

Abstract
In this review we discuss specific examples of regulation of cytokine genes and focus on a new mechanism involving post-transcriptional regulation via miRNAs. The post-transcriptional regulation of cytokine genes via the destabilizing activity of AU-rich elements [AREs] and miRNAs is a pre-requisite for regulating the half-life of many cytokines and achieving the temporal and spatial distributions required for regulation of these genes.
Keywords: miRNA, ARE, Cytokine, Chemokine, Evolution
1. Introduction
MicroRNAs (miRNAs) are endogenous, non-coding RNAs that are emerging as key players in regulating gene expression in eukaryotes. Currently ~695 human miRNAs are listed in the registry (miRBase Release 12.0 http://microrna.sanger.ac.uk/) [1,2] and they are believed to affect a large number of biological processes including cell differentiation and development. It is estimated that there are possibly more than 1000 human miRNAs. It has been proposed that miRNAs act as rheostats, modulating the expression of target genes to an optimal level rather than an on–off switch [3]. Recent studies suggest that miRNAs in addition to their more recognized role in development also play an important role in a variety of stress responses by modulating the translation and/or stability of multiple targeted transcripts [4].
Emerging studies suggest that miRNAs provide an added layer in orchestrating immune responses [5–7]. miRNAs function in shaping immunity by regulating the repertoire of genes expressed in immune cells and the magnitude and duration of responses to particular pathogens [8,9]. Expression profiling has shown distinct patterns of miRNA expression in different hematopoietic cell lineages [10] and single miRNAs can have substantial effects in regulating immune responses. For example, miR-155 knockdown, in mice, results in multiple defects in adaptive immunity and enhances IL-4 production [11–13]. The endonuclease enzyme Dicer that produces miRNAs and functions in RNA interference (RNAi) has been demonstrated to play a key role in the regulation of the immune system. Microarray studies in C. elegans containing a deletion in the Dicer gene showed enrichment for genes involved in innate immunity [6]. Furthermore defects in T lineage cells including incomplete IFN-γ repression, Treg cell development and function have also been demonstrated in mice lacking Dicer [14,15]. A study by Asirvatham et al. of 613 immune genes that utilized a computational approach to identify miRNA targets has identified ~275 newly predicted immune gene-miRNA interactions including transcription factors, chromatin modifiers and genes involved in immune signaling pathways and inflammation including cytokines [5].
In this review we discuss specific examples of regulation of cytokine genes and focus on mechanisms involving post-transcriptional regulation via miRNAs. The post-transcriptional regulation of cytokine genes via the destabilizing activity of AU-rich elements (AREs) and miRNAs is a pre-requisite for regulating the half-life of many cytokines and central to achieving temporal and spatial regulation of these genes [16].
2. Regulation mediated via AU-rich element (ARE) sites
The cis-acting structural RNA motifs of which the major class is the AU-rich elements can regulate the stability of mRNA and affect the translational efficiency in response to extracellular stimulation. Although AU-rich elements are mainly referred to as decay elements [17,18] they also regulate translation and mRNA export [19]. The best characterized AREs are found in cytokine transcripts and other early response genes but they are also found in a large and diverse repertoire of cellular transcripts (identified in ~4000 genes) [19,20]. The ARE typically contains an AUUUA pentamer repeated once or several times within a U-rich region of the 3′UTR [18]. The pentamer repeat and the uridine enrichment sites are two important characteristics of an ARE but are not always sufficient for the destabilizing effect. Zubiaga et al. concluded that the nonamer UUAUUUAUU is the minimal AU-rich motif that effectively destabilizes mRNA [21]. ARE sequences are frequently present in genes that encode proteins that regulate cell growth and responses to external stimuli including microorganisms, inflammation and environmental stimuli. The ARE sequences are conserved throughout evolution and have been shown to play a critical role in the inflammatory response of lower forms including fish [22].
ARE-mediated decay (AMD) is thought to regulate expression of up to 5–8% of all mRNAs [18]. A number of trans-acting factors called the ARE-binding proteins (ARE-BPs) regulate this process and can be functionally classified into two groups; (1) decay promoting/destabilizing ARE-BPs including tristetraprolin (TTP) [23], butyrate response factor 1, AU-rich binding factor 1 (AUF1), KH-type splicing regulatory protein (KHSRP) and (2) stabilizing ARE-BPs including Hu protein R (HUR) [24–27]. HuR and AUF-1 have also been shown to alter the stability of ARE containing mRNA in vivo [28]. HuR, a member of the embryonic lethal abnormal vision (ELAV) family of RNA binding proteins, is ubiquitously expressed in all proliferating cells [29]. The other members of the ELAV family of proteins also have high affinity for AU- or U- rich sequences and also exhibit poly(A)-binding activity in vitro [27]. AUF1 is a member of the hnRNP proteins that functions in vivo as an RNA-destabilizing factor in the ARE-mediated decay pathway [30].
TTP, a zinc-finger-containing inhibitory ARE-binding protein, has been shown to destabilize the expression of class II AREs that are present in many proto-oncogenes, growth factors and cytokines such as TNF-α and GM-CSF [31,32]. TIA-1 and TIAR are closely- related factors that inhibit the translation of TNF-α transcripts in macrophages [33] but not in T-lymphocytes [34]. TIA-1-induced translational repression is required for the translational arrest triggered by certain environmental stresses including heat, oxidative stress and energy deprivation [35]. TIA-1, TTP, HUR and other trans-acting factors are predicted to be miRNA targets and are further discussed below.
3. miRNA biogenesis and their regulation of target gene expression
miRNAs are small endogenous, evolutionarily conserved, double- stranded non-coding RNA molecules that serve as regulators of multiple genes. Many miRNA are conserved among vertebrate species while others have a more limited distribution [3,36]. It is likely that miRNAs are currently undergoing evolution (see below) particularly in the nervous and immune systems. Regulation by miRNAs in eukaryotes is usually mediated via the formation of imperfect hybrids with 3′UTR sequences of the target mRNAs. This induces translational repression and/or mRNA degradation. A recent study demonstrated that miR-2 inhibits translation initiation without affecting mRNA stability and that Drosophila miR-2 induces the formation of pseudo-polysomes that inhibit translation but only when translation initiates by means of the physiological cap structure [37]. In vitro translation systems have been used to address specific events and early kinetics of miRNA inhibition of translation and have implicated miRISC interference with m7G-cap recognition [38]. Other studies have shown that miRNP association with mRNA interferes with translation initiation complex formation [39] or that miRNA-binding interferes with elongation [40,41]. Interestingly, it has been shown that miRNAs, as a class, associate with actively translating mRNA [42]. Multiple mechanisms appear to contribute to miRNA-mediated gene regulation and their individual relevance is likely to vary depending on the gene, cell and condition [43].
The generation of microRNAs is dependent on the RNase III enzyme Dicer, the levels of which vary in different normal cells and in disease states. Dicer expression has been shown to change in development and the expression levels, in our studies and others, respond rapidly to certain stimuli including stresses and epigenetic regulators. We have demonstrated that Dicer protein expression but not mRNA in several human and murine tumor cell lines as well as primary human and mouse kidney cultures is inhibited by double-stranded RNA and Type I interferons. In contrast IFN-γ induces Dicer [44]. These findings identify interferons as potentially important regulators of Dicer expression. The dramatic and rapid regulation of Dicer observed may provide new insight into cellular responses to various stresses and potentially to infections.
Some miRNAs, like miR-16, are widely expressed while others are tissue or developmental stage specific [10,45,46]. The tissue-specific expression of miRNAs is suggestive of a role in cell differentiation and in maintaining cell identity; roles they may share with tissue-specific transcription factors. Certain miRNAs may be present in high levels; for example, in C. elegans, miR-2, miR-52, and miR-58 are present in an abundance of ~50,000 molecules per adult worm cell [47].
It is estimated that expression of ~8000 genes (30% of the human genome) can be regulated by miRNAs [48,49]. However, recent bioinformatics studies hint at the existence of tens of thousands of short non-coding RNAs similar to miRNAs with the potential to regulate expression of most or all human genes [50– 52]. Although it is uncertain whether all of the small RNAs are functional and some may be fragments of larger RNAs. Each miRNA may suppress multiple genes (on average ~200) and one mRNA can be targeted by multiple miRNAs [53–55].
Human miRNAs are processed from primary transcripts which may originate from introns, intergenic regions or non-coding RNAs [56–58]. RNA Pol II is responsible for the majority of miRNA transcripts however, RNA Pol III transcripts have recently been found to be processed into miRNAs [50]. About 25% of human miRNAs belong to intronic regions of protein coding genes [59]. Intronic miRNAs are usually co-expressed with their respective mRNAs as observed for human intronic miRNAs [60]. A mature miRNA emerges after a series of steps depicted in Fig. 1. The process begins with the transcription of a long primary transcript by RNA polymerase II. Functional mature miRNAs are excised by a pair of endonucleases (Drosha and Dicer) that are present in different compartments of the cell. The nuclear RNase III, Drosha, produces a precursor which is actively transported to the cytoplasm where Dicer cleaves pre-miRNAs to release mature ~22 nucleotide duplex miRNAs. A single strand of the small RNA is then loaded onto the RNA-induced silencing complex (RISC) that directs the miRNA to its target site on mRNA and leads to post-transcriptional repression (Fig. 1) [61,62]. An alternative pathway of miRNA biogenesis involves splicing from introns. These ‘mirtrons’ mimic the structural features of pre-miRNAs and are spliced out without Drosha processing. Thus far 14 mirtrons have been found in Drosophila and C. elegans [63]. The discovery of mirtrons raises the interesting question as to whether miRNAs could have evolved before Drosha/Pasha in eukaryotes [63]. Mirtrons have recently been identified in mammals as well; including 16 primate-specific mirtrons and 3 mirtrons that are well conserved in diverse mammals. The existence of well-conserved mirtrons indicates their relatively ancient incorporation into endogenous regulatory pathways [64].
Fig. 1.

A schematic representation of miRNA processing and activity. MicroRNAs are transcribed by RNA polymerase enzymes (mainly Pol III) as part of primary miRNA molecules. Two dsRNA-specific ribonucleases, Drosha and Dicer sequentially cleave the long-primary miRNA into the precursor hairpin miRNA and the mature miRNA (22 nt), respectively. A single strand of this mature miRNA enters the RNA-induced silencing complex (RISC) that mediates the miRNA function.
Analogous to protein coding genes, miRNA may also be subject to post-transcriptional regulation. A recent study demonstrated a role for SMAD proteins, regulators induced by TGF family members, in enhancing pre-miR-21 processing from its primary transcript [65]. This function appears to be mediated by specific SMAD association with Drosha and the p68 RNA helicase. It has also been shown that primary miRNA transcript processing can be blocked. For example, the developmentally-regulated RNA binding protein, Lin28, binds pri-let-7 transcripts and prevent their processing in ES cells [66]. Furthermore, RNA editing has been shown to present an additional mechanism of post-transcriptional regulation of miRNAs [67]. The ADAR editing enzymes are capable of converting specific adenosine residues to inosine in double-stranded RNA molecules. Edited miRNAs may be degraded by the RISC associated ribonuclease Tudor-SN or may be redirected to different targets [68].
