

Abstract
Corticotropin-releasing hormone, a major neuromodulator of the neuroendocrine stress response, is expressed in the immature hippocampus, where it enhances glutamate receptor-mediated excitation of principal cells. Since the peptide influences hippocampal synaptic efficacy, its secretion from peptidergic interneuronal terminals may augment hippocampal-mediated functions such as learning and memory. However, whereas information regarding the regulation of corticotropin-releasing hormone’s abundance in CNS regions involved with the neuroendocrine responses to stress has been forthcoming, the mechanisms regulating the peptide’s levels in the hippocampus have not yet been determined. Here we tested the hypothesis that, in the immature rat hippocampus, neuronal stimulation, rather than neuroendocrine challenge, influences the peptide’s expression. Messenger RNA levels of corticotropin-releasing hormone in hippocampal CA1, CA3 and the dentate gyrus, as well as in the hypothalamic paraventricular nucleus, were determined after cold, a physiological challenge that activates the hypothalamic pituitary adrenal system in immature rats, and after activation of hippocampal neurons by hyperthermia. These studies demonstrated that, while cold challenge enhanced corticotropin-releasing hormone messenger RNA levels in the hypothalamus, hippocampal expression of this neuropeptide was unchanged. Secondly, hyperthermia stimulated expression of hippocampal immediate-early genes, as well as of corticotropin-releasing hormone. Finally, the mechanism of hippocampal corticotropin-releasing hormone induction required neuronal stimulation and was abolished by barbiturate administration.
Taken together, these results indicate that neuronal stimulation may regulate hippocampal corticotropin-releasing hormone expression in the immature rat, whereas the peptide’s expression in the hypothalamus is influenced by neuroendocrine challenges.
Keywords: hippocampus, corticotropin-releasing factor, c-fos, rat, hypothalamus, stress
Corticotropin-releasing hormone (CRH) is a peptide with both neuroendocrine and neurotransmitter properties.57 The neuroendocrine effects of CRH, the key mediator of the stress response, originate from clusters of peptidergic cells in the hypothalamic paraventricular nucleus (PVN).24,34 CRH also functions as a neuromodulator in a number of limbic and autonomic brain circuits.21,22,24,43 CRH-producing neurons are widely but specifically distributed in the brain,49 including a substantial CRH-expressing neuronal population located in the central nucleus of the amygdala (ACe),22,53 a region considered as a major source for extra-endocrine CRH-mediated neurotransmission.24
Abundant target neurons for CRH, expressing cognate receptors, have been demonstrated in the hippocampus.2,13,15 In addition, physiological effects of CRH on hippocampal neurons have been found, including facilitation of memory retention and increased protein phosphorylation.10,33 Conversely, abnormalities of CRH-mediated limbic neurotransmission may contribute to neurological disorders such as depression43 or Alzheimer’s disease.10,33 The source of endogenous CRH that may act on hippocampal CRH receptors has been a focus of investigation. Scattered CRH-immunoreactive neurons had been described in the hippocampal formation,41,48,53 and recent double-labeling and ultrastructural studies have demonstrated a robust population of CRH-expressing GABAergic interneurons in the hippocampal formation of non-stressed developing rats.59 A subpopulation of these neurons, co-expressing parvalbumin, has been identified as basket and chandelier cells.59 Axon terminals of these CRH-immunoreactive interneurons, synapsing on cell bodies of hippocampal principal neurons, are thus strategically positioned to influence neurotransmission in the hippocampal circuit via release of both CRH and GABA.59
In addition, it has been demonstrated in both the mature and developing hippocampus that application of CRH may lead to excessive excitation of the hippocampal circuit, resulting in repetitive discharges of hippocampal neurons in vitro1,25,50 and in seizures in vivo.7,39 It has been suggested that excessive elevation of hippocampal CRH levels may contribute to abnormal neuronal excitation (seizures).6 Thus, regulation of CRH levels in peptidergic hippocampal neurons may be important for both normal and pathological activation of the hippocampal circuit,6,25,50 predicting that these levels may be subject to tight physiological control. However, the regulation of CRH expression in the hippocampus has not been elucidated. In the PVN and the ACe, transcription of the CRH gene is enhanced by stress (defined operationally as activation of the neuroendocrine hypothalamic–pituitary–adrenal axis), leading to increased steady-state levels of CRH mRNA.30,35,60 Concurrent with induction of CRH transcription in these regions, stress leads to robust expression of immediate-early genes,26,30,45 indicating general activation of the involved neurons.31,42 Whether CRH gene expression in the hippocampus is influenced by challenges that activate the neuroendocrine hypothalamic–pituitary–adrenal axis, or by physiological stimulation of hippocampal neurons (evident from electrophysiological, behavioral and molecular measures), has not been resolved. Here, we investigated the influence of two physiological stimuli (cold and hyperthermia) on immature rat hippocampal and hypothalamic CRH mRNA levels, demonstrating signal- and region-specific up-regulation of CRH expression. CRH mRNA induction correlated with c-fos expression, suggesting that activation of discrete signal-transduction pathways may be required for physiological signals to influence hippocampal CRH expression.
EXPERIMENTAL PROCEDURES
Animals and tissue preparation
Immature rats were used in this study for two reasons: (i) previous work from this laboratory has characterized hippocampal and hypothalamic CRH expression during this age,59,60 and (ii) hippocampal CRH expression may be particularly robust during the second postnatal week in the rat59 (and Chen et al., unpublished observations). Ten-day-old rats of both genders used in this study were offspring of timed pregnancy Sprague–Dawley-derived dams (Zivic-Miller, Zelienople, PA). Animals were maintained in NIH-approved facilities on a 12-h/12-h light–dark cycle with access to unlimited lab chow and water. Cages were monitored every 12 h for the presence of pups and the date of birth was considered as day 0. Litters were culled to 12 pups and mixed among experimental groups (n = 4–6 per group per experiment). When tissues were required for both immunocytochemistry and in situ hybridization, the experiments were repeated. Thus, effects of experimental manipulations were compared among litter-mates. Potential diurnal variability was addressed by initiating all experiments between 8.00 a.m. and 10.00 a.m., and cages were undisturbed for 24 h prior to initiation of experiments. All experimental procedures conformed to NIH guidelines. All efforts were made to minimize the number of animals used and their suffering.
To avoid potential confounders of different durations of separation from the mother on the animals’ stress response, both laboratory controls and experimental groups were subjected to the same duration (2–4 h) of maternal separation, and animals killed at later time-points were returned to home cages. Plasma corticosterone was measured for stress-free and laboratory controls, and at the end of hyperthermia or cold challenge using radioimmunoassay (ICN, Irvine, CA).23,60 Assay sensitivity was 0.1 μg/dl; interassay variability was 10–16%. Tissues were processed by one of two methods. For in situ hybridization (ISH), rapid decapitation preceded brain dissection on to powdered dry ice. Brains were stored at −80°C, and serial coronal sections (20 μm) were cut from the anterior commissure–medial preoptic region through to the level of the habenula (A 4.5 to A 1.2 mm) in the 10-day-old rat. Every 10th section was stained for neuroanatomical localization purposes; sections were stored at −80°C. For immunocytochemistry (ICC), animals were perfused under terminal anesthesia (pentobarbital, 100 mg/kg). Brains were cryoprotected and cut into 20-μm sections using a cryotome.
