen activator. Neurobiol Dis. 2010;38:376–385. doi: 10.1016/j.nbd.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 4.Sandoval KE, Witt KA. Blood–brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis. 2008;32:200–219. doi: 10.1016/j.nbd.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Barr TL, Latour LL, Lee KY, Schaewe TJ, Luby M, Chang GS, El-Zammar Z, Alam S, Hallenbeck JM, Kidwell CS, Warach S. Blood-brain barrier disruption in humans is independently associated with increased matrix metalloproteinase-9. Stroke. 2010;41:e123–128. doi: 10.1161/STROKEAHA.109.570515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosell A, Cuadrado E, Ortega-Aznar A, Hernández-Guillamon M, Lo EH, Montaner J. MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke. 2008;39:1121–1126. doi: 10.1161/STROKEAHA.107.500868. [DOI] [PubMed] [Google Scholar]
- 7.Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao H, Sapolsky RM, Steinberg GK. Phosphoinositide-3-kinase/akt survival signal pathways are implicated in neuronal survival after stroke. Mol Neurobiol. 2006;34:249–270. doi: 10.1385/MN:34:3:249. [DOI] [PubMed] [Google Scholar]
- 9.Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta. 2008;1784:159–185. doi: 10.1016/j.bbapap.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 10.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11:329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 11.Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP. Central role for G protein-coupled phosphoinositide 3-kinase γ in inflammation. Science. 2000;287:1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- 12.Puri KD, Doggett TA, Huang CY, Douangpanya J, Hayflick JS, Turner M, Penninger J, Diacovo TG. The role of endothelial PI3Kgamma activity in neutrophil trafficking. Blood. 2005;106:150–157. doi: 10.1182/blood-2005-01-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fougerat A, Gayral S, Gourdy P, Schambourg A, Rückle T, Schwarz MK, Rommel C, Hirsch E, Arnal JF, Salles JP, Perret B, Breton-Douillon M, Wymann MP, Laffargue M. Genetic and pharmacological targeting of phosphoinositide 3-kinase-gamma reduces atherosclerosis and favors plaque stability by modulating inflammatory processes. Circulation. 2008;117:1310–1317. doi: 10.1161/CIRCULATIONAHA.107.720466. [DOI] [PubMed] [Google Scholar]
- 14.Doukas J, Wrasidlo W, Noronha G, Dneprovskaia E, Fine R, Weis S, Hood J, Demaria A, Soll R, Cheresh D. Phosphoinositide 3-kinase gamma/delta inhibition limits infarct size after myocardial ischemia/reperfusion injury. Proc Natl Acad Sci U S A. 2006;103:19866–19871. doi: 10.1073/pnas.0606956103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barber DF, Bartolomé A, Hernandez C, Flores JM, Redondo C, Fernandez-Arias C, Camps M, Rückle T, Schwarz MK, Rodríguez S, Martinez-A C, Balomenos D, Rommel C, Carrera AC. PI3Kgamma inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat Med. 2005;11:933–935. doi: 10.1038/nm1291. [DOI] [PubMed] [Google Scholar]
- 16.Jin R, Yu S, Song Z, Quillin JW, Deasis DP, Penninger JM, Nanda A, Granger DN, Li G. Phosphoinositide 3-kinase-gamma expression is upregulated in brain microglia and contributes to ischemia-induced microglial activation in acute experimental stroke. Biochem Biophys Res Commun. 2010;399:458–464. doi: 10.1016/j.bbrc.2010.07.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, Wakeham A, Itie A, Bouchard D, Kozieradzki I, Joza N, Mak TW, Ohashi PS, Suzuki A, Penninger JM. Function of PI3Kγ in thymocyte development, T cell activation, and neutrophil migration. Science. 2000;287:1040–1046. doi: 10.1126/science.287.5455.1040. [DOI] [PubMed] [Google Scholar]
- 18.Terao S, Yilmaz G, Stokes KY, Russell J, Ishikawa M, Kawase T, Granger DN. Blood cell-derived RANTES mediates cerebral microvascular dysfunction, inflammation, and tissue injury after focal ischemia-reperfusion. Stroke. 2008;39:2560–2570. doi: 10.1161/STROKEAHA.107.513150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin G, Tsuji K, Xing C, Yang YG, Wang X, Lo EH. CD47 gene knockout protects against transient focal cerebral ischemia in mice. Exp Neurol. 2009;217:165–170. doi: 10.1016/j.expneurol.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamada H, Yu F, Nito C, Chan PH. Influence of hyperglycemia on oxidative stress and matrix metalloproteinase-9 activation after focal cerebral ischemia/reperfusion in rats: relation to blood-brain barrier dysfunction. Stroke. 2007;38:1044–1049. doi: 10.1161/01.STR.0000258041.75739.cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strbian D, Karjalainen-Lindsberg ML, Tatlisumak T, Lindsberg PJ. Cerebral mast cells regulate early ischemic brain swelling and neutrophil accumulation. J Cereb Blood Flow Metab. 2006;26:605–612. doi: 10.1038/sj.jcbfm.9600228. [DOI] [PubMed] [Google Scholar]