Classically it is viewed that transcription factors (TFs) regulate gene expression responsible for the various cell phenotypes and responses of cells to environmental stimuli. With the discovery of miRNAs, it is speculated that miRNAs, in combination with TFs, control the expression of thousands of mammalian genes and this is associated with ‘tuning’ the transcriptional network [69]. miRNAs are believed to form regulatory networks by integrating the cell-intrinsic regulators including transcription factors and epigenetic controls with cell-extrinsic signals from other cells and the environment. One type of network may consist of a TF and miRNAs that can co-regulate the expression of common target genes (Fig. 2). Transcription factors regulate the expression of miRNAs forming a feed-forward loop that aids in the efficient control of mRNA [70,71]. These and other miR-TF regulatory architectures are described in detail by Shalgi et al. [70]. For example, c-Myc activates E2F1, a transcription factor, and miR-17-5p and miR-20. The E2F1 directly binds the promoter of the miR-17-92 cluster regulating its transcription [72]. MiR-20, a component of the miR-17-92 cluster has been shown to regulate the translation of E2Fs [72]. This feed forward loop possesses an additional property in that it has a shorter response time to a signal compared to purely transcriptional loops. Recent in silico studies predict many miRNA: target regulatory relationships; for example, nine transcription factors that regulate the p53 gene were identified as targets of miRNAs [73].
Fig. 2.
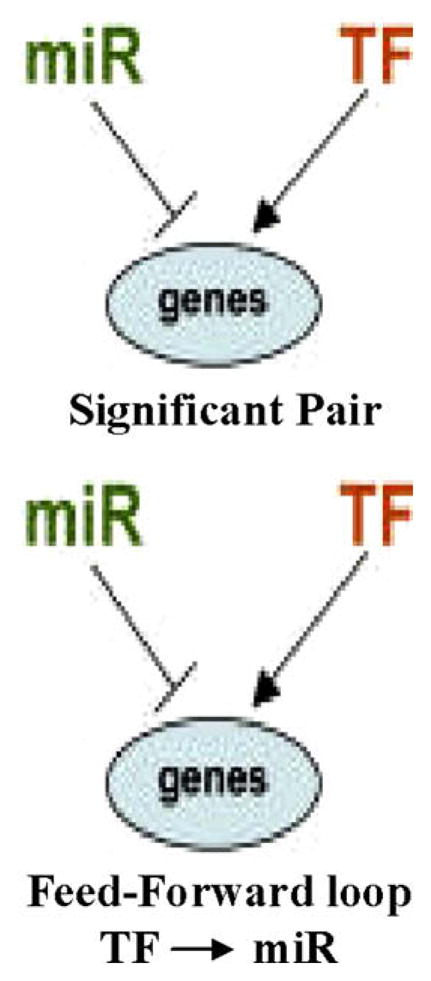
miRNA and transcription factors interact to form regulatory networks. The interactions between miRNAs and transcription factors have been shown to represent co-regulation network designs (adapted from Shalgi et al. [70]). As discussed in the text, feed-forward loops occur in high frequency in many biological networks and consist of a TF and miRNA that act as a switch. In the feed-forward loop diagrammed, a TF regulates its partner miRNA as well as target genes. Other more complex networks are also found, as described in Section 3 and [70].
miRNA-mediated gene regulation is conceptually similar in many ways to that mediated by transcription factors. Similar to transcription factors, miRNAs bind to defined cis-regulatory sequences. Also, miRNAs function as cell-type specific switches that determine gene expression [74]. For example, the lsy-6 miRNA constitutes both an essential and a sufficient switch for chemosensory cell differentiation [75]. The occurrence of numerous distinct putative miRNA binding sites indicates a combinatorial model in which, for a gene to be turned on or off within a given cell type, a combination of trans-acting factors (miRNA and TF) specific to that cell binds to cis-regulatory elements. The presence of multiple miRNA target sites in immune genes [5] and studies of miRNA expression in brain and hematopoietic lineage cells [10] suggest that, like TFs, ‘codes’ of miRNAs may exist. The co-operative nature of miRNA action, as observed in in vivo studies using transgenic worms, is similar to the cooperation reported for TFs [76–78]. However, there also exist differences between TF and miRNA action. For example, TFs can act as either activators or repressors, and because of their size and structure they can simultaneously interact with distinct tissue-specific proteins. miRNAs are primarily repressors, although recent experiments have suggested that miRNAs may activate certain genes in growth arrested cells [79]. It has also been recently shown that miRNAs can bind sites in promoters to activate transcription and that sites in 5′UTRs may be capable of enhancing translation [80,81]. Clearly, our understanding of sequences targeted by miRNAs and their consequences are continuing and preliminary data suggests differing activities (activation vs repression) depends on cellular conditions [82].
Relatively little is known of the relationship between miRNAs and epigenetics in normal cells, although it is generally thought that miRNAs have an effect on the epigenetic machinery and that miRNA expression can itself be epigenetically controlled. Defects in both the epigenetic and miRNA regulation of genes have been documented in various cancers. A number of recent studies indicate that abnormalities of the epigenome and the miRNome are not independent but reflect epigenetic regulation of miRNA expression and miRNA regulation of components of the epigenetic machinery [83]. For example, miRNA expression profiling studies in T24 human bladder cancer cells and LD419 human normal fibroblast cells identified 17 of the 313 miRNAs studied to be up-regulated by a DNA demethylating agent and histone deacetylase inhibitors [84]. Additional studies have confirmed the effect of methylation on miRNA expression [84–87]. It is noteworthy that the epigenetic regulation of miRNAs can be cell-type specific [88]. In addition, two computational studies have predicted chromatin factors as major miRNA targets [5,89].
4. In silico miRNA target predictions
Plant miRNA are completely (22 nt) complementary to their targets leading to degradation of the message. Some miRNAs also cleave target mRNAs when the sequences of the miRNA and their target gene are nearly perfectly complementary [90]. However, unlike plant miRNA, the majority of animal miRNAs are only partially complementary to their targets and, although degradation may occur, most often they repress translation. However, the detailed mechanisms of gene silencing remain elusive [43,76,91,92]. Doench and Sharp have reported that the ability of miRNAs to suppress translation depends on the free energy of binding between the first 8 nt of the 5′ end of miRNA and its target mRNA [53]. These miRNA inhibit the expression of target mRNAs that are complementary to position 2–7 of the 5′ end the miRNA, known as the seed region [93]. The relative lack of specificity of target regulation in animals represents an intrinsic problem in understanding how miRNAs work. As mentioned, miRNAs are an important mechanism of normal control of tissue-specific populations of actively translated mRNAs [94]. However, could they be harmful to the cell? This is an important area for future research.
During the past 5–6 years computational tools have been developed to understand the structure of miRNAs and to identify likely miRNA target sites in mRNA 3′UTRs. The basic principles for target site prediction are largely derived from experimental studies and rely on: (1) the extent of sequence complementarity; (2) favorable miRNA-target duplex thermodynamics; (3) conservation of target 3′UTR sites in evolutionarily-related genomes; (4) multiplicity and co-operativity of miRNA–mRNA duplex [49,51,95–98]. Each algorithm weights the contributions of these individual features differently. As studies have progressed, these algorithms have been updated and improved. A database, TarBase, has been assembled to catalog miR:target gene interactions with experimental validation [99]. The ongoing need for more experimental demonstration of specific miR:target gene functional interactions will make TarBase a critical tool to multiple groups as they continue to refine predictive algorithms. The biological validation of truly functional miRmRNA is critical in future studies.
The efficacy of in silico prediction of miRNA targets is often difficult to assess because relatively few validated miRNA targets are known. Nonetheless, recent data showing experimental support for many target predictions is encouraging. For example, comparison of the performance of five computational methods using a set of 84 miR:target gene interactions, involving 32 miRNAs [99] demonstrated that 3 algorithms (PicTar, TargetScanS, and miRANDA) identify nearly 65% of the conserved unbiased experimentally supported interactions. Each of the algorithms has its own false positive rate but, since each program also uses differential weighting of prediction criteria, comparison of independent predictions can enhance the specificity of miRNA target site prediction [100]. Importantly the union of these individual programs would predict ~92% of the conserved and experimentally verified miR:target gene interactions [100]. While this method can achieve high probability predictions, false negatives can become more of a problem as algorithms are merged and thus the selection of algorithms for consensus studies is critical [100]. The in silico approach has thus far been the main tool for identifying miRNA targets often allowing the subsequent identification of a biological role for a miRNA. The immune gene miRNA target prediction survey of Asirvatham et al. [5] used the stringent combination of three algorithms described by Sethupathy et al. [100] but also reported the complete individual algorithm outputs.
5. miRNA regulation of innate immune responses
Computational analyses [5] indicate preferential targeting of immune genes by miRNAs with major targets including transcription factors, co-factors and chromatin modifiers whereas innate immune pathway molecules including TLR, chemokines, and cytokines are generally poor or non-targets. Of the interleukin genes (IL1-29) studied, 9 had predicted miRNA binding sites (Fig. 3A). Of 20 interleukin receptor genes examined, only two had high probability miRNA target sites. Statistical analysis indicated that predicted miRNA target sites were under-represented in the 3′UTRs of cytokine and chemokine ligand and receptor genes relative to the genome as a whole. Although, not the topic of this review, chemokines are important in inflammation and recent work suggests that, like cytokines, chemokines and their receptors are poor targets of miRNAs. However, despite a relative lack of miRNA targets selected chemokines are good targets of miRNAs [5]. Chemokines, along with cytokines, function as major regulators of many of the cellular components of the tumor microenvironment [101]. For example, targeting of CXCR4 with a miRNA blocks the invasive and metastatic properties of breast cancer cells [102]. Of the interferon genes and their receptors analyzed, only IFN-γ was predicted to have a single miRNA binding site. The relative paucity of miRNA binding sites in the cytokine genes compared to the genome as a whole was surprising in light of the reported role of mRNA stability in the regulation of cytokine expression [103]. Importantly, these findings suggest that either the regulation of the cytokine/chemokine/TLR genes is not often mediated via their 3′UTRs or that elements other than miRNA targets in the 3′UTR are involved in regulating message stability. As mentioned above and discussed further below, the AU-rich elements located in the 3′UTR are important determinants of cytokine message stability. Additionally, miRNA regulation of a pathway may not be at the ligand or receptor level but rather at upstream or downstream steps in the pathway. This has been well shown for LPS activation of TLR4 via the NF-κB pathway [104]. A comparable mechanism has been noted for the p53 pathway in which the downstream gene p21 but not p53 is a high probability target for miRNA regulation [5]. Similarly, in the JAK–STAT pathway numerous components of this pathway rather than ligands or receptors are the predicted targets of miRNA regulation. Approximately 50 miRNAs are predicted to target the genes of the JAK–STAT pathway including the negative regulators SOCS and PIAS.
Fig. 3.

Cytokines may be regulated both directly and indirectly by miRNA. (A) Post-transcriptional regulation of the interleukin genes mediated via miRNA and ARE sites in the 3′UTR. Interleukin genes with predicted miRNA binding sites are indicated in the first box. Those with ARE sites [20] are listed in the second box where the text in red signifies predicted regulation by both miRNAs and AREs. (B) Indirect miRNA regulation of genes (e.g., cytokine genes) having AU-rich sites in their 3′UTR. miRNA potentially regulate the expression of the components of the ARE machinery. Additionally, MAPKs regulate the function of ARE-BPs and they may in turn be regulated by miRNA [5]. MiR-16 sites are found to overlap with some ARE sites and could potentially provide another mechanism by which miRNA regulate the expression of ARE-containing transcripts like SOCS3 (see Section 9).