Experimental design
The overall strategy was to test the differential effects of two physiological stimuli on neuronal activation (measured by immediate-early gene expression, behavior and hippocampal electrophysiology) and on CRH gene expression in major fields of the hippocampal formation, compared with expression of these genes in the PVN, the key neuroendocrine stress-control region. Cold exposure was selected as a well-characterized physiological stimulus that activates the neuroendocrine hypothalamic–pituitary–adrenal axis in the immature rat.23,60 Cold stress has been shown to lead to release of CRH from PVN neurons60 and to up-regulation of CRH mRNA expression in the PVN.60 Hyperthermia was chosen as a stimulus known to induce hippocampal neuronal stimulation in the immature rodent,5,18,56 shown in previous behavioral and electrophysiological studies.5,14,18 CRH mRNA levels were determined using ISH to generate a systematic spatio-temporal analysis of CRH gene activation, whereas induction of the immediate-early gene c-fos was measured using both ISH and ICC.
The cold challenge paradigm
This followed previously established protocols.3,23,60 In brief, immature rats (n = 6 for ISH analysis, n = 4 for ICC) were separated from the mother and subjected to maximally tolerated cold exposure, defined by development of rigor and diminished response to tactile stimulus. Maximum exposure required 40–50 min at 4°C and resulted in an average rectal temperature of 11°C.23,60 Following cold exposure, pups were placed on a euthermic pad and regained normal core temperature (33–34°C) in 10–15 min.
Hyperthermia induction
This has been described in detail elsewhere.5,14,18,56 In brief, the core temperature of immature rats (n = 42) was raised using a regulated stream of moderately heated air, aiming for, ~41°C (as during high fever). Rectal temperatures were measured at baseline and at 2-min intervals. Hyperthermia stimulates hippocampal neurons, leading to electrophysiological and behavioral seizures.5,14,18,56 Behavioral seizures were monitored in all animals and, for correlating behavioral and electrophysiological activation, electrodes were implanted on the day prior to the experiment in a separate group of rats, as described below. Following the hyperthermia period, animals were placed on a cool surface for 30 min, hydrated orally and returned to home cages. Those who were sedated due to pentobarbital pretreatment were hydrated orally and returned to their cages when their behavior normalized (typically <1 h). Hyperthermia (>39.5°C) was maintained for 27–28 min (Table 1).
Table 1.
Quantitative parameters of hyperthermia-induced behavioral activation (seizures)
| Group/parameter | Hyperthermia duration (min) | Seizure duration (min) |
|---|---|---|
| Hyperthermic seizures | 27.16 ± 0.56 (n = 31) | 25.00 ± 0.61 |
| Hyperthermic controls | 28.00 ± 0.76 (n = 8) | — |
| Normothermic controls | — | — |
Hyperthermia was induced using warm air, as described in the Experimental Procedures. Maximal core temperature of the experimental group (42.1 ± 0.12°C) did not differ significantly from that of hyperthermic controls (41.9 ± 0.18°C). Neuronal activation was prevented in hyperthermic controls via pre-administration of a short-acting barbiturate (see Fig. 4).
Controls
Two groups of controls were used. “Stress-free” animals (n = 6, used for ISH) were killed within 45 s of disturbance;23,59 “lab controls” (n = 5 for ISH, n = 4 for ICC) were kept in the laboratory, separated from their mothers for the same duration as the experimental groups, under normothermic conditions (core temperature 33–34°C). The groups differed in plasma corticosterone levels (Table 3), but not in CRH mRNA levels or their (negligible) c-fos expression.
Table 3.
Stimulus-specific increases of plasma corticosterone levels
| Treatment paradigm |
||||
|---|---|---|---|---|
| Lab controls | Hyperthermia | Cold challenge | Stress-free | |
| Plasma corticosterone | 2.17 ± 0.56 | 4.42 ± 1.13 | 6.58* ± 0.72 | 0.70 ± 0.08 |
Plasma corticosterone levels (expressed in μg/dl), a measure of the neuroendocrine stress response, were influenced by the cold challenge but not by hyperthermia. Stress-free controls were killed within 45 s of entry into the animal facility; laboratory controls were also separated from their mothers for the duration of the experimental manipulations. They were killed with the other groups, that were subjected to 30 min of hyperthermia or to cold. ANOVA showed a robust effect of treatment: P < 0.0001, sum of squares 118 (residual 35).
Significantly different from laboratory controls (P < 0.01, Bonferroni’s post hoc test).
Elimination of hyperthermia-induced neuronal stimulation
To determine whether CRH gene expression required hippocampal neuronal activation, an experimental group was subjected to hyperthermia in the presence of the short-acting barbiturate, pentobarbital, that disrupts polysynaptic neurotransmission via interaction with the GABAA receptor. Behavioral measures of hippocampal activation (seizures) were abolished by pentobarbital in all animals. To ascertain elimination of electrophysiological parameters of hyperthermia-induced neuronal stimulation, hippocampal electroencephalograms (EEGs) were obtained in both pentobarbital-pretreated and naive rats. In brief, one day prior to experiments, immature rats (n = 6) were implanted with bipolar electrodes directed at the dorsal hippocampus, using methods and coordinates described in detail previously.4,7,9,18 After a 24-h recuperation period, hippocampal EEGs were obtained in the presence or absence of pentobarbital (30 mg/kg, i.p.), both before and subsequent to induction of hyperthermia. Recordings were obtained from freely moving animals, using long flexible wires, as described elsewhere.7,9 Electrode placement was verified in all animals.
In situ hybridization histochemistry for c-fos and corticotropin-releasing hormone messenger RNAs
ISH was performed as described previously for oligonucleotide probes8,23,60 or cRNA probes.19,20 In brief, 20-μm coronal sections were collected on gelatin-coated slides and stored at −80°C. Sections were thawed, air-dried, fixed in paraformaldehyde, dehydrated and rehydrated through graded ethanols, exposed to 0.25% acetic anhydride in 0.1 M triethanolamine (pH 8) and dehydrated. Pre-hybridization (1 h) and hybridization steps were performed in a humidified chamber in a solution of 50% formamide, 5 × SET, 0.2% sodium dodecyl sulfate, 5 × Denhardt’s, 0.5 mg/ml salmon sperm DNA, 0.25 mg/ml yeast tRNA, 100 mM dithiothreitol and 10% dextran sulfate. Following pre-hybridization, sections were hybridized overnight. For detection of CRH mRNA, sections were incubated at 40°C and the hybridization buffer included a deoxy-oligonucleotide probe complementary to the coding region of CRH mRNA and 3′ end-labeled with [35S]dATP as described previously.8,60 Post-hybridization, sections were washed, most stringently at 0.03 × standard saline citrate). For detection of c-fos mRNA, sections were hybridized at 55°C with 1 × 106 c.p.m. of 35S-labeled ribonucleotide probe complementary to c-fos mRNA.16 After hybridization, sections were digested with 200 μg/ml RNase A (Calbiochem, La Jolla, CA) for 30 min at 37°C. Sections underwent serial washes of increasing stringency at 55°C, the most stringent at 0.03 × standard saline citrate for 1 h.19,20 Hybridized sections were apposed to film (Hyperfilm βMax, Amersham, IL) for seven to 14 days. Selected sections were also dipped in emulsion (NTB-2, Eastman Kodak, Rochester, NY) and exposed for four to six weeks.
Acquisition and quantitative analysis of in situ hybridization signal
Semi-quantitative analysis of CRH mRNA was performed following ISH, as described previously.19,23 Digitized images of each brain section were acquired using a StudioStar scanner (AGFA, resolution 1200 × 1200 dots per inch) and analysed using the ImageTool software program (University of Texas Health Science Center, San Antonio, TX, version 1.25). Densities were calibrated using 14C standards and are expressed in μCi/g after correcting for background by subtracting the density of the hybridization signal over the corpus callosum. All analyses were carried out without knowledge of treatment group. Initially, 20 coronal sections of the anterior (septal) and mid hippocampus, A 2.6 to A 2.0 mm in reference to bregma,50 were analysed. To optimize neuroanatomical matching and to minimize inter-ISH and inter-animal variability of background signal, the 10 best sections for each animal, based on background and anatomical match, were chosen by a “blinded” investigator, and these 10 sections were used in the analysis. Occasionally, less than 10 sections were available per animal, and this is indicated in the figure legends. Similarly, matched coronal sections encompassing the dorsomedial parvocellular PVN were also analysed.