- 22.Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- 23.Moran PM, Higgins LS, Cordell B, Moser PC. Age-related learning deficits in transgenic mice expressing the 751-amino acid isoform of human amyloid precursor protein. Proc Natl Acad Sci. 1995;92:5341–5345. doi: 10.1073/pnas.92.12.5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kunz A, Abe T, Hochrainer K, Shimamura M, Anrather J, Racchumi G, Zhou P, Iadecola C. Nuclear factor-kappaB activation and postischemic inflammation are suppressed in CD36-null mice after middle cerebral artery occlusion. J Neurosci. 2008;28:1649–1658. doi: 10.1523/JNEUROSCI.5205-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McColl BW, Rothwell NJ, Allan SM. Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. J Neurosci. 2008;28:9451–9462. doi: 10.1523/JNEUROSCI.2674-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suh SW, Gum ET, Hamby AM, Chan PH, Swanson RA. Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal NADPH oxidase. J Clin Invest. 2007;117:910–918. doi: 10.1172/JCI30077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cho S, Park EM, Febbraio M, Anrather J, Park L, Racchumi G, Silverstein RL, Iadecola C. The class B scavenger receptor CD36 mediates free radical production and tissue injury in cerebral ischemia. J Neurosci. 2005;25:2504–2512. doi: 10.1523/JNEUROSCI.0035-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang X, Polavarapu R, She H, Mao Z, Yepes M. Tissue-type plasminogen activator and the low-density lipoprotein receptor-related protein mediate cerebral ischemia-induced nuclear factor-kappaB pathway activation. Am J Pathol. 2007;171:1281–1290. doi: 10.2353/ajpath.2007.070472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wakisaka Y, Chu Y, Miller JD, Rosenberg GA, Heistad DD. Spontaneous intracerebral hemorrhage during acute and chronic hypertension in mice. J Cereb Blood Flow Metab. 2010;30:56–69. doi: 10.1038/jcbfm.2009.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and TIMPs are associated with blood–brain barrier opening after reperfusion in rat brain. Stroke. 1998;29:2189–2195. doi: 10.1161/01.str.29.10.2189. [DOI] [PubMed] [Google Scholar]
- 31.Ridder DA, Schwaninger M. NF-kappaB signaling in cerebral ischemia. Neuroscience. 2009;158:995–1006. doi: 10.1016/j.neuroscience.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 32.Kahles T, Luedike P, Endres M, Galla HJ, Steinmetz H, Busse R, Neumann-Haefelin T, Brandes RP. NADPH oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke. 2007;38:3000–3006. doi: 10.1161/STROKEAHA.107.489765. [DOI] [PubMed] [Google Scholar]
- 33.Chen H, Song YS, Chan PH. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J Cereb Blood Flow Metab. 2009;29:1262–1272. doi: 10.1038/jcbfm.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kleinschnitz C, Grund H, Wingler K, Armitage ME, Jones E, Mittal M, Barit D, Schwarz T, Geis C, Kraft P, Barthel K, Schuhmann MK, Herrmann AM, Meuth SG, Stoll G, Meurer S, Schrewe A, Becker L, Gailus-Durner V, Fuchs H, Klopstock T, de Angelis MH, Jandeleit-Dahm K, Shah AM, Weissmann N, Schmidt HH. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 2010;8:pii, e1000479. doi: 10.1371/journal.pbio.1000479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. Cereb Blood Flow Metab. 2007;27:697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]
- 36.Belayev L, Busto R, Zhao W, Ginsberg MD. Quantitative evaluation of blood–brain barrier permeability following middle cerebral artery occlusion in rats. Brain Res. 1996;739:88–96. doi: 10.1016/s0006-8993(96)00815-3. [DOI] [PubMed] [Google Scholar]
- 37.Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, Tsukita S. Size-selective loosening of the blood–brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161:653–660. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamann GF, Okada Y, Fitridge R, del Zoppo GJ. Microvascular basal lamina antigens disappear during cerebral ischemia and reperfusion. Stroke. 1995;11:2120–2126. doi: 10.1161/01.str.26.11.2120. [DOI] [PubMed] [Google Scholar]
- 39.Sugawara T, Chan PH. Reactive oxygen radicals and pathogenesis of neuronal death after cerebral ischemia. Antioxid Redox Signal. 2003;5:597–607. doi: 10.1089/152308603770310266. [DOI] [PubMed] [Google Scholar]
- 40.Frey RS, Gao X, Javaid K, Siddiqui SS, Rahman A, Malik AB. Phosphatidylinositol 3-kinase gamma signaling through protein kinase Czeta induces NADPH oxidase-mediated oxidant generation and NF-kappaB activation in endothelial cells. J Biol Chem. 2006;281:16128–16138. doi: 10.1074/jbc.M508810200. [DOI] [PubMed] [Google Scholar]
- 41.Lehmann K, Müller JP, Schlott B, Skroblin P, Barz D, Norgauer J, Wetzker R. PI3Kgamma controls oxidative bursts in neutrophils via interactions with PKCalpha and p47phox. Biochem J. 2009;419:603–610. doi: 10.1042/BJ20081268. [DOI] [PubMed] [Google Scholar]
- 42.Clark RK, Lee EV, Fish CJ, White RF, Price WJ, Jonak ZL, Feuerstein GZ, Barone FC. Development of tissue damage, inflammation and resolution following stroke: an immunohistochemical and quantitative planimetric study. Brain Res Bull. 1993;31:565–572. doi: 10.1016/0361-9230(93)90124-t. [DOI] [PubMed] [Google Scholar]
- 43.Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF-κB is activated and promotes cell death in focal cerebral ischemia. Nat Med. 1999;5:554–559. doi: 10.1038/8432. [DOI] [PubMed] [Google Scholar]
- 44.Crack PJ, Taylor JM, Ali U, Mansell A, Hertzog PJ. Potential contribution of NF-kappaB in neuronal cell death in the glutathione peroxidase-1 knockout mouse in response to ischemia-reperfusion injury. Stroke. 2006;37:1533–1538. doi: 10.1161/01.STR.0000221708.17159.64. [DOI] [PubMed] [Google Scholar]
- 45.Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, Chan PH, Park TS. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol. 2005;289:H558–568. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.