TGF-β was first discovered for its ability to stimulate the proliferation and transformation of mesenchymal cells [105] but has now been shown to have diverse effects [106,107]. TGF-β is produced by many cells including Treg cells and has been characterized as an immunosuppressant cytokine due to its inhibitory effect on effector T cell development and the promotion of Treg cells [108]. TGF-β directs the differentiation of both Foxp3+ Treg cells and TH17 cells in an inflammatory cytokine environment and provides a mechanism by which Treg cells terminate TH17 responses [109,110]. In silico analyses suggest that the TGF-β pathway is highly regulated by miRNAs. However TGF-β1 and the receptors TGF-βRII, Endoglin, and Cripto-1 do not have any predicted miRNA target sites whereas multiple miRNA binding sites are predicted in the downstream signaling components including all 7 SMAD genes and the co-repressor TGIF [5]. Seventeen of the 24 components in the TGF-β pathway are likely to have 3′UTR binding sites for 64 different miRNAs. Moreover the 17 target genes are all hubs with 8 or more different miRNA sites in their 3′UTR making them high probability sites of miRNA control [5].
6. Evolution of miRNAs and their targets
It is intriguing to speculate on the selective nature of miRNA regulation found in certain interleukin genes (see Fig. 3A). For example, the interleukin 1 family includes several closely-related genes; the major agonist proteins are IL-1α, IL-1β, and IL-18. IL-1α and IL-1β bind to the same receptors, induce similar biological functions [111] and are duplicated genes [112]. Computational analysis of the IL-1α and IL-1β 3′UTRs predict miRNA binding sites only in IL-1α [5] suggesting the possible loss of miRNA binding sites as a result of the duplication. The change in gene expression patterns resulting from gain or loss of miRNA binding sites could have a considerable effect on evolution. During gene duplication, the loss of or an alteration in target sites could result in variations in development that may be selectable during evolution [113]. Positive selection could act on miRNA target sites in genes whose down-regulation is advantageous and negative selection on target sites in genes whose expression is disadvantageous to survival [114]. From an evolutionary perspective, it has been shown that miRNA stem-loop structures contribute to the evolution of robustness in genetic regulation [115,116]. The mechanism by which miRNA acquire new functions is not well understood but one prominent possibility is to utilize the other strand (miRNA*) of the duplex, generated by Dicer, that is usually degraded [117]. Gene duplication and diversification have been implicated in the evolution of small RNA regulatory pathways and in the alterations in spatial and temporal miRNA expression as well as the evolution of gene regulation in multiple organisms [118,119]. Some excellent recent reviews of microRNAs that focus on their evolution are recommended [119–124].
Several years ago Bartel’s group [48] reported a selective avoidance pattern of genes with target sites for miRNAs. Certain genes were expressed at developmental stages before miRNA expression and their levels tended to fall as miRNA appeared. Thus, in general, miRNA target genes in animals were co-expressed at low levels in the same tissues as the miRNAs that target them. Moreover, genes that are co-expressed together with miRNAs in the same cell usually lack target sites. This suggests that animal genes may be under evolutionary pressure to react with or avoid miRNAs [48,114,125]. Chen and Rajewsky [126] have emphasized the comparison of the evolution of genes by transcriptional regulation with the post-transcriptional gene regulation mediated by miRNAs. However, although most animal miRNAs repress translation, several miRNAs cleave target mRNAs such as HoxB8 [127].
Typically vertebrate evolution of genes has been explained by genome duplications with subsequent diversification of the duplicated copies. Two massive gene duplications occurred during emergence of vertebrates [128]. A recent study of the causal relationship of gene duplication events to the advent of morphological complexity in vertebrates indicated that although duplications increase diversity they can not account for the origin of the novel families themselves [129]. Vertebrates have high levels of non-coding RNAs (ncRNA) compared to invertebrates [130] and among the ncRNA are miRNAs that have increased substantially with >250 new miRNAs added in the vertebrate lineage [130,131]. It seems likely that the enhanced transcription status created more hairpins and therefore potentially more miRNAs which increased its organismal complexity. Additionally, the maintenance of an optimal phenotype in the face of genetic perturbation (genetic robustness) mediated by miRNAs may be inherent in the stem-loop structure of miRNAs compared to random RNA sequences [116].
Although single nucleotide polymorphisms (SNP) have been shown to be rare in miRNA targets these sites are subject to polymorphism and selection that likely contribute to the evolution of regulatory systems and possibly to various diseases [132,133]. One example of an evolutionary relationship is seen in the IL-1 and IL-18 genes. There is differential regulation of IL-1α and IL-1β by miRNAs, with IL-1α being a target but not IL-1B. These predictions, if functionally verified, could open new avenues for regulating IL-1 in cancer therapy [111]. IL-18, which is more closely related to IL-1β than IL-1α, also does not have any predicted miRNA sites [134]. Speculatively, the lack of miRNA regulation of IL-1β and IL-18 could have evolved to regulate the overlapping functions of these genes. It should be mentioned in the context of evolution that miRNAs themselves could be duplicated and subsequently acquire separate functions.
Similar to IL-1, IL-12 has 2 chains but these do not arise from gene duplication. Interestingly, the IL-12α chain has no predicted miRNA targets but the IL-12β has a single high probability miRNA binding site [5]. The biologically active IL-12 is a heterodimer consisting of disulfide linked subunits, IL-12p35 (IL-12α) and IL-12p40 (IL-12β), that are products of distinct genes [135]. The p35 chain is ubiquitously expressed whereas the p40 chain is highly regulated and expressed in cells where bioactive IL-12 is strongly induced [136]. The expression of these transcripts is primarily regulated at the transcriptional level [137] and, to a certain extent, by reversible histone acetylations and deacetylations [138]. However, post-transcriptional regulation also appears critical for the control of IL-12β chain as demonstrated in a study where a decrease in the synthesis of IL-12β chain was related to decrease mRNA stability [139]. The temporary defect in the levels of IL-12β chain noted during neonatal dendritic cell (DC) development is also consistent with the post-transcriptional control of IL-12β [140]. Whether miRNA to IL-12β are present in the neonate and functionally regulate IL-12 is, to our knowledge, unknown. However, collectively these data suggest that miRNAs provide an important layer of post-transcriptional gene regulation in the immune system during development.
There are numerous examples in the literature [5,141] of duplicated or functionally closely-related genes in which miRNA regulation differentially targets the related genes. For example, estrogen receptor (ERα) is targeted by miRNA [5,141] but ERβ is not. Differential targeting was also noted between the methyl binding proteins MBD2 and MBD3 and other examples in computational studies [5]. Speculatively, miRNAs could have evolved to regulate the overlapping functions of closely-related duplicated genes. These may be ‘clues’ and future in-depth studies of these miR:gene interactions may enhance our understanding of these important pathways. However, this phenomenon is not restricted to duplicated genes and the functionally-related RAG1/2 and HRAS/KRAS genes are also examples where only one member of the gene pair is a miRNA target [5].
miRNAs that belong to the same family may regulate identical or similar target genes suggesting a functional redundancy of these miRNAs. For example, a cluster of miRNAs, miR-160-1, 160-2, 160-3, and 160-4, control the expression of an auxin response factor homolog in Physcomitrella [142]. IL-25 shares sequence similarity with IL-17 and these cytokines are likely to be targeted by a common set of miRNAs (miR-17/20/106) [5]. The CBP and p300 genes are functionally and structurally closely-related chromatin cofactors and, while each has its own distinct set of miRNAs, they share predicted binding sites for 10 miRNAs [5]. In contrast, for many other closely-related genes the predicted miRNAs are distinctive suggesting a high specificity of the miRNA:target interaction in spite of conservation of gene sequence and function.
As mentioned above the genes in a pathway can be regulated by a common set of miRNAs while in many others this has not been found. A computational study identified two functionally correlated miRNA:mRNA modules that are defined as groups of miRNAs and their mRNAs involved in similar biological processes [143]. In module I, three miRNAs (miR-127, -212, and -132) share putative target genes. The miRNAs in this module are considered tumor suppressors and their target genes are primarily oncogenes. Module II consists of two miRNAs (miR-98 and let-7f-2) and five target genes that are involved in cell cycle or metabolism [143]. An example of module I is predicted in the RNAi pathway where DICER1 and all four Argonaute (EIF2C) genes are targeted by a common miRNA (let-7d) [5]. Thus a single member of the let-7 family of eleven miRNAs could potentially modulate RNAi pathway components and the expression of the ~8000 genes predicted to be regulated by miRNAs.
7. miRNAs indirectly regulate the expression of cytokine genes
As mentioned above, miRNAs can potentially regulate cytokine expression by directly binding to target sites in the 3′UTRs of mRNAs or, as discussed below, by indirect mechanisms. Although computational analysis indicates that the 3′UTRs of most cytokines lack miRNA target sites, miRNAs could nevertheless regulate cytokine expression by targeting ARE-binding proteins (ARE machinery components) such as TTP, AUF1 and members of HUR family (Fig. 3B). A recent study demonstrated that the machinery components involved in the ARE-mediated decay pathway are heavily predicted targets of miRNAs [5]. As depicted in Fig. 3, miRNA repression of several ARE components may alter levels of inflammatory cytokines as well as other immune genes. The current ARE registry lists ~4000 genes as potential targets [18,20] although in only a few have these AREs been functionally documented. Many of the components of the ARE pathway, are regulated by phosphorylation mediated by the MAPK pathway [144–146]. For example TTP-mediated TNF-α decay is inhibited by the combined activation of ERK and p38 [147]. TTP is also regulated by the proteasome and its degradation is enhanced by inhibition of the MAPK pathways [145]. Thus the MAPK kinase and proteasome pathways interact to regulate TTP and other ARE-BP and thereby the half-life of a large group of mRNAs. Whether the specific MAPKs and proteasome components involved in ARE control are themselves regulated by miRNAs is unknown. It seems however that message stability of TNF-α and possibly other cytokines is mediated via ARE elements, MAPK, the proteasome and indirectly by miRNA which target TTP and other ARE-BP.
MRNA stability is regulated by multiple mechanisms including AU-rich elements present in the 3′UTR which are cis-acting destabilizing elements. It has been demonstrated that Dicer and Argonaute may be required for the ARE-mediated mRNA decay in Drosophila and HeLa cells [148]. Sequence analysis identified miR-16 as possessing complementary sequence to the canonical AUUUA and demonstrated a role for this miRNA in interaction with the ARE sequence. MiR-16 has a UAAAUAUU sequence and is conserved in mammals. MiR-16 is present in high levels in most cells and is potentially a ‘master miR’ involved in determining mRNA stability via AREs. The down-modulation and over-expression of miR-16 increased or decreased, respectively, the stability of a reporter RNA engineered to contain the ARE of TNF-α or Cox-2 [148]. The ARE binding proteins and miR-16 were required for recognition of ARE sequences and their cooperation may be essential in ARE-mediated mRNA degradation. Furthermore EIF2C/AGO family members may participate in the interaction between miR-16 and TTP [148]. Although the interaction of miR-16 with ARE sites in mRNA has been demonstrated in vitro, the exact mechanism of miRNA targeting of ARE sequences and potential role is yet to be defined. Although an association between miR-16, ARE, RISC, and other proteins have been observed, the role that each plays in determining how message stability is controlled by miRNAs is, as yet, uncertain [43,149].