Immunocytochemistry for Fos protein
ICC using an affinity purified polyclonal antiserum raised in rabbit directed against the Fos protein (Santa Cruz Biotech, Santa Cruz, CA) was performed on free-floating sections using routine avidin–biotin methodology.15,59 Localization of antibody binding was visualized using diaminobenzidine, as directed in the Vectastain Elite system (Vector, Burlingame, CA).
Analysis and statistical considerations
Analysis of ISH results is described above. For ICC, quantitative analysis was restricted to estimation of the number of cells expressing Fos protein in the hippocampal formation. While the significant limitations of quantitative analysis using ICC are recognized, the virtual all-or-none response of hippocampal c-fos signal in this study permitted evaluation of the presence or absence of c-fos expression in the tissue. The significance of observed quantitative differences among experimental groups was evaluated using one-way ANOVA (PRISM software program, GraphPad), looking at the effects of treatment. Different brain regions from each animal were analysed independently. Bonferroni’s post hoc test was used if required. When two groups were compared, such as laboratory controls and stress-free controls or laboratory controls and experimentals for a given region at a single time-point, the unpaired Student t-test was used. P < 0.05 was considered significant and values are presented as means with standard errors (S.E.M.).
RESULTS
C-fos expression, a marker of neuronal activation, is induced by hyperthermia, but not by cold exposure, in the hippocampal formation
Neuronal activation was determined using molecular measures, i.e. induction of the immediate-early gene c-fos, generally considered an adequate marker of cellular stimulation in the CNS.31,42 This was correlated with behavioral and electrophysiological measures as described below. Following hyperthermia, rapid expression of c-fos mRNA and of Fos protein in hippocampal CA1 and CA3 was evident, using ISH and ICC, respectively. The left panels of Fig. 1 demonstrate minimal c-fos mRNA signal in the hippocampus of a laboratory control animal (Fig. 1A). Hyperthermia resulted in robust c-fos mRNA signal over the hippocampus, but not over the PVN (Fig. 1C). The converse—expression of c-fos in the PVN but not the hippocampus—was observed following cold exposure (Fig. 1E). Immunocytochemical analysis of Fos protein expression revealed individual Fos-immunoreactive hippocampal neurons: Numerous hippocampal CA3 neurons expressed Fos protein in response to hyperthermia (Fig. 1D), but not following cold challenge (Fig. 1F). Little immediate-early gene expression, using either ISH or ICC, was observed in the hippocampi of laboratory control animals (Fig. 1A, B). The 1-h time-point was found in preliminary studies to coincide with peak hippocampal Fos expression in the immature rat.11 Further evidence for hyperthermia-related hippocampal neuronal stimulation was obtained via behavioral analyses and depth bipolar electrode recording (Table 1, Fig. 4A, top two tracings). Table 1 shows the duration and extent of hyperthermia in experimental and hyperthermic control groups. After a 2- to 4-min latency, hyperthermia induced behavioral manifestations consistent with limbic seizures in virtually all animals. These seizures consisted of sudden arrest of all movement and a strong flexion spasm, usually accompanied by oral automatisms, as described in detail previously and shown to correlate with seizure discharges in hippocampal EEGs.5,18 Electrophysiological recordings were obtained from a separate group of rats, as they involved electrode implantation and thus tissue invasion and irritation. As shown in Fig. 4A, hyperthermia led to dramatic alteration of the low-amplitude, non-rhythmic hippocampal activity (baseline, top left tracing). Repetitive semi-rhythmic, high-voltage discharges (arrowheads, top right tracing) coincided with the behavioral measures described above.
Fig. 1.
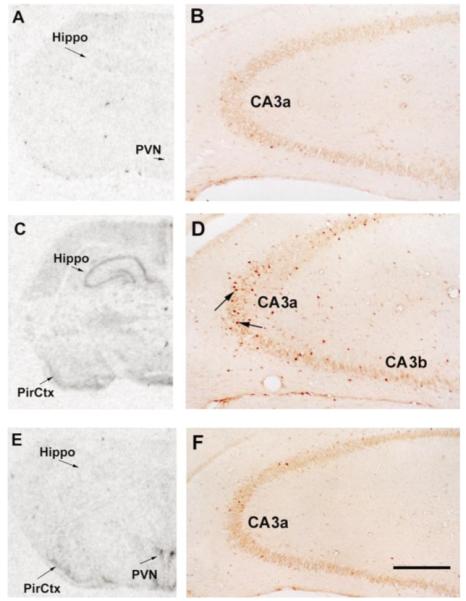
Differential induction of hippocampal c-fos mRNA and Fos protein expression by two distinct physiological stimuli. Nine- or ten-day-old rats were killed 1 h following hyperthermia (C, D) or cold stress (E, F) and compared with laboratory controls (A, B). Film autoradiograms following ISH for detection of c-fos mRNA are shown in A, C and E. Little c-fos mRNA signal was observed in animals maintained in the laboratory for 2–4 h (A). Hyperthermia induced c-fos mRNA expression in the hippocampus (Hippo) and piriform cortex (PirCtx; C). Following cold exposure, c-fos mRNA was expressed in the PVN and piriform cortex, but not in the hippocampus (E). Fos protein immunoreactivity was evident in individual hippocampal neurons following hyperthermia (D), but not after cold (F) or in laboratory controls (B). Scale bar in F = 2 mm (A, C, E), 0.4 mm (B, D, F).
Fig. 4.

Hippocampal electrophysiological activation and up-regulation of CRH mRNA expression are abolished by barbiturate pre-administration. (A) Representative EEG recordings obtained via bipolar depth electrodes from the hippocampi of immature rats. Top left: the baseline theta-frequency, low-amplitude hippocampal EEG activity. The arrow shows a movement artifact. Top right: hyperthermia induced repetitive high-amplitude semi-rhythmic discharges (arrowheads), typical of electrographic seizures involving the hippocampus. The animal was motionless during these events. Pentobarbital (Pentobarb; 30 mg/kg, i.p.), given immediately prior to the onset of hyperthermia, altered the baseline activity by eliminating the theta rhythm and decreasing the EEG amplitude (left, bottom tracing). Importantly, when pentobarbital-treated animals were subjected to hyperthermia, electrographic seizures were not observed on the hippocampal EEG (right, bottom tracing), and no behavioral seizures occurred. Vertical scale: 50 μV; horizontal: 1 s. (B) Semi-quantitative analysis showing the effects of hyperthermia and barbiturates (pentobarbital, Pb) on CRH mRNA levels in the hippocampus. Bars represent CRH mRNA hybridization signal over the hippocampal formation (combined CA1, CA3 and DG) 8.5 h after hyperthermia alone or with pentobarbital pretreatment. Hyperthermia resulted in a significant increase relative to both the control and the pentobarbital-treated rats. *P < 0.05 (Student’s t-test), comparing hyperthermics to the laboratory controls and the hyperthermic (pentobarbital-treated) controls. Hippocampal CRH mRNA levels of animals injected with pentobarbital alone did not differ significantly from those who received an i.p. injection of vehicle (88 ± 5.5%; P = 0.52, Student’s t-test). Samples are the mean ± S.E.M. of a total of 30 matched sections from at least three animals per group.