We identified two hundred genes with potential miR-16 target sites and of these three (SOCS2, AGO1, and RARB) have ARE sites in their 3′UTRs. Examination of the SOCS2 miR-16 target indicates that the ARE binding site lies at position 11–15 which are outside the 5′ seed region of miR-16 [5]. We speculate that the additional interactions provided by the ARE may be necessary for the functional activity of miR-16:SOCS2 interaction similar to that previously described for miR-16:TNF-α. This type of co-operative activity could enable miRNAs to further modulate the stability of message via ARE-mediated degradation. For example, TTP negatively regulates the expression of inflammatory cytokines such as TNF-α. During a stress response, negative regulation can be abrogated via the activation of MAPK leading to phosphorylation and inactivation of TTP thereby enhancing cytokine message stability and levels. As mentioned above, computational examination of the 3′UTRs of various immune genes did not identify likely miRNA target sites in any of the TLRs or the majority of cytokines or chemokines. However, ~50% of the cytokines studied have ARE sites [5] (Fig. 3A). Interestingly, overlapping sites for miR-16 binding and AREs were not identified in the cytokine genes analyzed with the exception of TNF-α.
8. P-bodies as sites of miRNA function
Several recent reports suggest that cytoplasmic compartments referred to as processing bodies (P-bodies or GW bodies) or, more broadly, stress granules are cellular sites where mRNA turnover occurs [150–153]. It is thought that miRNAs, following their generation by Dicer, bind to Argonaute proteins and identify their target mRNA by sequence complementarity in P-bodies. These cytoplasmic granules are major sites where cleavage and degradation of mRNA occurs. A model for miRNA function has been described in which miRNA targets associate with AGO1 via miRNA:mRNA base pairing interactions. The GW182 protein, a major component of P-bodies, interacts with AGO1 and recruits deadenylase and decapping enzymes leading to degradation of mRNA. In support of this model, it has been demonstrated that miRNA and their target mRNAs accumulate in P-bodies. The inhibition of essential structural components of P-bodies (GW182, TNRC6B, MOV-10, and Dcp-1/2) leads to both a loss of P-bodies and also an impairment of miRNA function [43,149,154–157]. P-bodies also serve as sites for storage of repressed mRNAs [155]. An important new thesis is that mRNAs may return to polysomes to synthesize new proteins and that certain cellular proteins may facilitate the exit of mRNA from P-bodies [153]. For example, the A3G protein co-localizes with miRNAs and argonaute proteins in stress granules and counteracts the translational repressive activity of several miRNAs (miR-10b, miR-16, miR-25, and let-7a). A3G facilitates the recruitment of miRNA-targeted mRNA to polysomes for translation leading to dissociation of miRNA-targeted mRNA from P-bodies [158]. However, the biological role of A3G has not been fully defined.
As indicated above, the entry of an mRNA into a P-body does not inevitably lead to its degradation and repression by miRNA can be a reversible process [152,153]. For example, the CAT-1 mRNA that is negatively regulated by miR-122a has been reported to escape both translational repression and P-body entrapment following amino acid starvation, oxidative stress or endoplasmic reticulum stress [151,159]. Following derepression, the CAT-1 mRNA is released from the P-bodies and is recruited by polysomes. Interestingly, HuR, an ARE binding protein, interferes with the function of miRNAs. HuR binds ARE containing-mRNAs and chaperones them to the cytosol [160]. This shifts the P-body-to-cytosol equilibrium of mRNAs thus prolonging message half-life. It has been proposed that the above mentioned and other ARE binding proteins (~12 in all) generally act as modifiers altering the potential of miRNAs to repress gene expression [151,159]. Finally the HUR gene family members may themselves be targets for miRNA regulation [5], another example of the complex interaction between message stability, AREs and miRNAs.
The ability to reverse the negative effect of miRNAs discussed above would potentially make miRNA regulation more dynamic and more responsive to specific cellular needs. In neuronal cells many mRNAs are transported for long distances along the dendrites as repressed mRNPs to become translated only on arrival at their final destination at the synapse [46,161]. The Limk-1 transcription factor mRNA is transported in a dormant state within dendrites to synaptic sites by its association with miR-134. In the absence of synaptic activity miR-134 inhibits Limk-1 translation. However, miR-134-mediated repression of Limk1 mRNA is reversed in response to extracellular stimuli which activate the mTOR signaling pathway [46]. Such a regulatory mechanism may exist in immune pathways; although, there are, to our knowledge, no known examples of immune genes that are derepressed by release from miRNA repression.
9. miRNAs, inflammation and cancer
Inflammation involves a well-coordinated response which includes the activation of several hundred genes including multiple cytokines, chemokines, matrix remodeling proteases, reactive oxygen, and nitrogen species and others. Although the relationship between inflammation and tumorigenesis is widely accepted, the cellular and molecular mechanisms involved are incompletely understood. One thesis suggests that a failure to precisely control the components of inflammation leads to persistent inflammation and the production of cells and growth/survival factors that enhance the growth of surrounding cells which become cancers (Fig. 4) [162]. The identification of miRNAs which are associated with cancer (oncomiRs) [163] and the involvement of miRNA in inflammation and immunity have further strengthened the potential link between miRNAs, inflammation and cancer [164]. The identification of miRNAs involved in inflammation and cancer and their functional relevance could provide new molecular targets for early detection, prevention and perhaps therapy for cancer.
Fig. 4.

miRNAs provide a link between inflammation and cancer. miRNAs have been identified as having roles in the regulation of innate and adaptive immunity. Inflammatory mediators and cytokines can regulate miRNA expression which may contribute to the regulation of multiple genes. The regulatory networks formed include both feed forward and feedback loops and contribute to inflammation and oncogenesis.
As mentioned earlier, specific miRNAs (miR-132, -146, and -155) are activated by inflammatory mediators such as NF-κB [8]. Two of the predicted targets of miR-146 (TRAF6 and IRAK1) are involved in LPS signaling and miR-146 may therefore regulate inflammation via a classical negative feedback pathway [104]. Moreover, TTP and AUF1 are predicted targets of miR-146 [5] and both are message destabilizing proteins. Thus inflammatory stimuli that induce the expression of NF-κB and miR-146 could indirectly enhance the expression of multiple proinflammatory cytokines. In mice, LPS also induces rapid transient changes in expression of other miRNAs (miR-21, -25, -27b, -100, -140, -142-3p, -181c, -187, -194, -214, -223, and -224) although the role of these miRNAs in inflammation is, as yet, uncertain [165]. In addition to LPS induction of miR-155, several other inflammatory mediators, including poly-(I:C) and IFN-β, were identified to up-regulate the expression of miR-155 thereby characterizing miR- 155 as a component of the primary response to various inflammatory mediators [56,166–170].
The observation that miRNAs are likely regulators of cytokine mRNAs [150] suggests that they might also be involved in diseases related to abnormal immune responses including certain inflammatory disorders. For example, miRNAs have been reported to provide a layer of regulation in the pathogenesis of chronic inflammatory skin diseases e.g., psoriasis and atopic eczema [171]. Psoriasis is characterized by a specific miRNA expression profile that is different from healthy skin and other chronic inflammatory skin diseases such as eczema. The keratinocyte-specific miRNA (miR-203) was observed to be up-regulated in psoriatic plaques leading to a concurrent down-regulation of suppressor of cytokine signaling-3 (SOCS-3) protein translation. SOCS-3 is a negative regulator of the IL-6 and IFN-γ-induced signaling pathways and therefore the suppression of SOCS3 may lead to an increase in inflammatory response in the skin [171]. High probability miRNA binding sites are present in all of the SOCS genes (SOCS1-7) [5]. Interestingly, these predictions suggest that members of the SOCS gene family are differentially targeted by miRNAs with SOCS7 having a single miRNA binding site, SOCS5 & SOCS6 multiple miRNA binding sites (>8) and SOCS-2, -3, -4 having 2–4 predicted miRNA binding sites. The identification of a common set of miRNA (let-7/miR-98) binding sites in the 3′UTR of SOCS1 and SOCS4 transcripts and also in IL-6, IL-10, and IL-13 suggests a potential functional redundancy of these miRNAs [5]. MiR-146a is known to be associated with psoriasis and also regulates the genes in the TNF-α signaling pathway [104,172]. The importance of TNF-α in psoriasis has been demonstrated by the efficiency of the anti-TNF-α therapies suggesting that miRNAs are potential therapeutic targets in the treatment of inflammatory diseases [171].
miRNAs may also be involved in pregnancy, a process requiring precisely controlled inflammation. The human chorioamniotic membranes participate in a variety of physiological and pathological processes during pregnancy. Infection and inflammation of the amniotic cavity can lead to both preterm delivery and adverse prenatal outcomes [173]. Functional genomics studies have identified a unique gene expression pattern during intra-amniotic infection [174]. Interestingly miRNA expression in the human chorioamniotic membrane changes with advancing gestational age suggesting a functional role for miRNAs. Thirteen miRNAs display decreased expression with advancing gestation and with histological chorioamnionitis. MiR-223 was experimentally identified in chorioamnionitis- related inflammation and its predicted target genes include several genes involved in inflammation and immune responses [175]. However, all other miRNAs identified as regulators of the inflammatory and anti-inflammatory cytokines remain potential candidate miRNAs responsible for inflammation in the chorioamniotic membranes.
The frequent identification of inflammatory cells in the tumor microenvironment led to the concept that these cells play a role in tumor progression, invasion and angiogenesis [176,177]. The proinflammatory cytokines and chemokines produced in the tumor microenvironment, including TNF-α, IL-1, IL-6, and IL-8, enhance cell proliferation, cell survival, cell migration and tumor angiogenesis [178]. Cytokine signaling could contribute to tumor progression by stimulating cell growth and differentiation and/or inhibiting the apoptosis of altered cells at the inflammatory site [179,180]. The proinflammatory cytokine IL-6 has multifaceted functions including regulation of immune responses, hematopoiesis and inflammation. It has also been shown to be involved in tumor progression, inhibition of apoptosis of cancer cells and fever-induced angiogenesis [181–183]. Elevated IL-6 levels were observed in patients with endometrial cancer, non-small cell lung carcinoma, colorectal cancer, renal cell carcinoma, breast, and ovarian cancer. The persistent activation of IL-6, as observed in certain cancers and chronic inflammatory conditions, induces the expression of miRNA let-7a which contributes to IL-6-mediated tumor cell survival by modulating Stat3 activity [162]. Computational analyses indicate the presence of a high probability miRNA binding site for let-7 in IL-6 and also a let-7 site in the 3′UTR of STAT3 [5]. The let-7 target site predicted in the 3′UTR of IL-6, together with IL-6-mediated increases in the expression of miR-21, let-7 and other miRNA, suggests the potential for a repressive feedback loop [162]. A well-known example of such a loop is observed in mammalian neuronal development in which the RE1-silencing transcription factor (REST)/NRSF, a transcriptional repressor of multiple protein-coding genes, activates miRNAs including miR-29 and -135b [184,185]. REST in turn has putative binding sites for several brain-related miRNAs including miR-29 and miR-135b in its 3′UTR. This would produce a double-negative feedback loop involving the REST complex and miRNAs [185]. The let-7 miRNA has also been reported to be poorly expressed in lung cancer [186,187] and over-expression of let-7 inhibits lung cancer cell growth in vitro suggesting that it may act as a tumor-suppressor [188]. Let-7 targets K-RAS and HMGA2 and its loss may allow activation of these oncogenes, enhance tumor progression, and may represent a link between inflammation and cancer [54,187,188]. Additionally tumor-derived TGF-β may facilitate suppression of the immune response to tumors [189]. As mentioned above, computational analysis indicates that the ligand TGFB1 is not a miRNA target but many of the signaling components of this pathway are heavily targeted by miRNAs. MiRNAs directed at these pathway components are therefore potential therapeutic targets for cancer treatment.