Corticotropin-releasing hormone messenger RNA in the hippocampal formation of the immature rat is up-regulated by hyperthermia, but not by cold challenge
Expression of CRH mRNA was evident over the hippocampal formation of 10-day-old rats (Fig. 2, top). The low-magnification autoradiograms show hybridization signal over the pyramidal cell layer of CA1 and particularly of CA3a fields of Ammon’s horn, and over the granule cell layer of the dentate gyrus (DG). Dark-field, higher magnification photomicrographs of emulsion-dipped hippocampal sections reveal silver grains over individual neurons (Fig. 2A). The location and morphology of these CRH-expressing neurons are consistent with their identification as interneurons.59 After hyperthermia (Fig. 2C), signal intensity over CRH-expressing neurons was increased, compared with laboratory controls (Fig. 2B). No change in CRH mRNA signal intensity was observed following cold challenge (Fig. 2D).
Fig. 2.
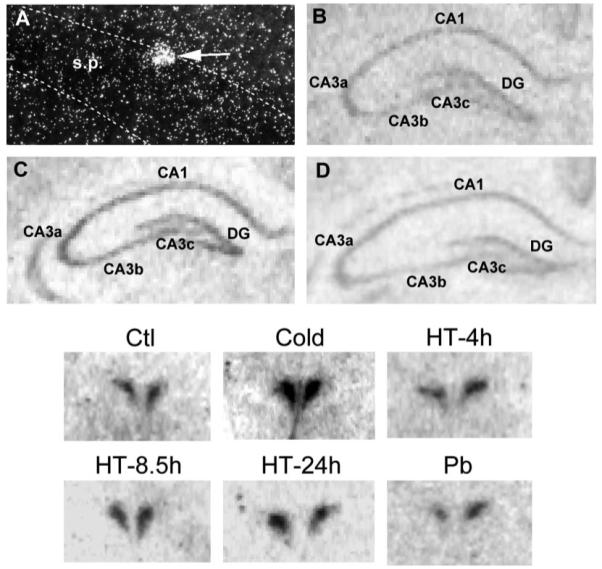
Differential CRH mRNA expression in the hippocampus and PVN of immature rats. Top: CRH mRNA signal was detected using ISH in coronal sections through the hippocampus of 10-day-old rats. (A) Dark-field photomicrograph of an emulsion-dipped section, showing silver grains over a CRH mRNA-expressing neuron (arrow) located at the edge of CA3 stratum pyramidale (s.p.). (B–D) Representative film autoradiograms demonstrating CRH mRNA expression in the hippocampus of laboratory control rats (B), compared with those subjected to hyperthermia (C) or to cold exposure (D). Hyperthermia resulted in significant increases of CRH mRNA hybridization signal over the entire hippocampal formation (CA1 and CA3 subfields, and the DG). Magnifications: × 200 (A), × 15 (B–D). Bottom: representative photomicrographs of CRH mRNA expression in the PVN, as influenced by cold challenge (Cold), hyperthermia (HT) or pentobarbital (Pb) administration. Under both stress-free and laboratory control conditions (see Experimental Procedures), CRH mRNA signal was robust in the parvicellular division of the PVN. These levels were markedly enhanced 4 h after cold. In contrast, CRH mRNA levels were not appreciably altered 4, 8.5 or 24 h after hyperthermia. In addition, this transcript was not influenced by barbiturate administration. Magnification: × 9.
Up-regulation of hippocampal corticotropin-releasing hormone gene expression by hyperthermia and associated neuronal stimulation is robust and sustained
Sequential time-course analysis demonstrated significant enhancement of steady-state CRH mRNA levels in all hippocampal fields investigated, that started within 1 h following hyperthermia (see Fig. 3). Thus, for CA1, control values (0.040 ± 0.004 μCi/g) increased to 0.054 ± 0.003 μCi/g by 1 h, and peaked (0.058 ± 0.003 μCi/g) during the 4- to 8.5-h time-points (P = 0.0004, ANOVA; P < 0.05, P < 0.01 and P < 0.001, laboratory controls vs 1, 4 and 8.5 h, respectively, Bonferroni’s post hoc test). Similar increases in CRH mRNA levels were also found in the DG (P = 0.0001, F = 5.47) and CA3 (P = 0.0001, F = 5.95). The enhancement of CRH gene expression was sustained: for the 8.5-h time-point, for example, CRH mRNA levels were 151%, 154% and 143% of controls for the DG granule cell layer, CA3 and CA1, respectively. The significant increase of CRH mRNA expression persisted in all fields of the hippocampal formation for at least 24 h (Fig. 3). By 48 h (the duration of the experiment), steady-state CRH mRNA levels were still significantly increased in the DG and CA3, but not in CA1. It should be noted that a 50% increase in steady-state CRH mRNA levels is highly significant physiologically.23,28,35 Indeed, adrenalectomy, with total elimination of the negative feedback to CRH expression in the hypothalamus, results in a similar enhancement of CRH mRNA levels (see Ref. 23 for discussion). In contrast to its effects on hippocampal CRH expression, hyperthermia-related neuronal activation did not alter CRH mRNA levels in the PVN (Fig. 3, bottom panel, and see autoradiograms in Fig. 2, bottom panel). ANOVA revealed no significant effect of treatment (P = 0.28, F = 1.33, SStreatment = 0.26, d.f. = 5). Although visual inspection suggested a reduction of CRH mRNA levels at the 1- and 48-h time-points, these trends did not reach statistical significance (Bonferroni’s multiple comparison tests for 1 h: t = 1.631, P > 0.05; control vs 48 h: t = 1.304, P > 0.05).
Fig. 3.
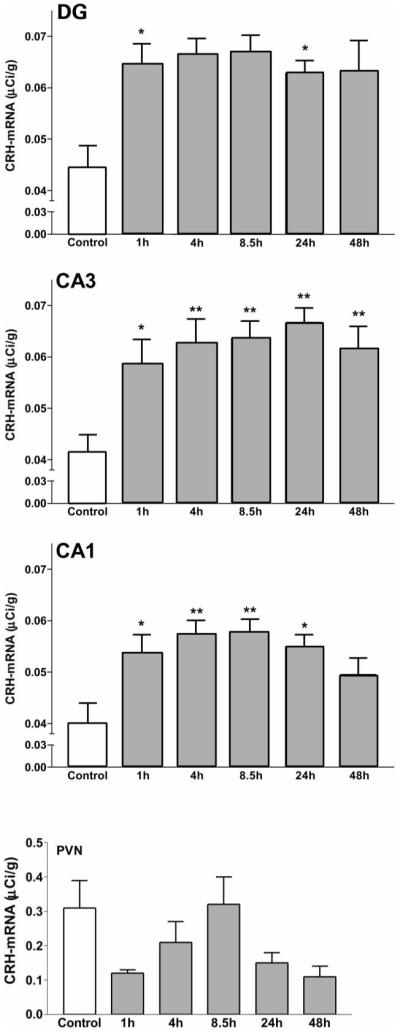
Time-course and regional distribution of the effects of neuronal activation on CRH mRNA expression in the hippocampal formation and PVN. Semi-quantitative analysis of CRH mRNA hybridization signal over the DG and hippocampal CA3 or CA1, as well as the PVN, at specific time-points following hyperthermia. Sections were subjected to ISH and analysis as described in the Experimental Procedures. For hippocampal CRH expression (top three panels), ANOVA revealed significant effects of hyperthermia (CA1: P = 0.0004, F = 4.84, d.f. = 5; CA3: P = 0.0001, F = 5.95; DG: P = 0.0001, F = 5.47). Bonferroni’s post hoc test analysis revealed a significant increase (*P < 0.01; **P < 0.05) of CRH mRNA levels at all time-points (DG, CA3) or for 24 h (CA1) relative to laboratory control levels. Each bar represents the mean ± S.E.M. of six to 10 matched sections from each of at least three rat pups, providing a sample size of 18–30. Bottom panel: in contrast to the hippocampus, hypothalamic CRH mRNA expression was not significantly influenced by hyperthermia (ANOVA for effect of treatment revealed P > 0.05). Also, although a trend for reduced CRH mRNA levels may be apparent for the 1- and 48-h time-points, it did not reach statistical significance (Bonferroni’s multiple comparison tests).