MiR-155 may also provide a link between inflammation and cancer. Enhanced expression of miR-155 promotes cancer and hence has been referred to as an ‘oncomiR’ [166–170]. MiR-155 has been shown to contribute to the homeostatic regulation of the normal immune system and this miRNA is specifically required for TH cell differentiation and germinal center development [11]. Computational analysis strongly suggests that miR-155 can act as an ‘immunomiR’ [5]; and miR-155 mutant mice display impaired B- and T-cell immunity and defects in antigen-presenting cells [11]. Several potentially important targets of miR-155 including BCL11A, CTLA4, GCN5, HDAC4, NFAT5, MECP2, SKI, SMARCA4, and SOCS1 have been predicted but not yet verified [5]. MiR-155 directly regulates the expression of Pu.1, a critical factor in terminal B-cell differentiation [12]. Therefore, miR-155 may act as an immunomiR as well as an oncomiR targeting genes in multiple general cellular pathways.
TNF-α, a pro-inflammatory cytokine secreted by tumor-associated inflammatory cells, has recently emerged as a major regulator linking inflammation to cancer pathogenesis. TNF binds to homotrimeric receptors, TNFRI and TNFRII, and can initiate an inflammatory cascade involving other cytokines, chemokines, growth factors and endothelial adhesion factors. TNF-α has both anti- and procancer effects [176]. It is regulated by both transcriptional and post-transcriptional mechanisms allowing for a rapid and transient production in response to a variety of stimuli [190]. The TNF-α sequence includes ARE sites and the importance of post-transcriptional regulation was demonstrated in mice with a germ line deletion in the TNF-α 3′UTR. These mice developed pathological conditions similar to rheumatoid arthritis and Crohn’s disease associated with a 3–10 fold increase in the TNF-α protein levels [191]. Asirvatham et al. [5] were unable to identify miRNA binding sites in the 3′UTR of TNF while the components of the ARE-mediated decay pathway were heavily targeted by miRNAs. It appears that ARE control of TNF-α expression may operate at multiple post-transcriptional levels including the miRNA regulation of ARE-BPs. Additionally the JNK and p38 pathways have been identified to regulate the ARE-mediated translational repression of TNF-α message [33,147] and the components of these pathways were also identified as miRNA targets suggesting an additional indirect miRNA regulation. miRNAs directed at these pathway components are therefore potential therapeutic targets for therapy.
NF-κB is an important transcription factor activated by TNF-α and has been shown to facilitate the development of inflammation and cancer due to its ability to induce pro-inflammatory cytokines such as IL-6, IL-8, and TNF-α and chemokines [178,192,193]. In this putative feedback loop, NF-κB is activated by and induces the expression of pro-inflammatory cytokines. The NF-κB induction of miR-146a/b acts as a negative regulator for TRAF6 and IRAK1, downstream signaling components of the TNF-α pathway. We have also identified a putative miR-9 binding site in the 3′UTR of the NFKB1 transcript [5]. The functional verification of this miR:-gene interaction could provide another site of miRNA regulation. As mentioned above, the TNFα/IKKβ signaling pathway has been suggested as a link between inflammation and cancer. TNF-α can, via IKKβ, activate mTOR signaling and this potentially oncogenic pathway may be relevant in both epithelial tumors and hematological malignancies. The elevated levels of TNF-α derived from macrophages and tumor cells have been reported to up-regulate mTOR through inactivation of the TOR suppressor TSC1 via IKKβ leading to tumor angiogenesis [194]. Activation of TOR is an important factor in the development of cancers having deficient or mutated TSC1/TSC2 [194]. Extending and integrating these findings could potentially provide new therapeutic strategies for inflammation-mediated cancers using IKK inhibitors and/or inhibitors (antagomirs) of miRNAs targeting TSC1/TSC2.
In recent years, the recognition of miRNAs and their functions has uncovered an additional regulatory layer of gene expression. Profiling and predictive studies are exploding in the literature and demonstrate the broad importance of miRNA regulation in biology and medicine. The near future is likely to bring a rapid expansion in verified miR:gene targets which will provide the prelude to therapeutic studies. Various commercial and academic labs are advancing the understanding of miR:gene regulation and designing pre-clinical applications [195]. Artificial miRNAs (amiRNAs) have been designed to target single genes or groups of related genes [196,197]. For instance, synthetic miRNAs designated miR-CCR1- 85, miR-CCR1-206, and miR-CCR1-656 have been designed and shown to function as down-regulators of CCR1 protein and reduce the invasiveness of hepatocellular carcinoma [HCC] cell lines. The miRNA-mediated down-modulation of invasiveness has been suggested as a therapeutic target for HCC [198].
Acknowledgments
The authors wish to thank Heinz Baumann for the review and helpful comments of the manuscript. This work was supported by NIH grants HD 17013 and CA 124971.
References
- 1.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase tools for miRNA genomics. Nucleic Acids Res. 2008;36:D154–8. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–4. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 4.Leung AK, Calabrese JM, Sharp PA. Quantitative analysis of argonaute protein reveals microRNA-dependent localization to stress granules. Proc Natl Acad Sci USA. 2006;103:18125–30. doi: 10.1073/pnas.0608845103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asirvatham AJ, Gregorie CJ, Hu Z, Magner WJ, Tomasi TB. MicroRNA targets in immune genes and the Dicer/argonaute and ARE machinery components. Mol Immunol. 2008;45:1995–2006. doi: 10.1016/j.molimm.2007.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Welker NC, Habiq JW, Bass BL. Genes misregulated in C. elegans deficient in Dicer, RDE-4, or RDE-1 are enriched for innate immunity genes. RNA. 2007;13:1090–102. doi: 10.1261/rna.542107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cobb BS, Hertweck A, Smith J, O’Connor E, Graf D, Cook T, et al. A role for Dicer in immune regulation. J Exp Med. 2006;203:2519–27. doi: 10.1084/jem.20061692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci USA. 2007;104:1604–9. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat Immunol. 2008;9:839–45. doi: 10.1038/ni.f.209. [DOI] [PubMed] [Google Scholar]
- 10.Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–6. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- 11.Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–8. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 12.Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S, et al. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity. 2007;6:847–59. doi: 10.1016/j.immuni.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–11. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muljo SA, Ansel KM, Kanellopoulou C, Livingston DM, Rao A, Rajewsky K. Aberrant T cell differentiation in the absence of Dicer. J Exp Med. 2005;202:261–9. doi: 10.1084/jem.20050678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cobb BS, Nesterova TB, Thompson E, Hertweck A, O’Connor E, Godwin J, et al. T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. J Exp Med. 2005;201:1367–73. doi: 10.1084/jem.20050572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ross J. mRNA stability in mammalian cells. Microbiol Rev. 1995;59:423–50. doi: 10.1128/mr.59.3.423-450.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilusz CJ, Wormington M, Peltz SW. The cap-to-tail guide to mRNA turnover. Nat Rev Mol Cell Biol. 2001;4:237–46. doi: 10.1038/35067025. [DOI] [PubMed] [Google Scholar]
- 18.Bakheet T, Frevel M, Williams BR, Greer W, Khabar KS. ARED: human AU-rich element-containing mRNA database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic Acids Res. 2001;29:246–54. doi: 10.1093/nar/29.1.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Espel E. The role of the AU-rich elements of mRNAs in controlling translation. Semin Cell Dev Biol. 2005;16:59–67. doi: 10.1016/j.semcdb.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 20.Bakheet T, Williams BR, Khabar KS. ARED 3.0: the large and diverse AU-rich transcriptome. Nucleic Acids Res. 2006;34:D111–4. doi: 10.1093/nar/gkj052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zubiaga AM, Belasco JG, Greenberg ME. The nonamer UUAUUUAUU is the key AU-rich sequence motif that mediates mRNA degradation. Mol Cell Biol. 1995;4:2219–30. doi: 10.1128/mcb.15.4.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roca FJ, Cayuela ML, Secombes CJ, Meseguer J, Mulero V. Post-transcriptional regulation of cytokine genes in fish: a role for conserved AU-rich elements located in the 3′-untranslated region of their mRNAs. Mol Immunol. 2007;44:472–8. doi: 10.1016/j.molimm.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 23.Cao H, Deterding LJ, Blackshear PJ. Phosphorylation site analysis of the anti-inflammatory and mRNA-destabilizing protein tristetraprolin. Expert Rev Proteomics. 2007;4:711–26. doi: 10.1586/14789450.4.6.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bevilacqua A, Ceriani MC, Capaccioli S, Nicolin A. Post-transcriptional regulation of gene expression by degradation of messenger RNAs. J Cell Physiol. 2003;195:356–72. doi: 10.1002/jcp.10272. [DOI] [PubMed] [Google Scholar]
- 25.Barreau C, Paillard L, Osborne HB. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 2006;33:7138–50. doi: 10.1093/nar/gki1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fan XC, Steitz JA. Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J. 1998;17:3448–60. doi: 10.1093/emboj/17.12.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma WJ, Cheng S, Campbell C, Wright A, Furneaux H. Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J Biol Chem. 1996;271:8144–51. doi: 10.1074/jbc.271.14.8144. [DOI] [PubMed] [Google Scholar]
- 28.Brewer G, Saccani S, Sarkar S, Lewis A, Pestka S. Increased interleukin-10 mRNA stability in melanoma cells is associated with decreased levels of A + U-rich element binding factor AUF1. J Interferon Cytokine Res. 2003;23:553–64. doi: 10.1089/107999003322485053. [DOI] [PubMed] [Google Scholar]
- 29.Peng SS, Chen CY, Xu N, Shyu AB. RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J. 1998;17:3461–70. doi: 10.1093/emboj/17.12.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loflin P, Chen CY, Shyu AB. Unraveling a cytoplasmic role for hnRNP D in the in vivo mRNA destabilization directed by the AU-rich element. Genes Dev. 1999;13:1884–97. doi: 10.1101/gad.13.14.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lai WS, Blackshear PJ. Interactions of CCCH zinc finger proteins with mRNA: tristetraprolin-mediated AU-rich element-dependent mRNA degradation can occur in the absence of a poly [A] tail. J Biol Chem. 2001;276:23144–54. doi: 10.1074/jbc.M100680200. [DOI] [PubMed] [Google Scholar]
- 32.Carballo E, Lai WS, Blackshear PJ. Evidence that tristetraprolin is a physiological regulator of granulocyte-macrophage colony-stimulating factor messenger RNA deadenylation and stability. Blood. 2000;95:1891–9. [PubMed] [Google Scholar]
- 33.Piecyk M, Wax S, Beck AR, Kedersha N, Gupta M, Maritim B, et al. TIA-1 is a translational silencer that selectively regulates the expression of TNF-alpha. EMBO J. 2000;19:4154–63. doi: 10.1093/emboj/19.15.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saito K, Chen S, Piecyk M, Anderson P. TIA-1 regulates the production of tumor necrosis factor alpha in macrophage but not in T-lymphocytes. Arthritis Rheum. 2001;44:2879–87. doi: 10.1002/1529-0131(200112)44:12<2879::aid-art476>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 35.Anderson P, Kedersha N. Stressful initiations. J Cell Sci. 2002;115:3227–34. doi: 10.1242/jcs.115.16.3227. [DOI] [PubMed] [Google Scholar]
- 36.Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet. 2005;37:766–70. doi: 10.1038/ng1590. [DOI] [PubMed] [Google Scholar]
- 37.Thermann R, Hentze MW. Drosophila miR2 induces pseudo-polysomes and inhibits translation initiation. Nature. 2007;447:875–8. doi: 10.1038/nature05878. [DOI] [PubMed] [Google Scholar]
- 38.Mathonnet G, Fabian MR, Svitkin YV, Parsyan A, Huck L, Murata T, et al. MicroRNA inhibition of translation initiation in vitro by targeting the capbinding complex eIF4F. Science. 2007;317:1764–7. doi: 10.1126/science.1146067. [DOI] [PubMed] [Google Scholar]
- 39.Wang B, Yanez A, Novina CD. MicroRNA-repressed mRNAs contain 40S but not 60S components. Proc Natl Acad Sci USA. 2008;105:5343–8. doi: 10.1073/pnas.0801102105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nottrott S, Simard MJ, Richter JD. Human let-7a miRNA blocks protein production on actively translating polyribosomes. Nat Struct Mol Biol. 2006;13:1108–14. doi: 10.1038/nsmb1173. [DOI] [PubMed] [Google Scholar]
- 41.Petersen CP, Bordeleau ME, Pelletier J, Sharp PA. Short RNAs repress translation after initiation in mammalian cells. Mol Cell. 2006;21:533–42. doi: 10.1016/j.molcel.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 42.Maroney PA, Yu Y, Fisher J, Nilsen TW. Evidence that microRNAs are associated with translating messenger RNAs in human cells. Nat Struct Mol Biol. 2006;13:1102–7. doi: 10.1038/nsmb1174. [DOI] [PubMed] [Google Scholar]
- 43.Wu L, Belasco JG. Let me count the ways: mechanisms of gene regulation by miRNAs and siRNAs. Mol Cell. 2008;29:1–7. doi: 10.1016/j.molcel.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 44.Wiesen Tomasi. Mol Immunol. in press. [Google Scholar]
- 45.Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE, et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature. 2004;432:226–30. doi: 10.1038/nature03076. [DOI] [PubMed] [Google Scholar]
- 46.Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, et al. A brain-specific microRNA regulates dendritic spine development. Nature. 2006;439:283–9. doi: 10.1038/nature04367. [DOI] [PubMed] [Google Scholar]
- 47.Lim LP, Lau NC, Weinstein EG, Abdelhakim A, Yekta S, Rhoades MW, et al. The microRNA of Caenorhabditis elegans. Genes Dev. 2003;17:991–1008. doi: 10.1101/gad.1074403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Farh KK, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, et al. The widespread impact of mammalian microRNAs on mRNA repression and evolution. Science. 2005;310:1817–21. doi: 10.1126/science.1121158. [DOI] [PubMed] [Google Scholar]
- 49.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 50.Bentwich I. Identifying human microRNAs. Curr Top Microbiol Immunol. 2008;320:257–69. doi: 10.1007/978-3-540-75157-1_12. [DOI] [PubMed] [Google Scholar]
- 51.Brameier M, Wiuf C. Ab initio identification of human microRNAs based on structure motifs. BMC Bioinform. 2007;8:478. doi: 10.1186/1471-2105-8-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dieci G, Fiorino G, Castelnuovo M, Teichmann M, Pagano A. The expanding RNA polymerase III transcriptome. Trends Genet. 2007;23:614–22. doi: 10.1016/j.tig.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 53.Doench JG, Sharp PA. Specificity of microRNA target selection in translational repression. Genes Dev. 2004;18:504–11. doi: 10.1101/gad.1184404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–9. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pillai RS, Bhattacharyya SN, Filipowicz W. Repression of protein synthesis by miRNAs: how many mechanisms? Trends Cell Biol. 2007;17:118–26. doi: 10.1016/j.tcb.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 56.Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14:1902–10. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Borel C, Gagnebin M, Gehrig C, Kriventseva EV, Zdobnov EM, Antonarakis SE. Mapping of small RNAs in the human ENCODE regions. Am J Hum Genet. 2008;82:971–81. doi: 10.1016/j.ajhg.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tyler DM, Okamura K, Chung WJ, Hagen JW, Berezikov E, Hannon GJ, et al. Functionally distinct regulatory RNAs generated by bidirectional transcription and processing of microRNA loci. Genes Dev. 2008;22:26–36. doi: 10.1101/gad.1615208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li SC, Tang P, Lin WC. Intronic microRNA: discovery and biological implications. DNA Cell Biol. 2007;26:195–207. doi: 10.1089/dna.2006.0558. [DOI] [PubMed] [Google Scholar]
- 60.Baskerville S, Bartel DP. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA. 2005;11:241–7. doi: 10.1261/rna.7240905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morlando M, Ballarino M, Gromak N, Pagano F, Bozzoni I, Proudfoot NJ. Primary microRNA transcripts are processed co-transcriptionally. Nat Struct Mol Biol. 2008 doi: 10.1038/nsmb.1475. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6:376–85. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- 63.Ruby JG, Jan CH, Bartel DP. Intronic microRNA precursors that bypass drosha processing. Nature. 2007;448:83–6. doi: 10.1038/nature05983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berezikov E, Chung W-J, Willis J, Cuppen E, Lai EC. Mammalian mirtron genes. Mol Cell. 2007;28:328–36. doi: 10.1016/j.molcel.2007.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454:56–61. doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of microRNA processing by Lin28. Science. 2008;320:97–100. doi: 10.1126/science.1154040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, Shiekhattar R, et al. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol. 2006;13:13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Faller M, Guo F. MicroRNA biogenesis: there’s more than one way to skin a cat. Biochim Biophys Acta. 2008 doi: 10.1016/j.bbagrm.2008.08.005. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou F, Ferguson J, Chang JT. Inter- and intra-combinatorial regulation by transcription factors and microRNAs. BMC Genomics. 2007;8:396. doi: 10.1186/1471-2164-8-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shalgi R, Lieber D, Oren M, Pilpel Y. Global and Local Architecture of the Mammalian microRNA-transcription factor regulatory network. PLoS Comput Biol. 2007;3:e131. doi: 10.1371/journal.pcbi.0030131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Petrocca F, Visone R, Onelli MR, Shah MH, Nicoloso MS, de Martino I, et al. E2F1-regulated microRNAs impair TGFβ-dependent cell-cycle arrest and apoptosis in gastric cancer. Cancer Cell. 2008;13:272–86. doi: 10.1016/j.ccr.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 72.Sylvestre Y, De Guire V, Querido E, Mukhopadhyay UK, Bourdeau V, Major F, et al. An E2F/miR-20a autoregulatory feedback loop. J Biol Chem. 2007;282:2135–43. doi: 10.1074/jbc.M608939200. [DOI] [PubMed] [Google Scholar]
- 73.Yang Y, Tantoso E, Chua GH, Yeo ZX, Ng FS, Wong ST, et al. In silico analysis of p53 using the p53 knowledgebase: mutations, polymorphisms, microRNAs and pathways. In Silico Biol. 2007;7:61–75. [PubMed] [Google Scholar]
- 74.Wenick AS, Hobert O. Genomic cis-regulatory architecture and trans-acting regulators of a single interneuron-specific gene battery in C. elegans. Dev Cell. 2004;6:757–70. doi: 10.1016/j.devcel.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 75.Johnston RJ, Hobert O. A microRNA controlling left/right neuronal asymmetry in Caenorhabditis elegans. Nature. 2003;426:845–9. doi: 10.1038/nature02255. [DOI] [PubMed] [Google Scholar]
- 76.Doench JG, Petersen CP, Sharp PA. siRNAs can function as miRNAs. Genes Dev. 2003;17:438–42. doi: 10.1101/gad.1064703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chang S, Johnston RJ, Jr, Frøkjaer-Jensen C, Lockery S, Hobert O. MicroRNAs act sequentially and asymmetrically to control chemosensory laterality in the nematode. Nature. 2004;430:785–9. doi: 10.1038/nature02752. [DOI] [PubMed] [Google Scholar]
- 78.Seggerson K, Tang L, Moss EG. Two genetic circuits repress the Caenorhabditis elegans heterochronic gene lin-28 after translation initiation. Dev Biol. 2002;243:215–25. doi: 10.1006/dbio.2001.0563. [DOI] [PubMed] [Google Scholar]
- 79.Vasudevan S, Tong Y, Steitz JA. Switching from repression to activation: microRNAs can up-regulate translation. Science. 2007;318:1931–4. doi: 10.1126/science.1149460. [DOI] [PubMed] [Google Scholar]
- 80.Place RF, Li LC, Pookot D, Noonan EJ, Dahiya R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci USA. 2008;105:1608–13. doi: 10.1073/pnas.0707594105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ørom UA, Nielsen FC, Lund AH. MicroRNA-10a binds the 5′UTR of ribosomal protein mRNAs and enhances their translation. Mol Cell. 2008;30:460–71. doi: 10.1016/j.molcel.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 82.Buchan JR, Parker R. Molecular biology. The two faces of miRNA. Science. 2007;318:1877–8. doi: 10.1126/science.1152623. [DOI] [PubMed] [Google Scholar]
- 83.Fabbri M. MicroRNAs and cancer epigenetics. Curr Opin Investig Drugs. 2008;9:583–90. [PubMed] [Google Scholar]
- 84.Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–43. doi: 10.1016/j.ccr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 85.Lujambio A, Ropero S, Ballestar E, Fraga MF, Cerrato C, Setien F, et al. Genetic unmasking of an epigenetically silenced micro-RNA in human cancer cells. Cancer Res. 2007;67:1424–9. doi: 10.1158/0008-5472.CAN-06-4218. [DOI] [PubMed] [Google Scholar]
- 86.Brueckner B, Stresemann C, Kuner R, Mund C, Musch T, Meister M, et al. The human let-7a-3 locus contains an epigenetically regulated microRNA gene with oncogenic function. Cancer Res. 2007;67:1419–23. doi: 10.1158/0008-5472.CAN-06-4074. [DOI] [PubMed] [Google Scholar]
- 87.Han L, Witmer PD, Casey E, Valle D, Sukumar S. DNA methylation regulates microRNA expression. Cancer Biol Ther. 2007;6:1284–8. doi: 10.4161/cbt.6.8.4486. [DOI] [PubMed] [Google Scholar]