Cold challenge does not influence corticotropin-releasing hormone messenger RNA levels in the hippocampal formation
Based on the findings above and on the established time-course of the effects of cold challenge on CRH mRNA levels, the 4-h time-point was chosen, to coincide with maximal up-regulation of CRH mRNA in the hypothalamus.60 In contrast to the effects of hyperthermia-related neuronal stimulation, cold challenge did not alter steady-state CRH mRNA levels significantly in any hippocampal field. Specifically, CRH mRNA levels in CA1 were 0.040 ± 0.004 μCi/g in laboratory controls and 0.046 ± 0.004 μCi/g in the experimental group (P > 0.05, Student’s t-test). The autoradiograms in Fig. 2 (top) illustrate that CRH mRNA signal over the hippocampus of cold challenged animals was not higher than that over the hippocampus from laboratory controls, and a semi-quantitative analysis of hippocampal CRH mRNA expression is shown in Table 2 (the values for total hippocampus are a mean of values obtained separately for CA1, CA3 and the DG). In contrast, activation of the neuroendocrine stress axis by cold (as also evident from plasma corticosterone levels; Table 3) increased CRH mRNA levels in the PVN (Fig. 2, bottom), as shown before.
Table 2.
Stimulus-specific increases of hippocampal corticotropin-releasing hormone messenger RNA expression
| Treatment paradigm |
|||
|---|---|---|---|
| Control | Hyperthermia | Cold challenge | |
| CRH mRNA levels | 100 ± 4.8 | 148 ± 4.5* | 97 ± 4.6 |
Relative levels of CRH mRNA hybridization signal over the hippocampal formation (combined DG, CA3 and CA1) 4 h following hyperthermia or cold challenge. CRH mRNA expression was selectively up-regulated following hyperthermia (Student’s t-test, comparing hyperthermia to laboratory controls, P < 0.001), but not following cold challenge. Samples are the mean ± S.E.M. of a total of 23–40 matched sections from at least three rats per group, and values are expressed as the percentage of control levels. Note: CRH mRNA levels of stress-free and laboratory controls did not differ, and the groups were combined.
Neuronal stimulation is required for enhancement of corticotropin-releasing hormone gene expression in the hippocampus
The requirement for robust hippocampal neuronal activation by a given stimulus to influence CRH gene expression in this region is further illustrated in Fig. 4. Administration of sodium pentobarbital, a blocker of polysynaptic neurotransmission via interaction with the GABAA receptor, eliminated the behavioral, electrophysiological and molecular (c-fos expression; not shown) parameters of hippocampal neuronal stimulation: It suppressed both behavioral seizures (Table 1) and the epileptic discharges recorded from the hippocampus, as shown in Fig. 4A: the top tracings demonstrate the electrophysiological recordings from depth bipolar electrodes placed in the dorsal hippocampus of immature rats. Hyperthermia induced intense neuronal stimulation, i.e. synchronous rhythmic EEG discharges5,14,18 that were accompanied by behavioral seizures.18,56 The bottom tracings show that administration of the short-acting barbiturate, pentobarbital, prior to induction of hyperthermia eliminated the electrographic evidence of neuronal stimulation. Pentobarbital did not alter the duration or the intensity (maximal temperature) of the hyperthermia per se (Table 1). However, the compound eliminated all three parameters of hippocampal neuronal stimulation, as well as the hyperthermia-induced up-regulation of CRH mRNA levels (Fig. 4B). For example, in CA3, at the time of peak hyperthermia effect on CRH expression (8.5 h), CRH mRNA levels were 0.041 ± 0.004 μCi/g in normothermic controls, 0.064 ± 0.003 μCi/g in the experimental group and 0.041 ± 0.002 μCi/g in pentobarbital-treated hyperthermic controls (P < 0.001 vs the experimental group). The suppression of hyperthermia-induced up-regulation of CRH expression by pentobarbital was similar in all three fields of the hippocampal formation, and these values are combined in Fig. 4B.
DISCUSSION
The major findings of these experiments are: (i) hippocampal CRH mRNA levels were enhanced by an elevation of core temperature that resulted in neuronal stimulation and immediate-early gene expression; (ii) induction of hippocampal CRH gene expression required neuronal stimulation and was abolished by barbiturate administration; (iii) whereas cold, a powerful activator of the neuroendocrine stress response in the immature rat, enhanced CRH mRNA levels in the PVN, hippocampal CRH expression was unaffected. Taken together, these results demonstrate that CRH expression is differentially regulated by physiologically relevant stimuli, that may lead to sustained up-regulation of the peptide’s mRNA levels. Furthermore, regulation of hippocampal CRH expression is independent of that in the hypothalamus and is influenced by hippocampal neuronal stimulation.
CRH is abundantly expressed in CNS regions shown to participate in the neuroendocrine and behavioral responses to stress. For example, the role of CRH, released from terminals of hypothalamic PVN neurons, in mediating the neuroendocrine stress response has been established,47,57,60 and local release of CRH in the ACe, influencing stress-induced behaviors, has been demonstrated.40,54 Consistent with the functional role of hypothalamic and amygdala CRH in modulating stress responses, up-regulation of CRH expression in these two regions by challenges that activate the neuroendocrine stress response has been documented.23,29 Teleologically, this mechanism for regulation of CRH levels should be advantageous, resulting in larger releasable pools of CRH upon subsequent challenges.
In the hippocampus, the presence of CRH-expressing inter-neurons has been documented in both the mature and developing rat,41,48,53,59 but the physiological functions of the peptide and the regulation of CRH levels at hippocampal synapses have not been fully established. Specifically, whether hippocampal CRH participates in the neuroendocrine stress response has been questioned.38 Here, we show that a prototypic physiological challenge that activates the hypothalamic–pituitary–adrenal axis (cold exposure of the immature rat) does not alter CRH expression in the hippocampus. This is in contrast to up-regulation of CRH in the PVN (Fig. 2, bottom panels).
If challenges that activate the hypothalamic–pituitary–adrenal axis do not up-regulate hippocampal CRH expression, what mechanisms control the peptide’s levels in this region? Up-regulation of CRH mRNA in some hippocampal fields of the mature rat has been shown following kindling52 or kainate administration.44 These experimental manipulations share intense excitation of hippocampal neurons and are associated with activation of numerous genes.12,27,46 In the current studies, moderate hyperthermia (within the physiological range seen in febrile immature humans) led to motor stimulation and to electrophysiological activation of immature rat hippocampal neurons.5,14,56 Taken together with the studies mentioned above, this may be interpreted to indicate that intense stimulation of hippocampal neurons (demonstrated in the current study by both c-fos induction and EEG) leads to enhanced CRH expression in this region.
Activity-dependent regulation of CRH expression in the hippocampus is consistent with emerging information regarding the putative physiological functions of this peptide in the hippocampal formation. Application of CRH has been shown to enhance hippocampal pyramidal cell firing1 and synaptic efficacy.32 More recently, in vitro studies have shown that CRH modulates glutamatergic mechanisms to increase excitatory neurotransmission in the hippocampal circuit.6,25 Thus, under physiological circumstances, release of CRH may modulate hippocampal neurotransmission, augmenting glutamate-mediated alterations of hippocampal function. This may influence both normal hippocampal processes such as long-term potentiation and, when excessive, may promote abnormal excitation1,6,25 and excitotoxicity.9,36,37
The neuroanatomical basis for the influence of CRH on hippocampal neurotransmission has been explored. Previous work from the authors’ laboratory has demonstrated a substantial CRH-expressing interneuronal population in the hippocampal formation of the immature rat.59 These cells are located primarily within, and bordering on, the pyramidal cell layer of CA3 and CA1. The majority of these neurons, co-expressing CRH and parvalbumin, were identified as basket and axo-axonic (chandelier) cells, synapsing on somata and axon initial segments of pyramidal cells, respectively.59 They are thus strategically positioned to influence pyramidal cell firing. In addition, CRH receptors, particularly corticotropin-releasing factor-1, shown to mediate the excitation-promoting actions of CRH,4 are abundantly expressed in hippocampal pyramidal cells.2,13,15 These data support the notion that, in the hippocampus, CRH modulates synaptic neurotransmission; in turn, expression of the CRH gene is controlled by hippocampal circuit activation.