- 88.Diederichs S, Haber DA. Sequence variations of microRNAs in human cancer: alterations in predicted secondary structure do not affect processing. Cancer Res. 2006;66:6097–104. doi: 10.1158/0008-5472.CAN-06-0537. [DOI] [PubMed] [Google Scholar]
- 89.Chuang JC, Jones PA. Epigenetics and microRNAs. Pediatr Res. 2007;61:24R–9R. doi: 10.1203/pdr.0b013e3180457684. [DOI] [PubMed] [Google Scholar]
- 90.Bagga S, Bracht J, Hunter S, Massirer K, Holtz J, Eachus R, et al. Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell. 2005;122:553–63. doi: 10.1016/j.cell.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 91.Zeng Y, Yi R, Cullen BR. MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proc Natl Acad Sci USA. 2003;100:9779–84. doi: 10.1073/pnas.1630797100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Engels BM, Hutvagner G. Principles and effects of microRNA-mediated post-transcriptional gene regulation. Oncogene. 2006;25:6163–9. doi: 10.1038/sj.onc.1209909. [DOI] [PubMed] [Google Scholar]
- 93.Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–73. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 94.Rana TM. Illuminating the silence: understanding the structure and function of small RNAs. Nat Rev Mol Cell Biol. 2007;8:23–36. doi: 10.1038/nrm2085. [DOI] [PubMed] [Google Scholar]
- 95.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–98. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 96.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 99.Sethupathy P, Corda B, Hatzigeorgiou AG. TarBase: a comprehensive database of experimentally supported animal microRNA targets. RNA. 2006;12:192–7. doi: 10.1261/rna.2239606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sethupathy P, Megraw M, Hatzigeorgiou AG. A guide through present computational approaches for the identification of mammalian microRNA targets. Nat Methods. 2006;3:881–6. doi: 10.1038/nmeth954. [DOI] [PubMed] [Google Scholar]
- 101.Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–83. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liang Z, Wu H, Reddy S, Zhu A, Wang S, Blevins D, et al. Blockade of invasion and metastasis of breast cancer cells via targeting CXCR4 with an artificial microRNA. Biochem Biophys Res Commun. 2007;363:542–6. doi: 10.1016/j.bbrc.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 103.Khabar KS, Young HA. Post-transcriptional control of the interferon system. Biochimie. 2007;89:761–9. doi: 10.1016/j.biochi.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. 2006;103:12481–6. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Frolik CA, Dart LL, Meyers CA, Smith DM, Sporn MB. Purification and initial characterization of a type beta transforming growth factor from human placenta. Proc Natl Acad Sci USA. 1983;80:3676–80. doi: 10.1073/pnas.80.12.3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hannon GJ, Beach D. P15INK4B is a potential effector of TGF-β-induced cell cycle arrest. Nature. 1994;371:257–61. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- 107.Datto MB, Li Y, Panus JF, Howe DJ, Xiong Y, Wang XF. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc Natl Acad Sci USA. 1995;92:5545–9. doi: 10.1073/pnas.92.12.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGFbeta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–86. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGF-β in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–89. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 110.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–4. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 111.Apte RN, Dotan S, Elkabets M, White MR, Reich E, Carmi Y, et al. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 2006;25:387–408. doi: 10.1007/s10555-006-9004-4. [DOI] [PubMed] [Google Scholar]
- 112.Haskill SG, Martin L, Van Le J, Morris A, Peace GJ, Jaffe C, et al. cDNA cloning of an intracellular form of the human interleukin-1 receptor antagonist associated with epithelium. Proc Natl Acad Sci USA. 1991;88:3681–5. doi: 10.1073/pnas.88.9.3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Plasterk RH. MicroRNAs in animal development. Cell. 2006;124:877–81. doi: 10.1016/j.cell.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 114.Stark A, Brennecke J, Bushati N, Russell RB, Cohen SM. Animal microRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell. 2005;123:1133–46. doi: 10.1016/j.cell.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 115.Hornstein E, Shomron N. Canalization of development by microRNAs. Nat Genet. 2006;38(Suppl):S20–4. doi: 10.1038/ng1803. [DOI] [PubMed] [Google Scholar]
- 116.Borenstein E, Ruppin E. Direct evolution of genetic robustness in microRNA. Proc Natl Acad Sci USA. 2006;103:6593–8. doi: 10.1073/pnas.0510600103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Okamura K, Phillips MD, Tyler DM, Duan H, Chou YT, Lai EC. The regulatory activity of microRNA* species has substantial influence on microRNA and 3′UTR evolution. Nat Struct Mol Biol. 2008;15:354–63. doi: 10.1038/nsmb.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Maher C, Stein L, Ware D. Evolution of Arabidopsis microRNA families through duplication events. Genome Res. 2006;16:510–9. doi: 10.1101/gr.4680506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chapman EJ, Carrington JC. Specialization and evolution of endogenous small RNA pathways. Nat Rev Genet. 2007;8:884–96. doi: 10.1038/nrg2179. [DOI] [PubMed] [Google Scholar]
- 120.Liu N, Okamura K, Tyler DM, Phillips MD, Chung WJ, Lai EC. The evolution and functional diversification of animal microRNA genes. Cell Res. 2008;18:985–96. doi: 10.1038/cr.2008.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Boyd SD. Everything you wanted to know about small RNA but were afraid to ask. Lab Invest. 2008;88:569–78. doi: 10.1038/labinvest.2008.32. [DOI] [PubMed] [Google Scholar]
- 122.Li LC. The multifaceted small RNAs. RNA Biol. 2008;5:61–4. doi: 10.4161/rna.5.2.5989. [DOI] [PubMed] [Google Scholar]
- 123.Hawkins PG, Morris KV. RNA and transcriptional modulation of gene expression. Cell Cycle. 2008;7:602–7. doi: 10.4161/cc.7.5.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jones WD. MicroRNA mutant turns back the evolutionary clock for fly olfaction. Bioessays. 2008;30:621–3. doi: 10.1002/bies.20780. [DOI] [PubMed] [Google Scholar]
- 125.Rajewsky N. microRNA target predictions in animals. Nat Genet. 2006;38(Suppl):S8–S13. doi: 10.1038/ng1798. [DOI] [PubMed] [Google Scholar]
- 126.Chen K, Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet. 2007;8:93–103. doi: 10.1038/nrg1990. [DOI] [PubMed] [Google Scholar]
- 127.Yekta S, Shih IH, Bartel DP. MicroRNA-directed cleavage of HOXB8 mRNA. Science. 2004;304:594–6. doi: 10.1126/science.1097434. [DOI] [PubMed] [Google Scholar]
- 128.Dehal P, Boore JL. Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol. 2005;3:e314. doi: 10.1371/journal.pbio.0030314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Heimberg AM, Sempere LF, Moy VN, Donoghue PC, Peterson KJ. MicroRNAs and the advent of vertebrate morphological complexity. Proc Natl Acad Sci USA. 2008;105:2946–50. doi: 10.1073/pnas.0712259105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sempere LF, Cole CN, McPeek MA, Peterson KJ. The phylogenetic distribution of metazoan microRNAs: insights into evolutionary complexity and constraint. J Exp Zoolog B Mol Dev Evol. 2006;306:575–88. doi: 10.1002/jez.b.21118. [DOI] [PubMed] [Google Scholar]
- 131.Hertel J, Lindemeyer M, Missal K, Fried C, Tanzer A, Flamm C, et al. Students of Bioinformatics Computer Labs 2004 and 2005. The expansion of the metazoan microRNA repertoire. BMC Genomics. 2006;7:25. doi: 10.1186/1471-2164-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chen K, Rajewsky N. Natural selection on human microRNA binding sites inferred from SNP data. Nat Genet. 2006;38:1452–6. doi: 10.1038/ng1910. [DOI] [PubMed] [Google Scholar]
- 133.Georges M, Coppieters W, Charlier C. Polymorphic miRNA-mediated gene regulation: contribution to phenotypic variation and disease. Curr Opin Genet Dev. 2007;17:166–76. doi: 10.1016/j.gde.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 134.Dinarello CA. Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am J Clin Nutr. 2006;83:447S–55S. doi: 10.1093/ajcn/83.2.447S. [DOI] [PubMed] [Google Scholar]
- 135.Gubler U, Chua AO, Schoenhaut DS, Dwyer CM, McComas W, Motyka R, et al. Coexpression of two distinct genes is required to generate secreted bioactive cytotoxic lymphocyte maturation factor. Proc Natl Acad Sci USA. 1991;88:4143–7. doi: 10.1073/pnas.88.10.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Trinchieri G. Interleukin-12: a cytokine at the interface of inflammation and immunity. Adv Immunol. 1998;70:83–243. doi: 10.1016/s0065-2776(08)60387-9. [DOI] [PubMed] [Google Scholar]
- 137.Trinchieri G. Interleukin-12: a proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Ann Rev Immunol. 1995;13:251–76. doi: 10.1146/annurev.iy.13.040195.001343. [DOI] [PubMed] [Google Scholar]
- 138.Lu J, Sun H, Wang X, Liu C, Xu X, Li F, et al. Interleukin-12 p40 promoter activity is regulated by the reversible acetylation mediated by HDAC1 and p300. Cytokine. 2005;31:46–51. doi: 10.1016/j.cyto.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 139.Lee SM, Seun Y, Chang L, Bruner V, Qian J, Indes J, et al. Decreased interleukin-12 [IL-12] from activated cord versus adult peripheral blood mononuclear cells and upregulation of interferon-γ, natural killer and lymphokine-activated killer activity by IL12 in cord blood mononuclear cells. Blood. 1996;88:945–54. [PubMed] [Google Scholar]
- 140.Goriely S, Vincart B, Stordeur P, Vekemans J, Willems F, Goldman M, et al. Deficient IL-12(p35) gene expression by dendritic cells derived from neonatal monocytes. J Immunol. 2001;166:2141–6. doi: 10.4049/jimmunol.166.3.2141. [DOI] [PubMed] [Google Scholar]
- 141.Zhao JJ, Lin J, Yang H, Kong W, He L, Ma X, et al. MicroRNA-221/222 negatively regulates ERα and associates with tamoxifen resistance in breast cancer. J Biol Chem. 2008 doi: 10.1074/jbc.M806041200. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 142.Axtell MJ, Bartel DP. Antiquity of microRNAs and their targets in land plants. Plant Cell. 2005;17:1658–73. doi: 10.1105/tpc.105.032185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Joung JG, Hwang KB, Nam JW, Kim SJ, Zhang BT. Discovery of microRNA-mRNA modules via population-based probabilistic learning. Bioinformatics. 2007;23:1141–7. doi: 10.1093/bioinformatics/btm045. [DOI] [PubMed] [Google Scholar]
- 144.Hitti E, Iakovleva T, Brook M, Deppenmeier S, Gruber AD, Radzioch D, et al. Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine-rich element. Mol Cell Biol. 2006;26:2399–407. doi: 10.1128/MCB.26.6.2399-2407.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Brook M, Tchen CR, Santalucia T, McIlrath J, Arthur JS, Saklatvala J, et al. Posttranslational regulation of tristetraprolin subcellular localization and protein stability by p38 mitogen-activated protein kinase and extracellular signal-regulated kinase pathways. Mol Cell Biol. 2006;26:2408–18. doi: 10.1128/MCB.26.6.2408-2418.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tchen CR, Brook M, Saklatvala J, Clark AR. The stability of tristetraprolin mRNA is regulated by mitogen-activated protein kinase p38 and by tristetraprolin itself. J Biol Chem. 2004;279:32393–400. doi: 10.1074/jbc.M402059200. [DOI] [PubMed] [Google Scholar]