The requirement for stimulation of hippocampal neurons4,14,56 for up-regulation of hippocampal CRH expression, suggested in the current study, is further illustrated by the effects of barbiturates: hyperthermia-induced hippocampal CRH mRNA expression was abolished (in CA1) or attenuated (in CA3) by barbiturate pretreatment, providing clues for the mechanisms for CRH mRNA activation in the hippocampus: Pentobarbital interferes with polysynaptic neurotransmission by modulating GABAA receptors. Indeed, it eliminated the hyperthermia-induced intense hippocampal neuronal stimulation, as evident from behavioral and electrophysiological criteria (Fig. 4A, and Refs 4, 14, 18 and 56). Thus, the effect of the barbiturate is consistent with the notion that neuronal stimulation, distinct from any hormonal or autonomic consequences of elevated core temperature, is required for hyperthermia-induced up-regulation of hippocampal CRH mRNA expression.
The current study also highlights the region-specific regulation of CRH expression. Hypothalamic CRH mediates the hormonal stress response, and CRH mRNA levels in the PVN are up-regulated by numerous acute physiological stressors.35,58 Accordingly, cold challenge activated both neuroendocrine responses (such as enhanced plasma corticosterone; Table 3) and CRH expression in the PVN.23,60 However, this physiological, neuroendocrine challenge did not influence CRH mRNA levels in any hippocampal region.
In the immature rat, prolonged separation from the mother can augment neuroendocrine responses3,34,51 and immediate-early gene expression51 to physiological challenges. To control for potential effects of maternal separation on hypothalamic51 and hippocampal CRH gene expression, laboratory controls were employed, that were separated from their mothers for the same duration as were animals subjected to hyperthermia or cold challenge. Therefore, the enhanced CRH mRNA levels in hippocampal neurons stimulated by hyperthermia, compared with CRH mRNA in laboratory controls, cannot be due to effects of maternal separation.
The up-regulation of hippocampal CRH mRNA levels noted in the current study was quite sustained, lasting for 24–48 h. The methods used here cannot distinguish with certainty whether this fact resulted from prolonged effects of the intense, hyperthermia-induced neuronal stimulation, or reflect the relatively long half-life of CRH mRNA. Using specific probes directed against heteronuclear (unedited) CRH RNA, our6 and other17 groups have shown a rapid activation of the CRH gene in the immature rat hypothalamus after appropriate challenges. The half-life of heteronuclear CRH RNA is short, permitting resolution of gene activation and inactivation in the range of minutes.6,17 However, steady-state CRH mRNA levels, with their slow turnover, may not fully reflect the kinetics of CRH gene activation in the immature rat.
It may be interesting to compare the results of the current study to the few reports about the regulation of CRH expression in the hippocampus of the adult rat. Of note, in mature experimental animals, levels of hippocampal CRH expression, unlike those in the hypothalamus,35,58 have not been found to correlate with stress.38 It may be argued that the threshold to induction of hippocampal CRH expression is higher than that required for up-regulation of the CRH gene in the PVN. Indeed, the presence of differential thresholds for the effects of a given stimulus on CRH function has recently been demonstrated.55 However, the cold challenge employed here60 is a powerful activator of the neuroendocrine stress response, resulting in markedly elevated plasma corticosterone3,23,60 (Table 3). Therefore, the absence of an effect of cold challenge on hippocampal CRH expression, combined with the rapid and prolonged up-regulation of CRH mRNA levels in the hippocampus by hyperthermia, indicates that enhanced CRH gene transcription in the hippocampus requires factors other than provocation of physiological, hormonal responses to stress.
Finally, potential interactions between hippocampal and hypothalamic CRH expression should be noted: the hippocampus is recognized to have negative effects on hypothalamic parvocellular neurons expressing CRH.24 Therefore, it could be argued that decreasing hippocampal neuronal activity by pentobarbital may disinhibit CRH expression in the PVN. However, CRH mRNA levels in the PVN were not changed by pentobarbital administration alone (0.56 ± 0.19 vs 0.42 ± 0.05 μCi/g in vehicle-injected controls; P = 0.37).
CONCLUSION
In summary, this study demonstrates a robust and sustained stimulus-specific up-regulation of CRH gene expression in the hippocampus of the immature rat. Given the established and emerging functions of hippocampal CRH, these findings suggest that modulation of hippocampal CRH levels may be a physiological mechanism for influencing hippocampal neurotransmission.
Acknowledgements
This research was supported by NS28912, NS35439 and HD28413. We thank Dr R. Bender for critical review of the manuscript, M. Eghbal-Ahmadi for technical assistance and M. Hinojosa for expert editorial assistance.
Abbreviations
- ACe
amygdaloid central nucleus
- ANOVA
analysis of variance
- CA
Ammon’s horn of the hippocampus
- CRH
corticotropin-releasing hormone
- DG
dentate gyrus
- EEG
electroencephalogram
- ICC
immunocytochemistry
- ISH
in situ hybridization
- PVN
paraventricular nucleus of the hypothalamus
REFERENCES
- 1.Aldenhoff JB, Gruol DL, Rivier J, Vale W, Siggins GR. Corticotropin releasing factor decreases postburst hyperpolarizations and excites hippocampal neurons. Science. 1983;221:875–877. doi: 10.1126/science.6603658. [DOI] [PubMed] [Google Scholar]
- 2.Avishai-Eliner S, Yi SJ, Baram TZ. Developmental profile of messenger RNA for the corticotropin-releasing hormone receptor in the rat limbic system. Dev. Brain Res. 1996;91:159–163. doi: 10.1016/0165-3806(95)00158-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Avishai-Eliner S, Yi SJ, Newth CJ, Baram TZ. Effects of maternal and sibling deprivation on basal and stress induced hypothalamic–pituitary–adrenal components in the infant rat. Neurosci. Lett. 1995;192:49–52. doi: 10.1016/0304-3940(95)11606-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baram TZ, Chalmers DT, Chen C, Koutsoukos Y, DeSouza EB. The CRF1 receptor mediates the excitatory actions of corticotropin releasing factor (CRF) in the developing rat brain: in vivo evidence using novel, selective, non-peptide CRF receptor antagonist. Brain Res. 1997;770:89–95. doi: 10.1016/s0006-8993(97)00759-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baram TZ, Gerth A, Schultz L. Febrile seizures: an appropriate-aged model suitable for long-term studies. Brain Res. dev. Brain Res. 1997;98:265–270. doi: 10.1016/s0165-3806(96)00190-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baram TZ, Hatalski CG. Neuropeptide-mediated excitability: a key triggering mechanism for seizure generation in the developing brain. Trends Neurosci. 1998;21:471–476. doi: 10.1016/s0166-2236(98)01275-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baram TZ, Hirsch E, Snead OC, Schultz L. Corticotropin-releasing hormone-induced seizures in infant rats originate in the amygdala. Ann. Neurol. 1992;31:488–494. doi: 10.1002/ana.410310505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baram TZ, Lerner SP. Ontogeny of corticotropin releasing hormone gene expression in rat hypothalamus—comparison with somatostatin. Int. J. devl Neurosci. 1991;9:473–478. doi: 10.1016/0736-5748(91)90033-i. [DOI] [PubMed] [Google Scholar]
- 9.Baram TZ, Ribak CE. Peptide-induced infant status epilepticus causes neuronal death and synaptic reorganization. NeuroReport. 1995;6:277–280. doi: 10.1097/00001756-199501000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Behan DP, Heinrichs SC, Troncoso JC, Liu XJ, Kawas CH, Ling N, DeSouza EB. Displacement of corticotropin releasing factor from its binding protein as a possible treatment for Alzheimer’s disease. Nature. 1995;378:284–287. doi: 10.1038/378284a0. [DOI] [PubMed] [Google Scholar]
- 11.Brunson KL, Schultz L, Baram TZ. Induction of c-fos expression in specific populations of limbic neurons in the immature rat after CRH administration. Soc. Neurosci. Abstr. 1998;28:733.14. [Google Scholar]
- 12.Burazin TC, Gundlach AL. Rapid and transient increases in cellular immediate early gene and neuropeptide mRNAs in cortical and limbic areas after amygdaloid kindling seizures in the rat. Epilepsy Res. 1996;26:281–293. doi: 10.1016/s0920-1211(96)00060-5. [DOI] [PubMed] [Google Scholar]
- 13.Chalmers DT, Lovenberg TW, DeSouza EB. Localization of novel corticotropin-releasing factor receptor (CRF2) mRNA expression to specific subcortical nuclei in rat brain: comparison with CRF1 receptor mRNA expression. J. Neurosci. 1995;15:6340–6350. doi: 10.1523/JNEUROSCI.15-10-06340.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen K, Baram TZ, Soltesz I. Febrile seizures in the developing brain result in persistent modification of neuronal excitability in limbic circuits. Nat. Med. 1999;5:888–894. doi: 10.1038/11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y, Brunson K, Mueller MG, Cariaga W, Baram TZ. Immunocytochemical distribution of corticotropin-releasing hormone receptor type-1 (CRF1)-like immunoreactivity in the mouse brain: light microscopy analysis using an antibody directed against the C-terminus. J. comp. Neurol. 2000;420:305–323. doi: 10.1002/(sici)1096-9861(20000508)420:3<305::aid-cne3>3.0.co;2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Curran T, Gordon MB, Rubino KL, Sambucetti LC. Isolation and characterization of the c-fos (rat) cDNA and analysis of post-translational modification in vitro. Oncogene. 1987;2:79–84. [PubMed] [Google Scholar]
- 17.Dent GW, Smith MA, Levine S. Rapid induction of corticotropin releasing hormone gene transcription in the paraventricular nucleus of the developing rat. Endocrinology. 2000;141:1593–1598. doi: 10.1210/endo.141.5.7455. [DOI] [PubMed] [Google Scholar]
- 18.Dube C, Chen K, Eghbal-Ahmadi M, Brunson K, Soltesz I, Baram TZ. Prolonged febrile seizures in immature rat model enhance hippocampal excitability long term. Ann. Neurol. 2000;47:336–344. [PMC free article] [PubMed] [Google Scholar]
- 19.Eghbal-Ahmadi M, Avishai-Eliner S, Hatalski CG, Baram TZ. Differential regulation of the expression of corticotropin-releasing factor receptor type 2 (CRF2) in hypothalamus and amygdala of the immature rat by sensory input and food intake. J. Neurosci. 1999;19:3982–3991. doi: 10.1523/JNEUROSCI.19-10-03982.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eghbal-Ahmadi M, Hatalski CG, Avishai-Eliner S, Baram TZ. Corticotropin releasing factor receptor type II (CRF2) messenger ribonucleic acid levels in the hypothalamic ventromedial nucleus of the infant rat are reduced by maternal deprivation. Endocrinology. 1997;138:5048–5051. doi: 10.1210/endo.138.11.5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fox EA, Gruol DL. Corticotropin-releasing factor suppresses the after-hyperpolarization in cerebellar Purkinje neurons. Neurosci. Lett. 1993;149:103–107. doi: 10.1016/0304-3940(93)90358-r. [DOI] [PubMed] [Google Scholar]
- 22.Gray TS, Bingaman EW. The amygdala: corticotropin-releasing factor, steroids, and stress. Crit. Rev. Neurobiol. 1996;10:155–168. doi: 10.1615/critrevneurobiol.v10.i2.10. [DOI] [PubMed] [Google Scholar]
- 23.Hatalski CG, Guirguis C, Baram TZ. Corticotropin releasing factor mRNA expression in the hypothalamic paraventricular nucleus and the central nucleus of the amygdala is modulated by repeated acute stress in the immature rat. J. Neuroendocr. 1998;10:663–669. doi: 10.1046/j.1365-2826.1998.00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herman JP, Cullinan WE. Neurocircuitry of stress: central control of the hypothalamo-pituitary–adrenocortical axis. Trends Neurosci. 1997;20:78–84. doi: 10.1016/s0166-2236(96)10069-2. [DOI] [PubMed] [Google Scholar]
- 25.Hollrigel GS, Chen K, Baram TZ, Soltesz I. The pro-convulsant actions of corticotropin-releasing hormone in the hippocampus of infant rats. Neuroscience. 1998;84:71–79. doi: 10.1016/s0306-4522(97)00499-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Honkaniemi J, Fuxe K, Rechardt L, Koistinaho J, Isola J, Gustafsson JA, Okret S, Pelto-Huikko M. Colocalization of fos- and glucocorticoid receptor-like immunoreactivities in the rat amygdaloid complex after immobilization stress. J. Neuroendocr. 1992;4:547–555. doi: 10.1111/j.1365-2826.1992.tb00203.x. [DOI] [PubMed] [Google Scholar]
- 27.Honkaniemi J, Sharp FR. Prolonged expression of zinc finger immediate-early gene mRNAs and decreased protein synthesis following kainic acid induced seizures. Eur. J. Neurosci. 1999;11:10–17. doi: 10.1046/j.1460-9568.1999.00401.x. [DOI] [PubMed] [Google Scholar]
- 28.Imaki T, Nahan JL, Rivier C, Sawchenko PE, Vale W. Differential regulation of corticotropin-releasing factor mRNA in rat brain regions by glucocorticoids and stress. J. Neurosci. 1991;11:585–599. doi: 10.1523/JNEUROSCI.11-03-00585.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalin NH, Takahashi LK, Chen FL. Restraint stress increases corticotropin-releasing hormone mRNA content in the amygdala and paraventricular nucleus. Brain Res. 1994;656:182–186. doi: 10.1016/0006-8993(94)91382-x. [DOI] [PubMed] [Google Scholar]
- 30.Kovacs KJ, Sawchenko PE. Sequence of stress-induced alterations in indices of synaptic and transcriptional activation in parvocellular neurosecretory neurons. J. Neurosci. 1996;16:262–273. doi: 10.1523/JNEUROSCI.16-01-00262.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Labiner DM, Butler LS, Cao Z, Hosford DA, Shin C, McNamara JO. Induction of c-fos mRNA by kindled seizures: complex relationship with neuronal burst firing. J. Neurosci. 1993;13:744–751. doi: 10.1523/JNEUROSCI.13-02-00744.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee EH, Huang AM, Tsuei KS, Lee WY. Enhanced hippocampal corticotropin-releasing factor gene expression associated with memory consolidation and memory storage in rats. Chin. J. Physiol. 1996;39:197–203. [PubMed] [Google Scholar]