- 147.Deleault KM, Skinner SJ, Brooks SA. Tristetraprolin regulates TNF-alpha mRNA stability via a proteasome dependent mechanism involving the combined action of the ERK and p38 pathways. Mol Immunol. 2008;45:13–24. doi: 10.1016/j.molimm.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 148.Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, et al. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell. 2005;120:623–34. doi: 10.1016/j.cell.2004.12.038. [DOI] [PubMed] [Google Scholar]
- 149.Lodish H, Zhou B, Liu G, Chen CZ. Micromanagement of the immune system by microRNAs. Nat Rev Immunol. 2008;8:120–30. doi: 10.1038/nri2252. [DOI] [PubMed] [Google Scholar]
- 150.Anderson P. Post-transcriptional control of cytokine production. Nat Immunol. 2008;9:353–9. doi: 10.1038/ni1584. [DOI] [PubMed] [Google Scholar]
- 151.Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W. Stress-induced reversal of microRNA repression and mRNA P-body localization in human cells. Cold Spring Harb Symp Quant Biol. 2006;71:513–21. doi: 10.1101/sqb.2006.71.038. [DOI] [PubMed] [Google Scholar]
- 152.Sheth U, Parker R. Targeting of aberrant mRNAs to cytoplasmic processing bodies. Cell. 2006;125:1095–109. doi: 10.1016/j.cell.2006.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Eulalio A, Behm-Ansmant I, Izaurralde E. P bodies: at the crossroads of post-transcriptional pathways. Nat Rev Mol Cell Biol. 2007;8:9–22. doi: 10.1038/nrm2080. [DOI] [PubMed] [Google Scholar]
- 154.Jakymiw A, Lian S, Eystathioy T, Li S, Satoh M, Hamel JC, et al. Disruption of GW bodies impairs mammalian RNA interference. Nat Cell Biol. 2005;7:1267–74. doi: 10.1038/ncb1334. [DOI] [PubMed] [Google Scholar]
- 155.Liu J, Rivas FV, Wohlschlegel J, Yates JR, 3rd, Parker R, Hannon GJ. A role for the P-body component GW182 in microRNA function. Nat Cell Biol. 2005;7:1261–6. doi: 10.1038/ncb1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Meister G, Landthaler M, Peters L, Chen PY, Urlaub H, Lührmann R, et al. Identification of novel argonaute-associated proteins. Curr Biol. 2005;15:2149–55. doi: 10.1016/j.cub.2005.10.048. [DOI] [PubMed] [Google Scholar]
- 157.Rehwinkel J, Behm-Ansmant I, Gatfield D, Izaurralde E. A crucial role for GW182 and the DCP1:DCP2 decapping complex in miRNA-mediated gene silencing. RNA. 2005;11:1640–7. doi: 10.1261/rna.2191905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Huang J, Liang Z, Yang B, Tian H, Ma J, Zhang H. Derepression of micro-RNA-mediated protein translation inhibition by apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3G [APOBEC3G] and its family members. J Biol Chem. 2007;282:33632–40. doi: 10.1074/jbc.M705116200. [DOI] [PubMed] [Google Scholar]
- 159.Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell. 2006;125:1111–24. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 160.Gallouzi IE, Steitz JA. Delineation of mRNA export pathways by the use of cell-permeable peptides. Science. 2001;294:1895–901. doi: 10.1126/science.1064693. [DOI] [PubMed] [Google Scholar]
- 161.Sutton MA, Schuman EM. Local translational control in dendrites and its role in long-term synaptic plasticity. J Neurobiol. 2005;64:116–31. doi: 10.1002/neu.20152. [DOI] [PubMed] [Google Scholar]
- 162.Meng F, Henson R, Wehbe-Janek H, Smith H, Ueno Y, Patel T. The MicroRNA let-7a modulates interleukin-6-dependent STAT-3 survival signaling in malignant human cholangiocytes. J Biol Chem. 2007;282:8256–64. doi: 10.1074/jbc.M607712200. [DOI] [PubMed] [Google Scholar]
- 163.Calin GA, Croce CM. MicroRNA-cancer connection: the beginning of a new tale. Cancer Res. 2006;66:7390–4. doi: 10.1158/0008-5472.CAN-06-0800. [DOI] [PubMed] [Google Scholar]
- 164.Hussain PS, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer. 2007;121:2373–80. doi: 10.1002/ijc.23173. [DOI] [PubMed] [Google Scholar]
- 165.Moschos SA, Williams AE, Perry MM, Birrell MA, Belvisi MG, Lindsay MA. Expression profiling in vivo demonstrates rapid changes in lung microRNA levels following lipopolysaccharide-induced inflammation but not in the anti-inflammatory action of glucocorticoids. BMC Genomics. 2007;17:240. doi: 10.1186/1471-2164-8-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Clurman BE, Hayward WS. Multiple proto-oncogene activations in avian leukosis virus-induced lymphomas: evidence for stage-specific events. Mol Cell Biol. 1989;9:2657–64. doi: 10.1128/mcb.9.6.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tam W, Ben-Yehuda D, Hayward WS. bic, a novel gene activated by proviral insertions in avian leukosis virus-induced lymphomas, is likely to function through its noncoding RNA. Mol Cell Biol. 1997;17:1490–502. doi: 10.1128/mcb.17.3.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci USA. 2005;102:3627–32. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA. 2006;103:2257–61. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Jiang J, Lee EJ, Schmittgen TD. Increased expression of microRNA-155 in Epstein-Barr virus transformed lymphoblastoid cell lines. Genes Chromosomes Cancer. 2006;45:103–6. doi: 10.1002/gcc.20264. [DOI] [PubMed] [Google Scholar]
- 171.Sonkoly E, Wei T, Janson PC, Sääf A, Lundeberg L, Tengvall-Linder M, et al. MicroRNAs: novel regulators involved in the pathogenesis of psoriasis? PLoS ONE. 2007;2:e610. doi: 10.1371/journal.pone.0000610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sonkoly E, Ståhle M, Pivarcsi A. MicroRNAs: novel regulators in skin inflammation. Clin Exp Dermatol. 2008;33:312–5. doi: 10.1111/j.1365-2230.2008.02804.x. [DOI] [PubMed] [Google Scholar]
- 173.Dudley DJ. Pre-term labor: an intra-uterine inflammatory response syndrome? J Reprod Immunol. 1997;36:93–109. doi: 10.1016/s0165-0378(97)00065-x. [DOI] [PubMed] [Google Scholar]
- 174.Marvin KW, Keelan JA, Eykholt RL, Sato TA, Mitchell MD. Use of cDNA arrays to generate differential expression profiles for inflammatory genes in human gestational membranes delivered at term and preterm. Mol Hum Reprod. 2002;8:399–408. doi: 10.1093/molehr/8.4.399. [DOI] [PubMed] [Google Scholar]
- 175.Montenegro D, Romero R, Pineles BL, Tarca AL, Kim YM, Draghici S, et al. Differential expression of microRNAs with progression of gestation and inflammation in the human chorioamniotic membranes. Am J Obstet Gynecol. 2007;197:289.e1–289.e6. doi: 10.1016/j.ajog.2007.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Balkwill F. Tumor necrosis factor or tumor promoting factor? Cytokine Growth Factor Rev. 2002;13:135–41. doi: 10.1016/s1359-6101(01)00020-x. [DOI] [PubMed] [Google Scholar]
- 177.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Karin M, Greten FR. NF-κB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–59. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 179.Pollard JW. Tumour-educated macrophages promote tumor progression and metastasis. Nat Rev Cancer. 2004;4:71–8. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 180.Hudson JD, Shoaibi MA, Maestro R, Carnero A, Hannon GJ, Beach DH. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J Exp Med. 1999;190:1375–82. doi: 10.1084/jem.190.10.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Frassanito MA, Cusmai A, Iodice G, Dammacco F. Autocrine interleukin-6 production and highly malignant multiple myeloma: relation with resistance to drug-induced apoptosis. Blood. 2001;97:483–9. doi: 10.1182/blood.v97.2.483. [DOI] [PubMed] [Google Scholar]
- 182.Becker G, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A, et al. TGF-β suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004;21:491–501. doi: 10.1016/j.immuni.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 183.Vardam TD, Zhou L, Appenheimer MM, Chen Q, Wang WC, Baumann H, et al. Regulation of a lymphocyte-endothelial-IL-6 trans-signaling axis by fever-range thermal stress: hot spot of immune surveillance. Cytokine. 2007;39:84–96. doi: 10.1016/j.cyto.2007.07.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Conaco C, Otto S, Han JJ, Mandel G. Reciprocal actions of REST and a microRNA promote neuronal identity. Proc Natl Acad Sci USA. 2006;103:2422–7. doi: 10.1073/pnas.0511041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wu J, Xie X. Comparative sequence analysis reveals an intricate network among REST, CREB and miRNA in mediating neuronal gene expression. Genome Biol. 2006;7:R85. doi: 10.1186/gb-2006-7-9-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64:3753–6. doi: 10.1158/0008-5472.CAN-04-0637. [DOI] [PubMed] [Google Scholar]
- 187.Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–47. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 188.Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007;21:1025–30. doi: 10.1101/gad.1540407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Smyth MJ, Cretney E, Kershaw MH, Hayakawa Y. Cytokines in cancer immunity and immunotherapy. Immunol Rev. 2004;202:275–93. doi: 10.1111/j.0105-2896.2004.00199.x. [DOI] [PubMed] [Google Scholar]
- 190.Anderson P. Post-transcriptional regulation of tumour necrosis factor alpha production. Ann Rheum Dis. 2000;59(Suppl):i3–5. doi: 10.1136/ard.59.suppl_1.i3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10:387–98. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- 192.Li H, Lin X. Positive and negative signaling components involved in TNFα-induced NF-κB activation. Cytokine. 2008;41:1–8. doi: 10.1016/j.cyto.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 193.Karin M. Nuclear factor-κB in cancer development and progression. Nature. 2006;441:431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 194.Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, et al. IKKβ suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130:440–55. doi: 10.1016/j.cell.2007.05.058. [DOI] [PubMed] [Google Scholar]
- 195.Lee YS, Dutta A. MicroRNAs in Cancer. Annu Rev Pathol. 2008 doi: 10.1146/annurev.pathol.4.110807.092222. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Schwab R, Ossowski S, Riester M, Warthmann N, Weigel D. Highly specific gene silencing by artificial microRNAs in Arabidopsis. Plant Cell. 2006;18:1121–33. doi: 10.1105/tpc.105.039834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Son J, Uchil PD, Kim YB, Shankar P, Kumar P, Lee SK. Effective suppression of HIV-1 by artificial bispecific miRNA targeting conserved sequences with tolerance for wobble base-pairing. Biochem Biophys Res Commun. 2008;374:214–8. doi: 10.1016/j.bbrc.2008.06.125. [DOI] [PubMed] [Google Scholar]
- 198.Wu X, Fan J, Wang X, Zhou J, Qiu S, Yu Y, et al. Downregulation of CCR1 inhibits human hepatocellular carcinoma cell invasion. Biochem Biophys Res Commun. 2007;355:866–71. doi: 10.1016/j.bbrc.2007.01.199. [DOI] [PubMed] [Google Scholar]