- 33.Lee EH, Hung HC, Lu KT, Chen WH, Chen HY. Protein synthesis in the hippocampus associated with memory facilitation by corticotropin-releasing factor in rats. Peptides. 1992;13:927–937. doi: 10.1016/0196-9781(92)90051-4. [DOI] [PubMed] [Google Scholar]
- 34.Levine S, Huchton DM, Wiener SG, Rosenfeld P. Time course of the effect of maternal deprivation on the hypothalamic–pituitary–adrenal axis in the infant rat. Devl Psychobiol. 1992;24:547–558. doi: 10.1002/dev.420240803. [DOI] [PubMed] [Google Scholar]
- 35.Lightman SL, Harbuz MS. Expression of corticotropin-releasing factor mRNA in response to stress. Ciba Foundation Symp. 1993;172:173–187. doi: 10.1002/9780470514368.ch9. [DOI] [PubMed] [Google Scholar]
- 36.Lyons MK, Anderson RE, Meyer FB. Corticotropin releasing factor antagonist reduces ischemic hippocampal neuronal injury. Brain Res. 1991;545:339–342. doi: 10.1016/0006-8993(91)91310-w. [DOI] [PubMed] [Google Scholar]
- 37.Maecker H, Desai A, Dash R, Rivier J, Vale W, Sapolsky R. Astressin, a novel and potent CRF antagonist, is neuroprotective in the hippocampus when administered after a seizure. Brain Res. 1997;744:166–170. doi: 10.1016/s0006-8993(96)01207-3. [DOI] [PubMed] [Google Scholar]
- 38.Makino S, Schulkin J, Smith MA, Pacak K, Palkovits M, Gold PW. Regulation of corticotropin-releasing hormone receptor messenger ribonucleic acid in the rat brain and pituitary by glucocorticoids and stress. Endocrinology. 1995;136:4517–4525. doi: 10.1210/endo.136.10.7664672. [DOI] [PubMed] [Google Scholar]
- 39.Marrosu F, Fratta W, Carcangiu P, Giagheddu M, Gessa GL. Localized epileptiform activity induced by murine CRF in rats. Epilepsia. 1988;29:369–373. doi: 10.1111/j.1528-1157.1988.tb03733.x. [DOI] [PubMed] [Google Scholar]
- 40.Merali Z, McIntosh J, Kent P, Michaud D, Anisman H. Aversive and appetitive events evoke the release of corticotropin-releasing hormone and bombesin-like peptides at the central nucleus of the amygdala. J. Neurosci. 1998;18:4758–4766. doi: 10.1523/JNEUROSCI.18-12-04758.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Merchenthaler I. Corticotropin releasing factor-like immunoreactivity in the rat central nervous system. Extrahypothalamic distribution. Peptides. 1984;5:53–69. doi: 10.1016/0196-9781(84)90265-1. [DOI] [PubMed] [Google Scholar]
- 42.Morgan JI, Curran T. Proto-oncogene transcription factors and epilepsy. Trends pharmac. Sci. 1991;12:343–349. doi: 10.1016/0165-6147(91)90594-i. [DOI] [PubMed] [Google Scholar]
- 43.Nemeroff CB. New vistas in neuropeptide research in neuropsychiatry: focus on corticotropin-releasing factor. Neuropsychopharmacology. 1992;6:69–75. [PubMed] [Google Scholar]
- 44.Piekut DT, Phipps B. Corticotropin-releasing factor induction in rat piriform cortex following kainate-elicited seizures. Neurosci. Lett. 1996;209:45–48. doi: 10.1016/0304-3940(96)12608-2. [DOI] [PubMed] [Google Scholar]
- 45.Rassnick S, Hoffman GE, Rabin BS, Sved AF. Injection of corticotropin-releasing hormone into the locus coeruleus or foot shock increases neuronal Fos expression. Neuroscience. 1998;85:259–268. doi: 10.1016/s0306-4522(97)00574-5. [DOI] [PubMed] [Google Scholar]
- 46.Rice AC, DeLorenzo RJ. Kindling induces long-term changes in gene expression. In: Corcoran M, Moshe SL, editors. Kindling 5. Plenum; New York: 1998. [Google Scholar]
- 47.Rivier J, Spiess J, Vale W. Characterization of rat hypothalamic corticotropin-releasing factor. Proc. natn. Acad. Sci. USA. 1983;80:4851–4855. doi: 10.1073/pnas.80.15.4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sakanaka M, Shibasaki T, Lederis K. Corticotropin releasing factor-like immunoreactivity in the rat brain as revealed by a modified cobalt–glucose oxidase–diaminobenzidine method. J. comp. Neurol. 1987;260:256–298. doi: 10.1002/cne.902600209. [DOI] [PubMed] [Google Scholar]
- 49.Sawchenko PE, Imaki T, Potter E, Kovacs K, Imaki J, Vale W. The functional neuroanatomy of corticotropin-releasing factor. Ciba Foundation Symp. 1993;172:5–21. doi: 10.1002/9780470514368.ch2. [DOI] [PubMed] [Google Scholar]
- 50.Smith BN, Dudek FE. Age-related epileptogenic effects of corticotropin-releasing hormone in the isolated CA1 region of rat hippocampal slices. J. Neurophysiol. 1994;72:2328–2333. doi: 10.1152/jn.1994.72.5.2328. [DOI] [PubMed] [Google Scholar]
- 51.Smith MA, Kim S-Y, Van Oers H, Levine S. Maternal deprivation and stress induce immediate early genes in the infant rat brain. Endocrinology. 1997;138:4622–4628. doi: 10.1210/endo.138.11.5529. [DOI] [PubMed] [Google Scholar]
- 52.Smith MA, Weiss SR, Berry RL, Zhang LX, Clark M, Massenburg G, Post RM. Amygdala-kindled seizures increase the expression of corticotropin-releasing factor (CRF) and CRF-binding protein in GABAergic interneurons of the dentate hilus. Brain Res. 1997;745:248–256. doi: 10.1016/s0006-8993(96)01157-2. [DOI] [PubMed] [Google Scholar]
- 53.Swanson LW, Sawchenko PE, Rivier J, Vale WW. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology. 1983;36:165–186. doi: 10.1159/000123454. [DOI] [PubMed] [Google Scholar]
- 54.Swiergiel AH, Takahashi LK, Kalin NH. Attenuation of stress-induced behavior by antagonism of corticotropin-releasing factor receptors in the central amygdala in the rat. Brain Res. 1993;623:229–234. doi: 10.1016/0006-8993(93)91432-r. [DOI] [PubMed] [Google Scholar]
- 55.Tanimura SM, Watts AG. Corticosterone can facilitate as well as inhibit corticotropin-releasing hormone gene expression in the rat hypothalamic paraventricular nucleus. Endocrinology. 1998;139:3830–3836. doi: 10.1210/endo.139.9.6192. [DOI] [PubMed] [Google Scholar]
- 56.Toth Z, Yan XX, Haftoglou S, Ribak CE, Baram TZ. Seizure-induced neuronal injury: vulnerability to febrile seizures in an immature rat model. J. Neurosci. 1998;18:4285–4294. doi: 10.1523/JNEUROSCI.18-11-04285.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213:1394–1397. doi: 10.1126/science.6267699. [DOI] [PubMed] [Google Scholar]
- 58.Watts AG. The impact of physiological stimuli on the expression of corticotropin-releasing hormone (CRH) and other neuropeptide genes. Front. Neuroendocr. 1996;17:281–326. doi: 10.1006/frne.1996.0008. [DOI] [PubMed] [Google Scholar]
- 59.Yan XX, Toth Z, Schultz L, Ribak CE, Baram TZ. Corticotropin-releasing hormone (CRH) containing neurons in the immature rat hippocampal formation: light and electron microscopic features and colocalization with glutamate decarboxylase and parvalbumin. Hippocampus. 1998;8:231–243. doi: 10.1002/(SICI)1098-1063(1998)8:3<231::AID-HIPO6>3.0.CO;2-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yi SJ, Baram TZ. Corticotropin-releasing hormone mediates the response to cold stress in the neonatal rat without compensatory enhancement of the peptide’s gene expression. Endocrinology. 1994;135:2364–2368. doi: 10.1210/endo.135.6.7988418. [DOI] [PMC free article] [PubMed] [Google Scholar]